Abstract
Water samples were collected and analyzed for conductivity, pH, temperature and trihalomethanes (THMs) during the fall of 2014 at two monitored municipal drinking water source ponds. Both spot (or grab) and time weighted average (TWA) sampling methods were assessed over the same two day sampling time period. For spot sampling, replicate samples were taken at each site and analyzed within 12 h of sampling by both Headspace (HS)- and direct (DI)- solid phase microextraction (SPME) sampling/extraction methods followed by Gas Chromatography/Mass Spectrometry (GC/MS). For TWA, a two day passive on-site TWA sampling was carried out at the same sampling points in the ponds. All SPME sampling methods undertaken used a 65-µm PDMS/DVB SPME fiber, which was found optimal for THM sampling. Sampling conditions were optimized in the laboratory using calibration standards of chloroform, bromoform, bromodichloromethane, dibromochloromethane, 1,2-dibromoethane and 1,2-dichloroethane, prepared in aqueous solutions from analytical grade samples. Calibration curves for all methods with R2 values ranging from 0.985–0.998 (N = 5) over the quantitation linear range of 3–800 ppb were achieved. The different sampling methods were compared for quantification of the water samples, and results showed that DI- and TWA- sampling methods gave better data and analytical metrics. Addition of 10% wt./vol. of (NH4)2SO4 salt to the sampling vial was found to aid extraction of THMs by increasing GC peaks areas by about 10%, which resulted in lower detection limits for all techniques studied. However, for on-site TWA analysis of THMs in natural waters, the calibration standard(s) ionic strength conditions, must be carefully matched to natural water conditions to properly quantitate THM concentrations. The data obtained from the TWA method may better reflect actual natural water conditions.
1. Introduction
Trihalomethanes (THMs) are normally considered to be of primary concern when naturally occurring organic compounds react with disinfection compounds in a water treatment process. In such cases, the compounds are referred to as disinfection byproducts (DBPs), of which the most prevalent are haloacetic acids (HAAs), haloacetonitriles (HANs), haloketones, chloral hydrate and chloropicrin [1]. However, THMs and substituted haloalkanes can be naturally produced in soils through the interaction of hydrogen peroxide decay products, if iron (III) is present [2], and then transported by rainfall or runoff, to the water column whereby they may continue to react to form THM precursors in natural waters [3]. Further, THMs can be formed naturally in the water column itself, through the interaction of natural bromide and/or chloride concentrations, especially when high dissolved organic matter (DOM) content is present. In North America, organic input from leaf foliage and other decay materials entering surface waters can be significant during the fall of the year, where THM concentrations have been found as high as 300 μg·L−1 (ppb) without disinfection treatment [4]. In unpolluted natural waters, bromine and the bromide ion concentrations are generally less the 1 ppm, and about 100 ppb in rainwater [5,6]. Typical chloride concentrations in surface waters are in the 10–30 ppm range, but due to component volatility, concentration variance from one water system to another is quite possible. Hydrogen peroxide has been found at concentrations from 1–30 ppb in surface waters. For rainwater, the hydrogen peroxide concentration can vary from 50 ppb to low parts per trillion (ppt) depending upon amount of sunlight exposure [6,7].
Brominated THMs are produced through bromination of humic acids in the same way chlorinated THMs are produced; usually from the reaction of HOBr or HOCl (respectively) with acetyl groups in humic acids, which are contained in breakdown products of natural organic matter like lignin [8]. The bromide ion can undergo a one-electron transfer from chlorine to form aqueous bromine, as demonstrated in Equations (1)–(3).
Cl2(aq) + 2Br−(aq) → 2Cl−(aq) + Br2(aq)
H2O + Br2(aq) ↔ HOBr(aq) + HBr(aq)
H2O + HBr(aq) ↔ BrO−(aq) + 3H+(aq) + 3e′
The bromite ion being more reactive than the chlorite, suggests that hypobromite will be a better brominating reagent than hypochlorite for chlorination [9]. Hence, brominated THM production might be preferred over their chloro adduct in unpolluted waters [10]. The exact reaction mechanism is not understood, mainly due to the complexity of the DOM involved. Use of humic model compounds, interpreted from fractions that have been isolated through ion exchange separations of humic acids [10], suggest that the reaction may follow the haloform reaction route, as demonstrated by 3,5-hydrobenzoic acid under basic conditions of pH > 6.8 in Figure 1 [9,11].
The original THM classification for bromodichloromethane, bromoform, chlorodibromomethane and chloroform has been extended to include a variety of different substituted halogenated compounds. Given the variety of THM precursors possible, the type of THM production may be altered by changes in pH; high pH favors THM adducts produced by way of Figure 1, low pH favors halonitrile THM adduct production. An increase in temperature also tends to increase the rate of THM production [12].
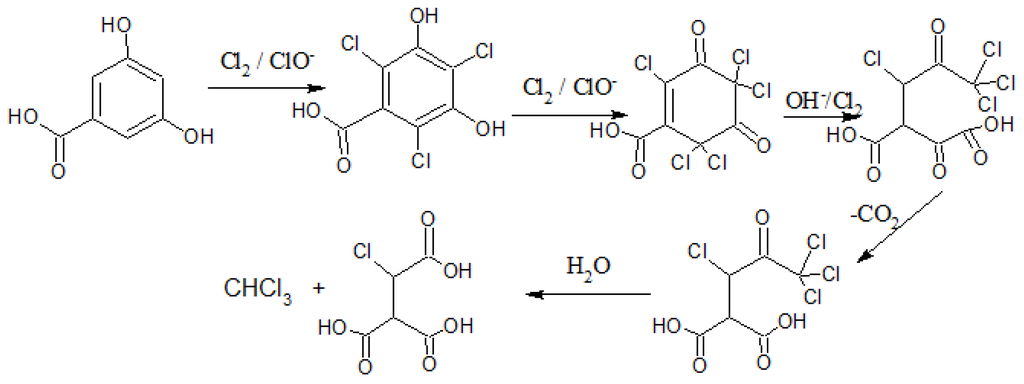
Figure 1.
Example of a possible haloform reaction of 3,5-hydroxybenzoic acid at pH > 7.
Figure 1.
Example of a possible haloform reaction of 3,5-hydroxybenzoic acid at pH > 7.
The interest in THMs in the water column are generally due to their deleterious health aspects, where many of the THMs are suspected carcinogens belonging to Group B2 or C of the International Agency for Research on Cancer (IARC) [13]. Thus it is necessary to monitor drinking water sources for THM contamination. The US Environmental Protection Agency (EPA) and European Union (EU) (respectively) have set maximum allowable (drinking water) limits for total THMs at 80 and 100 ppb (μg·L−1), where >90 ppb can have adverse effects on liver and kidney functions in humans [14,15]. However, prolonged THM intake concentration (exposure) limits have not been agreed upon. Currently, the Newfoundland and Labrador provincial government of Canada monitors 473 public water supplies in the island part of the province, where THM ranges from negligible to 300 ppb (μg·L−1) [3]. Current analytical techniques include: LLE, purge-and-trap and photometric detection methods [15], as in EPA501.2 (using liquid–liquid extraction (LLE)), and THM monitoring methods, as in EPA501.3 (using gas chromatography-mass spectrometry (GC/MS)), are typical. Samples must be analyzed within 10 days of sample collection [5] and, hence, it is important to utilize detection methods that are fast and robust for representative determination of THM concentrations in the natural waters.
In most occasions, spot (or grab) sampling is usually employed to sample natural waters, where the samples are analyzed in batch analysis later in the lab. Although spot sampling with subsequent THM extraction and analysis can give reasonable indicative data, the technique acquires information of an analyte concentration only at a particular point in time at a specific location. Although a large amount of spot water samples can be obtained, the extraction analysis cannot account for the effects of physical conditions such as episodic contamination or gradient flow. Comparatively, time weight averaging (TWA) sampling in a passive mode may be utilized to extract small amounts of analytes over longer time intervals, which can represent long-term concentrations more accurately than spot sampling.
The literature has shown that use of solid phase microextraction (SPME) technique affords the possibility of sampling THM analytes by either method (spot [16,17,18] and TWA [19,20,21]) with easy incorporation to subsequent GC/MS analysis. Work by Stack [22] and Valencia [23] have shown the utility of headspace- (HS-) SPME with GC/MS for measurement of THMs from swimming pools and chlorinated drinking waters. SPME is based upon analytes in the sample interacting with a selected polymeric fiber in a multiphase equilibration process, with analytes migrating along the phases until equilibrium is reached. This method of sampling can be used to extract analytes from a sealed vial by HS- or by direct immersion- (DI-) SPME sample extraction. As such, the mass of an analyte extracted by the SPME device fiber is related to the overall equilibrium of the analyte in the phases. Because the total mass of an analyte should be constant throughout the extraction, the mass balance formula for the analyte is given in Equation (4) [24].
where Co is the initial concentration of analyte in the sample, Cf, Ch, and Cs are the equilibrium concentrations of the analyte in the fiber coating, headspace, and sample, respectively, and where Vf, Vh, and Vs are the volumes of the fiber film coating, headspace, and sample.
For HS-SPME extraction, if the coating/gas distribution constant is taken to be Kfh = Cf/Ch, and the gas/sample matrix distribution constant is taken to be Khs = Ch/Cs, the mass of the analyte absorbed by the coating can be expressed [24] in Equation (5).
For DI-SPME extraction, there will be a single distribution constant to describe the equilibrium between the fiber and the sample, Kfs. As such, an equation to describe the amount of analyte adsorbed onto the fiber, n, shown in Equation (6):
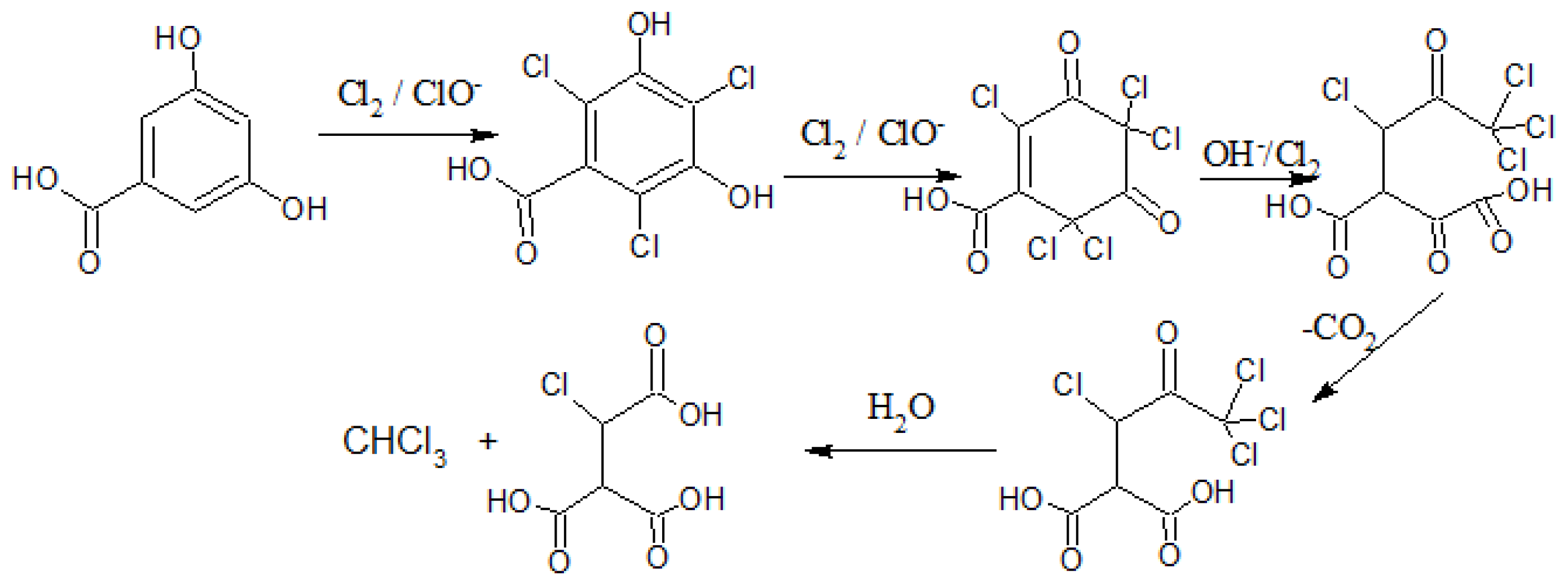
For TWA-SPME sampling, the coated fiber is retracted into the needle casing at a known distance from the end of the sheath during the sampling period. Under such conditions, the SPME device can be used as a TWA diffusive sampler (see Figure 2) whereby the sampler is allowed to stay in contact with the water [21,24]. During the process of diffusion, a linear concentration profile exists across the sampling distance Z, as shown in Figure 2b and where the sampling rate of the device, SR, can be computed using Equation (7) [24]:
where D is the diffusion coefficient of the analyte, A is the needle opening’s surface area, and Z is the diffusion path length, that is, the distance the fiber is retracted into the needle. The amount of analyte loaded onto the fiber coating, n, during the sampling time, t, can be found using Equation (8):
where C(t) is a time dependent concentration of the analyte at the needle opening. Using Equation (8), the sampling rate for a particular compound can be experimentally determined by Equation (9), using a known concentration and the SPME fiber retraction distance from the casing end.
Figure 2.
Diffusion volume (z) of a solid phase microextraction (SPME) needle and fiber. (a) Showing possible retraction distance of the fiber within the needle casing; (b) Enlargement of the resulting diffusion volume for Diffusion time weighted average (TWA) analysis.
Figure 2.
Diffusion volume (z) of a solid phase microextraction (SPME) needle and fiber. (a) Showing possible retraction distance of the fiber within the needle casing; (b) Enlargement of the resulting diffusion volume for Diffusion time weighted average (TWA) analysis.
The SR for a compound can be used to quantify unknown concentrations by simply finding the amount of analyte n on the fiber over time t, using Equation (10) [21]:
A current review [21] has discussed the utility of TWA and similar techniques and cites analyses of a variety of volatile organics [19] and polyaromatic hydrocarbons (PAHs) [20] from aqueous matrices. This paper explores the sampling, extraction and analysis of THMs from non-disinfected water samples. It compares spot sampling by both direct (DI-) and headspace (HS-) SPME and extraction with subsequent GC/MS analysis, to passive TWA sampling GC/MS methods using similar SPME fibers. Based on calibration standards and real sample analysis, the approaches to the analyses of volatile THM adducts are compared and discussed.
2. Experimental Section
2.1. Materials
The THM reference standards (with purity): bromoform (96%), bromodichloromethane (98%), and dibromochloromethane (98%) were purchased from Sigma-Aldrich (Oakville, ON, Canada) and chloroform (99.9%), 1,2-dibromoethane (99%), and 1,2-dichloroethane (99.5%) were supplied by Fisher Scientific (Nepean, ON, Canada). Ultrapure water, generated by a Barnstead water purifier (Dubuque, IA, USA), was used as the solvent for all standards and also as the solvent for the continuous flow system. Co-solvents like methanol were of Analar grade and used without further purification and obtained from Fisher Scientific, Nepean, ON, Canada. SPME fibers: 7 and 100 µm poly(dimethylsiloxane), (PDMS); 75 and 85 µm Carboxen(CAR)/PDMS; 65 µm PDMS/poly(divinylbenzene) (DVB); 65 µm Carbowax(CW)/DVB; 50 µm DVB/CAR/PDMS were purchased from Supelco, Belefonte, PA, USA and were conditioned under nitrogen as per manufacturer instructions. Fibers were further conditioned at 250 °C for 15 min prior to an analysis. Analytical analysis utilized screw top Teflon-Teflon coated septum-sealed capped amber HS-vials (Canadian Scientific, Peterborough, ON, Canada).
2.2. Sample Descriptions and Sampling Area
Water and TWA sampling were carried out at the two municipal surface water sites for the city of Corner Brook, NL, Canada: Trout Pond (Lat: 48.92567°N; Long: −57.89312°E) and Three Mile Pond (Lat: 48.93987°N; Long: −58.00831°E). The sample sites selected, had been monitored for THMs over a number of years, where values varied between 110 and 160 ppb, and hence THM detection could be expected. Trout Pond samples were taken from left side of the dock extending in the south quadrant of the pond, where there were trees, shrubs, and short grass on the bank. The bottom topography was formed from sand, pebble, and cobble sized material. Three Mile Pond samples were taken from the north-northeast section of the pond in shallow water, where there was a high density of trees and short grass on bank. The bottom topography was silt with an intermediate density of decaying vegetation. All samples were collected at the same depth (1/3 m below water surface) in triplicate and were sampled over a two days period during the fall of the year. Samples were analyzed on site for conductivity, dissolved solids, pH, temperature, and within 12 h of collection for trihalomethanes (THMs). For spot sampling, triplicate real water samples were collected each day over the two days period in the two ponds during the fall of the year. For TWA sampling, triplicate SPME fibers were positioned and left attached on a stand for a two days period that was placed in the pond at the same depth as was used for spot sampling.
2.3. Instrumentation
A Saturn 2000 and/or Saturn Star 3 GC/ion trap MS (Varian, Mississauga, ON, Canada) instruments with identical setups were used for analysis, where both ionization and mass analysis occur in the same chamber. The GCs were equipped with a 1079 split/splitless injector with SPME glass inserts (Varian) and a 5%-diphenyl-95%-dimethylpolysiloxane HP-5MS fused-silica capillary column (Agilent, Palo Alto, CA, USA) with dimensions: 30 m × 0.25 mm ID with a 0.25 µm phase. UHP helium carrier gas was set to a flow of 0.5 mL·min−1. THM standards and SPME fiber extractions were injected into the GC at 190 °C under 10 psi constant pressure and used an experimentally optimized column temperature program: 40 °C, held for 4 min, to 160 °C at a rate of 30 °C·min−1, and held for 4 min, then ramped to 210 °C at a rate of 50 °C·min−1, and held for 1 min, giving a total run time of 10 min. For the mass spectrometer (MS), the electron multiplier was operating at 2000 V and samples were analyzed by electron impact (70 eV) over an acquired mass range of 40–650 m/z. The transfer line, source, and ion trap temperatures were 170 °C, 100 °C and 120 °C, respectively. Quantification of the THMs from real water samples were performed by selected ion monitoring with a 100-ms dwell time, for limit of detection (LOD) and calibration studies. Selected ions used were m/z: bromoform (Mol. Wt. 252.75) 252, 173, 96; bromodichloromethane (Mol. Wt. 163.83); 163, 128, 93, 84; dibromochloromethane (Mol. Wt. 208.29); 208, 172, 128, 93; 1,2-dibromoethane (Mol. Wt. 187.86); 187, 108, 106; chloroform (Mol. Wt. 119.38): 119, 87, 85; and 1,2-dichloroethane (Mol. Wt. 98.96) 98, 63, 61.
2.4. SPME Extractions
Headspace (HS-SPME) analysis was conducted using seven SPME fibers, in order to assess best THM compound affinities to the different fiber compositions. HS-SPME analysis was conducted using a 40 mL amber glass vial that contained a total volume of 32 mL (of a 2 ppm THM standard and a stir bar) that was sealed with a screw top Teflon-Teflon coated septum cap and which was placed on a stirring mantle. The different fibers were tested toward extraction of target THM standards, under a variety of temperatures (25, 30, 40, 50, 60 and 70 °C), stirring rates (500, 600, 700, 750, 800 and 900 rpm), extraction times of (1, 2, 5, 8, 10, 15, 20, 25, 30, 40 min.) and with and without salt (10% wt./vol. of (NH4)2SO4) to determine optimal conditions. Ascorbic acid was used to re-adjust the pH back to the real sample conditions. Before and after each run, the SPME fiber was conditioned for 2 min at 220 °C. No carry over was noted after such conditioning. The extraction of all THM solutions (standards, spiked THM solutions) for calibration curve to cover the range (0.02, 0.05, 0.1, 0.2, 0.5, 1, 2 and 5 ppm (mg·L−1)) and comparison determinations were performed using the optimized conditions found from the above tests and were subsequently analyzed by optimized GC/MS method. For HS-SPME extractions, the fiber was placed in the sample headspace and for DI-SPME extractions, the fiber was placed directly into the stirred liquid.
2.5. SPME TWA Extractions
A continuous flow system (Figure 3) was first used to analyze and to optimize THM standard solution conditions for TWA sampling. The system was composed of a mixing compartment, and a series of connecting glassware components with sampling ports positioned to hold SPME fibers. The glassware was connected together using heat shrink tubing. The flow system also had an inlet and an exit port whereby a solvent would run through. The flow was controlled using a Dynamax model SD-200 solvent delivery system (Varian Instruments, Mississauga, ON, Canada) connected to the inlet port, and the flow rate was set to 10.00 ± 0.1 mL·min−1 at 25 °C ± 1.0 °C. Sample flowed out of the exit port through a loop and back into in the sampling reservoir. Sampling of the flow system was achieved through the use of six sampling ports in the glassware where the six SPME fibers could be inserted and tested simultaneously. The fiber needles had the springs removed in order to enable the fibers to be extended or retracted into the needle casing to specific distances, and held there for the duration of the sampling period. The continuous flow system could be connected to separate reservoirs that contained different mixes of THM standards. Sampling rates of 1, 2, 3, and 5 days, different fiber retracted positions (5, 10, 15, 20, 25 and 30 mm for different diffusion sampling lengths) and where the fiber was fully extended were studied and optimized to give best results for the six THM standards. A 1-ppm (mg·L−1) concentration was used to assess and optimize conditions. A concentration range of 0.02, 0.05, 0.1, 0.2, 0.5, 1, 2 and 5 ppm of the six THM standards were used to establish the calibration curve for TWA analysis.
Based upon optimized conditions found from the continuous flow system results, TWA sampling of THMs from real water samples was undertaken in two ways: (1) by (triplicate) TWA-SPME sampling of a collected water sample from sealed 40 mL vials using similar optimized HS-SPME conditions but with the optimized TWA sampling time and fiber retraction distance; and (2) by (triplicate) on-site sampling. For (2), each of the SPME fibers was positioned at the optimized retraction distance into their SPME needle casing (see Figure 2a). Triplicate fibers were then attached to a stand and placed into the pond. In both ponds, the fiber stand was positioned at the same pond point and water depth (1/3 m below water surface) where other water samples (1) were collected. After the optimized extraction time (2 days) the fiber stand was then retrieved and the fibers were taken for subsequent GC/MS analysis under optimized GC/MS conditions.

Figure 3.
A home built continuous flow system used TWA optimization.
Figure 3.
A home built continuous flow system used TWA optimization.

2.6. Zero Sink Test to Determine TWA Fiber Extraction Stability for THMs
To test the fiber ability to retain THM analytes during a TWA analysis, a zero sink test was performed. The SPME fiber was placed at the optimized distance in the needle casing. Then TWA headspace sampling fiber performance was quantified by comparing the GC peak areas obtained by: (1) TWA headspace sampling carried out for 90 min and then analyzed by GC/MS; to (2) TWA headspace sampling carried out for three 30 min intervals of sampling (90 min total) separated by 10 min intervals of TWA headspace sampling of a vial that contained only nitrogen gas, after which the analysis was run. Comparing the peak areas obtained from (1) and (2) results showed minimal loss (<2% peak areas for all six target THMs), suggesting that the THM-fiber extraction was stable. For such cases, the fiber is said to act as a zero sink for THMs compounds.
2.7. Method Validation
In this study, for each of the methods, the optimized parameters to extract THM components were evaluated to assess: calibration linearity, reproducibility and limits of detection and quantification. THM standards were prepared by serial dilution to give a set of aqueous standards for analysis that included concentrations ranging lower and higher of the required analytical calibration range (3 to 800 µg·L−1). For precision, triplicate analysis on each sample for each method was carried out to estimate component stability on the fiber and reproducibility of the HS-sampling scheme employed. The analysis was repeated randomly over the sampling period and the results were compared, via GC peak areas. Periodically, a spiked standard (10 mg·mL−1 of 1–2, dibromopropane) was used as an internal calibration marker to assess any bias in runs or methods. No variation (<2%) or loss of any of the 6 THM component peak areas from the fiber were noted. See Table 1. In addition, for TWA studies, a zero-sink test was performed to check for THM component loss from the fiber after absorption and before injection, though negligible change was found. Results show that acceptable correlation coefficients (R2: 0.966–0.998) were obtained using the 65 µm PDMS/DVB fiber for particular THM components covering a linear range of over two orders of magnitude. A linear regression method of least squares was used to ascertain linearity. From Table 2 it is seen that overall the calibration curves achieved for the DI-SPME method gave better R2 values and slightly lower detection limits than for HS- or TWA- methods. However, for all methods the R2 obtained using a 10-min (8 min extraction and 2 min desorption) period were satisfactory for quantitation at occurring levels and were similar to those obtained by EPA501.3 method [15]. The LOD and limit of quantitation (LOQ) were calculated from the S/N, where LOD = 3 × S/N, using a 10 µg/L standards [16]. The LODs vary for individual THMs and methods but generally fall within a 2.6–5.1 µg·L−1, which are low enough to detect THMs in natural water samples, (Table 1). The results obtained for total THMs were similar to Newfoundland and Labrador, Canada provincial data from previous years for the fresh water ponds [4].

Table 1.
Analysis of Trout and 3 Mile Ponds by Direct (DI)-, headsapace (HS)-, time weighted averaging (TWA)- Vial and Headspace- time weighted averaging (HS-TWA) On-site solid phase microextraction-gas chromatography/ mass spectrometry (SPME-GC/MS) Sampling of THM Standards.
| Detected THMs (µg·L−1) ± 1 | GC Retention Times (min) | DI-, HS- TWA-Vial Sampling a | HS-TWA Vial Sampling b | HS-TWA on-Site Sampling c | |||
|---|---|---|---|---|---|---|---|
| Trout Pond | 3 Mile Pond | Trout Pond | 3 Mile Pond | Trout Pond | 3 Mile Pond | ||
| Chloroform | 1.88 | 52 | 48 | 60 | 55 | 58 | 52 |
| Bromoform | 6.01 | 26 | 24 | 37 | 33 | 37 | 29 |
| Bromodichloromethane | 2.92 | 19 | 16 | 25 | 19 | 26 | 20 |
| Dibromochloromethane | 4.59 | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. |
| 1,2-dibromoethane | 6.26 | 14 | 10 | 25 | 17 | 25 | 21 |
| 1,2-dichloroethane | 2.21 | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. |
| Water Parameters | |||||||
| Total THM std. Detected | 111 | 98 | 147 | 120 | 146 | 122 | |
| Temp (°C) ± 0.5 °C | 25 | 25 | 25 | 25 | 9, 10 d | 12, 12 d | |
| pH ± 0.1 | 6.4 | 6.7 | 6.4 | 6.7 | 6.4 | 6.7 | |
| Conductivity (µS) ± 0.5 | -- e | -- e | 62 | 66 | 62 | 66 | |
| Dissolved solids (mg·L−1) ± 1 | -- e | -- e | 29 | 30 | 29 | 30 | |
N = 3. Results based upon calibration curves constructed by the appropriate SPME method for the extraction of THMS standards from sealed vials and followed by GC/MS analysis. For HS-TWA methods, the fiber was detracted 20 mm into the fiber casing and sampling was undertaken for two days (optimum conditions). a DI-, HS- and TWA-sampling methods with salt added under optimized conditions (<2% differences detected between these methods); b HS-TWA from sealed vial with no salt added but otherwise under optimized conditions; c HS-TWA sampling was positioned at the same depth (1/3 m below water surface) where water samples were collected for spot sampling; d Temperature data taken from City of Corner Brook, NL in 2013; e Sample was adjusted with 10% wt./vol. of (NH4)2SO4 salt with additional ascorbic acid for pH adjustment to the vial before extraction, hence not determined. n.d. = not detected. Note: Newfoundland and Labrador, Canada government total THM ppb data (by LLE-UV/VIS) was 158.15 ppb and 117.07 ppb for Trout Pond and 3 Mile Pond, respectively, in Winter 2013. [4].
Table 2.
Comparison of Headspace (HS-), Direct (DI-) and Headspace-Time Weighted Averaging (HS-TWA) SPME-GC/MS Sampling for Target THM Standards from Aqueous Samples.
| Compound | LOQ (µg·L−1) | DL (µg·L−1) | Calibration Equation | R2 |
|---|---|---|---|---|
| HS-SPME Extraction a | ||||
| Chloroform | 12 | 3.6 | y = 20.14x + 2.8 | 0.988 |
| Bromoform | 12 | 3.7 | y = 21.29x + 2.46 | 0.989 |
| Bromodichloromethane | 12 | 3.7 | y = 30.53x + 0.06 | 0.975 |
| Dibromochloromethane | 12 | 3.5 | y = 131x + 1109 | 0.993 |
| 1,2-dibromoethane | 13 | 3.8 | y = 30.54x + 0.4 | 0.998 |
| 1,2-dichloroethane | 13 | 3.9 | y = 20.27x + 0.4 | 0.966 |
| DI-SPME Extraction a | ||||
| Chloroform | 9 | 2.6 | y = 35.9x + 277 | 0.998 |
| Bromoform | 9 | 2.7 | y = 8.9x + 2549 | 0.989 |
| Bromodichloromethane | 9 | 2.7 | y = 19.84x + 3.12 | 0.985 |
| Dibromochloromethane | 8 | 2.5 | y = 17.2x + 3.1 | 0.993 |
| 1,2-dibromoethane | 11 | 3.3 | y = 307.1x + 1132 | 0.998 |
| 1,2-dichloroethane | 12 | 3.6 | y = 13.58x + 4.02 | 0.978 |
| HS-TWA Extraction a | ||||
| Chloroform | 12 | 3.5 | y = 10.97x + 3.62 | 0.989 |
| Bromoform | 12 | 3.5 | y = 11.78x + 3.84 | 0.988 |
| Bromodichloromethane | 11 | 3.2 | y = 11.89x + 3.82 | 0.984 |
| Dibromochloromethane | 12 | 3.4 | y = 11.54x + 4.78 | 0.990 |
| 1,2-dibromoethane | 13 | 3.8 | y = 128x + 593 | 0.997 |
| 1,2-dichloroethane | 12 | 3.7 | y = 10.24x + 4.28 | 0.968 |
| HS-TWA Extraction b | ||||
| Chloroform | 15 | 4.4 | y = 6.63x + 4.82 | 0.988 |
| Bromoform | 15 | 4.6 | y = 4.35x + 4.64 | 0.989 |
| Bromodichloromethane | 16 | 4.7 | y = 5.08x + 4.4 | 0.987 |
| Dibromochloromethane | 16 | 4.8 | y = 7.6x + 2.7 | 0.995 |
| 1,2-dibromoethane | 17 | 5.1 | y = 62.3x + 27848 | 0.996 |
| 1,2-dichloroethane | 16 | 4.8 | y = 14.15x + 4.78 | 0.985 |
DL = detection limit. Note: data averaged from triplicate analysis. DI- , HS- and TWA- analysis from aqueous sample THM standards using optimized conditions with salt added a and with no salt added b. Quantified Linear Range (3–800 µg·L−1) used for all extraction methods.
3. Results and Discussion
3.1. Fiber Selection
Three GC/MS methods using SPME fibers (HS-, DI- and HS-TWA-) are compared for simultaneous sampling and extraction of six TMH compounds from natural aqueous solutions. All sampling and extraction parameters for each of the methods were optimized using headspace vials. Further, results are compared to HS-TWA sampling from on-site natural waters. Commercial SPME fiber products available cover a wide range of polarities and volatilities for achieving optimal extraction efficiencies for a variety of compounds. Based upon the literature [19,20,21,22,23], the SPME fibers: 7 and 100 µm PDMS; 75 and 85 µm CAR/PDMS, 65 µm PDMS/DVB; 65 µm CW/DVB; and 50 µm DVB/CAR/PDMS were investigated for the extraction of THMs. PDMS is a good choice as it has the advantage of being able to extract both non-polar and polar compounds by adjusting the extraction conditions [22]. PDMS/DVB polymers are suitable in analyzing more volatile compounds [24]. Figure 4 shows typical performance obtained for the fibers tested under HS-SPME sampling and extraction method towards six THM compounds. Compared with the other fibers, both the 50 µm DVB/CAR/PDMS and the 65 µm PDMS/DVB fiber provided better extraction efficiency for all six target THM standards. This work was carried out using available 65 µm PDMS/DVB fibers for all analysis methods. Other work has suggested that the tri-phase fiber has shown promise toward THM analyses when considering compounds with greater potential range of polarities, such which might occur when analyzing disinfection by products (DBPs) found by halo-disinfection treatments [16].
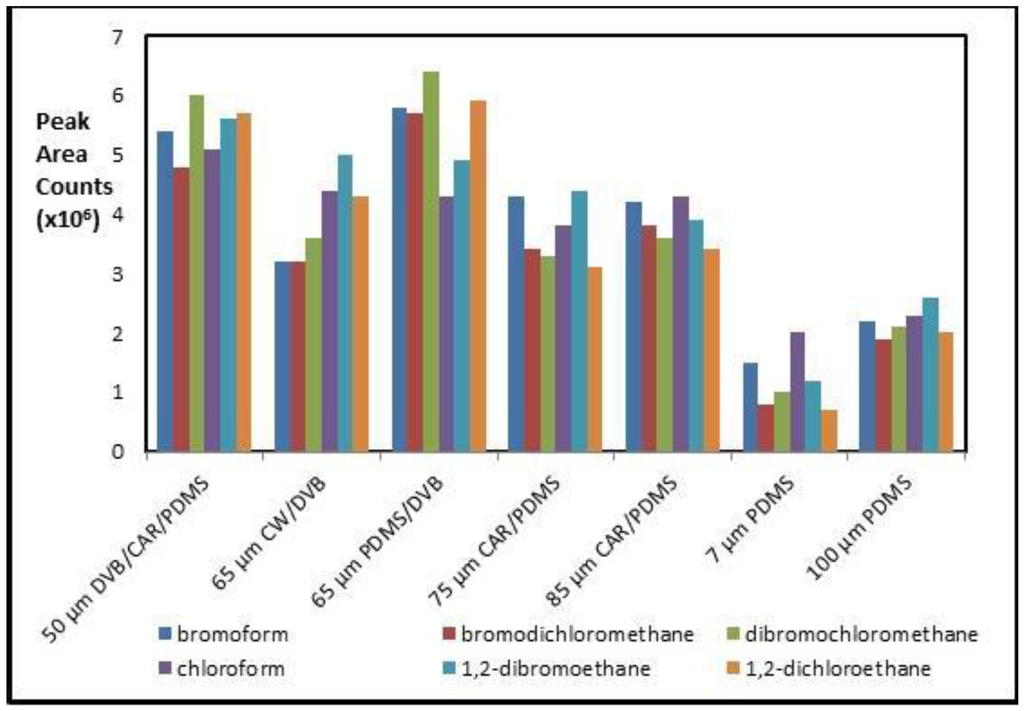
Figure 4.
A plot of Gas Chromatography (GC) peak area versus different tested SPME fibers towards the 6 targeted THMs. Optimized conditions: 10% wt./vol of Na2SO4 in 20 mL water sample, extraction time: 8 min, extraction temperature: 25 °C, desorption time: 2 min, desorption temperature: 190 °C.
Figure 4.
A plot of Gas Chromatography (GC) peak area versus different tested SPME fibers towards the 6 targeted THMs. Optimized conditions: 10% wt./vol of Na2SO4 in 20 mL water sample, extraction time: 8 min, extraction temperature: 25 °C, desorption time: 2 min, desorption temperature: 190 °C.
3.2. Extraction Parameter Optimization and Analysis Method
Fiber desorption conditions in the GC injection port can affect the sensitivity of THM target compounds. Desorption temperature is limited by the stability of the fiber coating and by analyte lability. The 65 µm PDMS/DVB fiber stability is a non issue since the high temperature limit for the fiber coating is 270 °C. However, since THMs have a range of polarities, significant thermal degradation can occur in their analysis at temperatures above 200 °C [25]. Therefore a desorption temperature of 190 °C was used in the GC injector port. To minimize other fiber contamination from the sample matrix, and/or analyte carry over, a fiber conditioning step was included, by heating the fiber at 220 °C for two minutes before and after an analysis run. A blank run acquired after showed no carry over or breakdown products.
Following parameter optimization, the conditions: 750 rpm stirring rate, 8 min extraction time at 25 °C, with 10% salt addition and 2-min desorption time at 190°, were found to give the best overall extraction conditions and peak areas for all six THMs for DI-SPME, HS-SPME extraction conditions. Slight differences in the time to extract were found for direct immersion and for headspace sampling. In headspace sampling, the equilibrium is reached faster than for the direct aqueous sampling (by about 4 min) presumably through a faster exchange rate in the gas phase. However, the extra time was left in this sampling method to keep the conditions the same for comparison with the direct immersion method. See Figure 5b for HS-SPME versus Figure 5a for DI-SPME extractions. The extra time did not change the amount acquired peak areas and this suggests that mass transfer to the fiber had reached equilibrium. Hence, Figure 5b was used for analysis for both DI- and HS-SPME method. A temperature of 25 °C was chosen to enable temperature maintenance in the lab by using a water jacket around the vial. Since absorption onto the fiber is an exothermic process, temperature changes affect diffusion rates and equilibrium times [21]. In headspace sampling, the rate of diffusion is faster in the gaseous headspace than in aqueous solution. For both HS- and DI- sampling methods, the temperature was purposely kept low to minimize such differences. Hence a temperature of 25 °C ± 1 °C was used for quantification and for linear calibration.
For TWA-SPME methods, a home built stop flow system (Figure 3) that contained six septum-sealed sampling ports where the six SPME fibers could be inserted and tested was used. The water flow (held at 10.00 mL/min flow rate at 25 °C) was controlled using a Dynamax model SD-200 solvent delivery system. This device was used to optimize TWA extraction rate (1, 2, 3 and 5 days) and to test fiber retracted distance into the SPME needle casing (5, 10, 15, 20, 25 and 30 mm) for optimal diffusion sampling length for HS-TWA method. A mixture of the THM standards at 1 ppm concentration in water methanol (90%:10%) solution were used to assess these parameters. Figure 5b shows typical extraction profile variations achieved over the sampling time of 1, 2 and 3 days periods for four of the THM standards. From these analyses, a two days sampling period for HS-TWA extraction and with a fiber retraction distance of 20 mm into the needle casing was found to give optimal maximum extraction of all six THM standards. For water analyses, the expected combined THM concentrations would be far below 1-ppm concentrations, as indicated from previously analyzed THM data for theses ponds [4]. Hence, possible saturation of the fiber with analytes would not be an issue when using this 2 days extraction time. These conditions were used for both onsite and vial sampling HS-TWA methods. For HS-TWA on-site sampling, the SPME fiber was placed in a holder at the appropriate sampling depth in the pond. For HS-TWA vial sampling these parameters were combined with the other optimized HS conditions and assumed to be optimal.
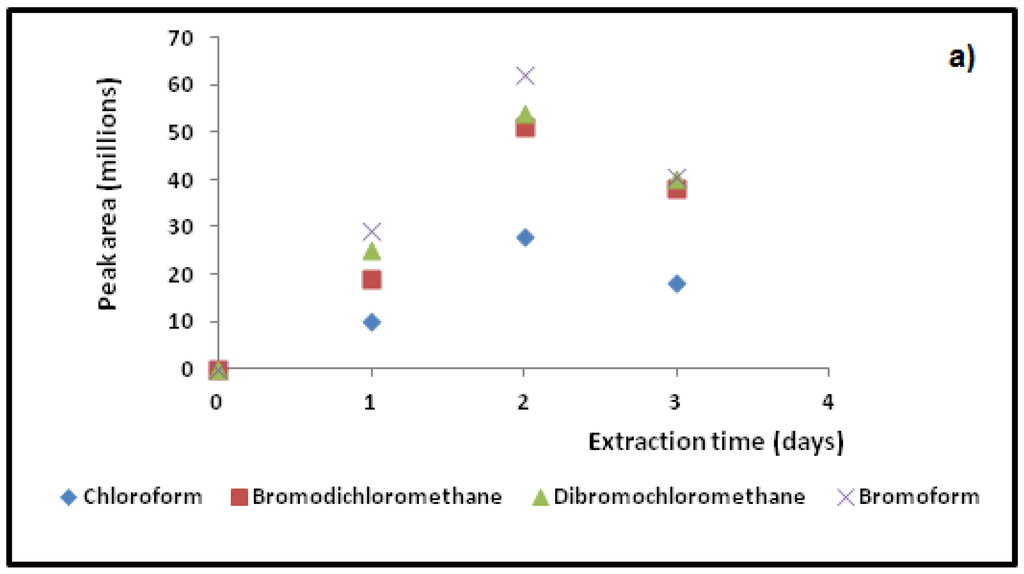
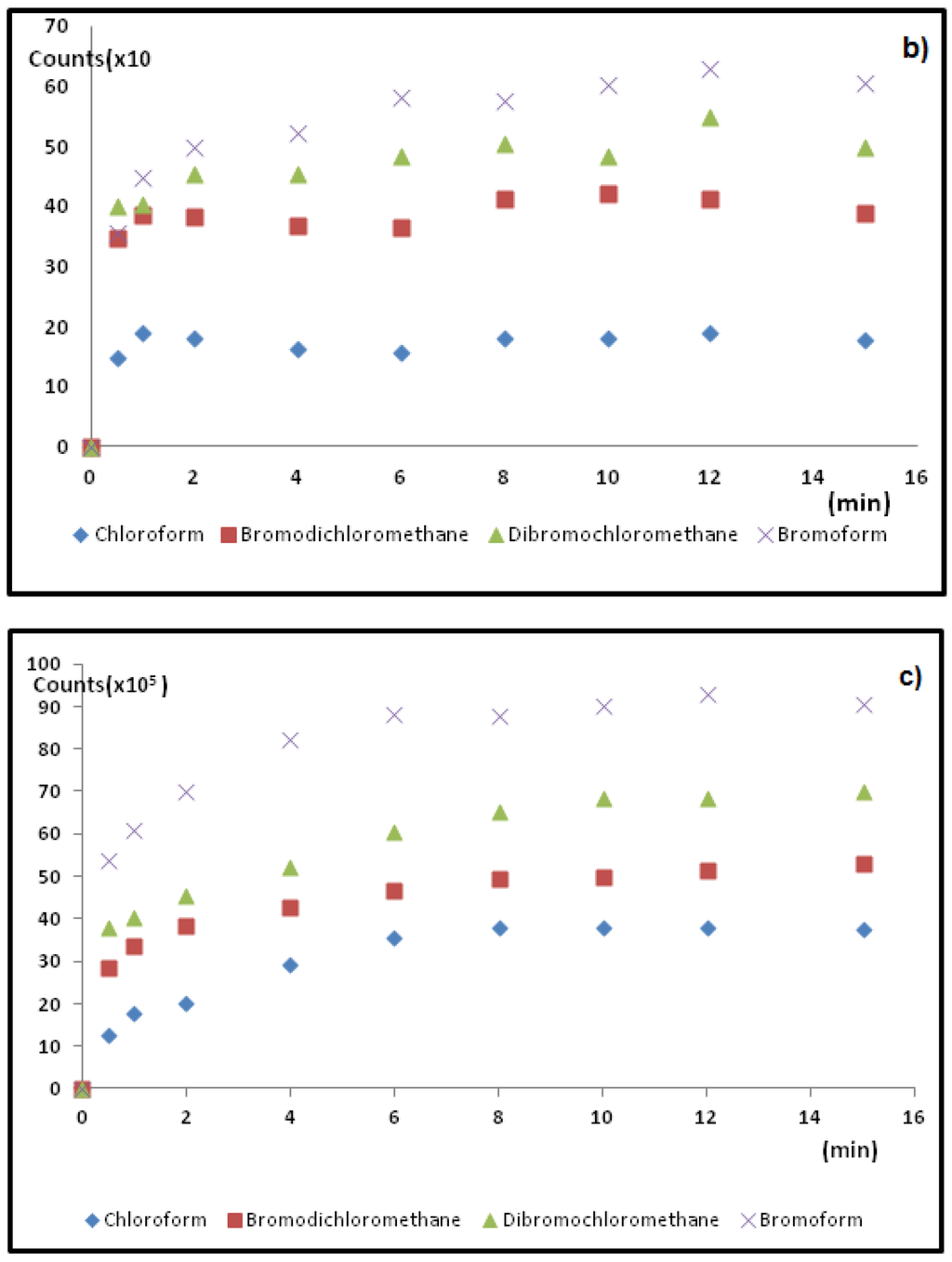
Figure 5.
Typical SPME fiber extraction profiles used for (a) HS-TWA SPME; (b) DI-SPME; and (c) HS-SPME methods to determine optimal sampling times for four target THMs.
Figure 5.
Typical SPME fiber extraction profiles used for (a) HS-TWA SPME; (b) DI-SPME; and (c) HS-SPME methods to determine optimal sampling times for four target THMs.
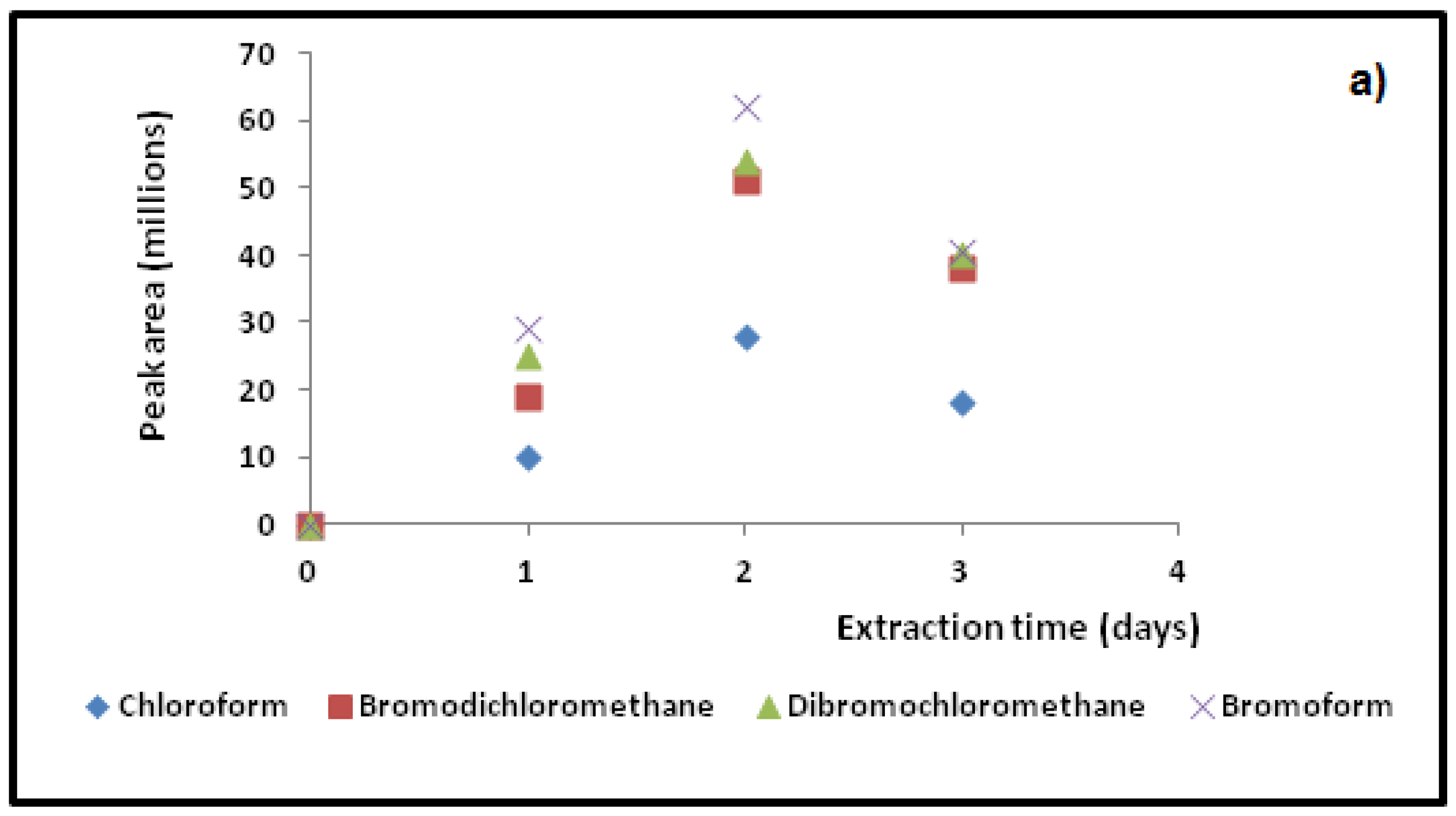
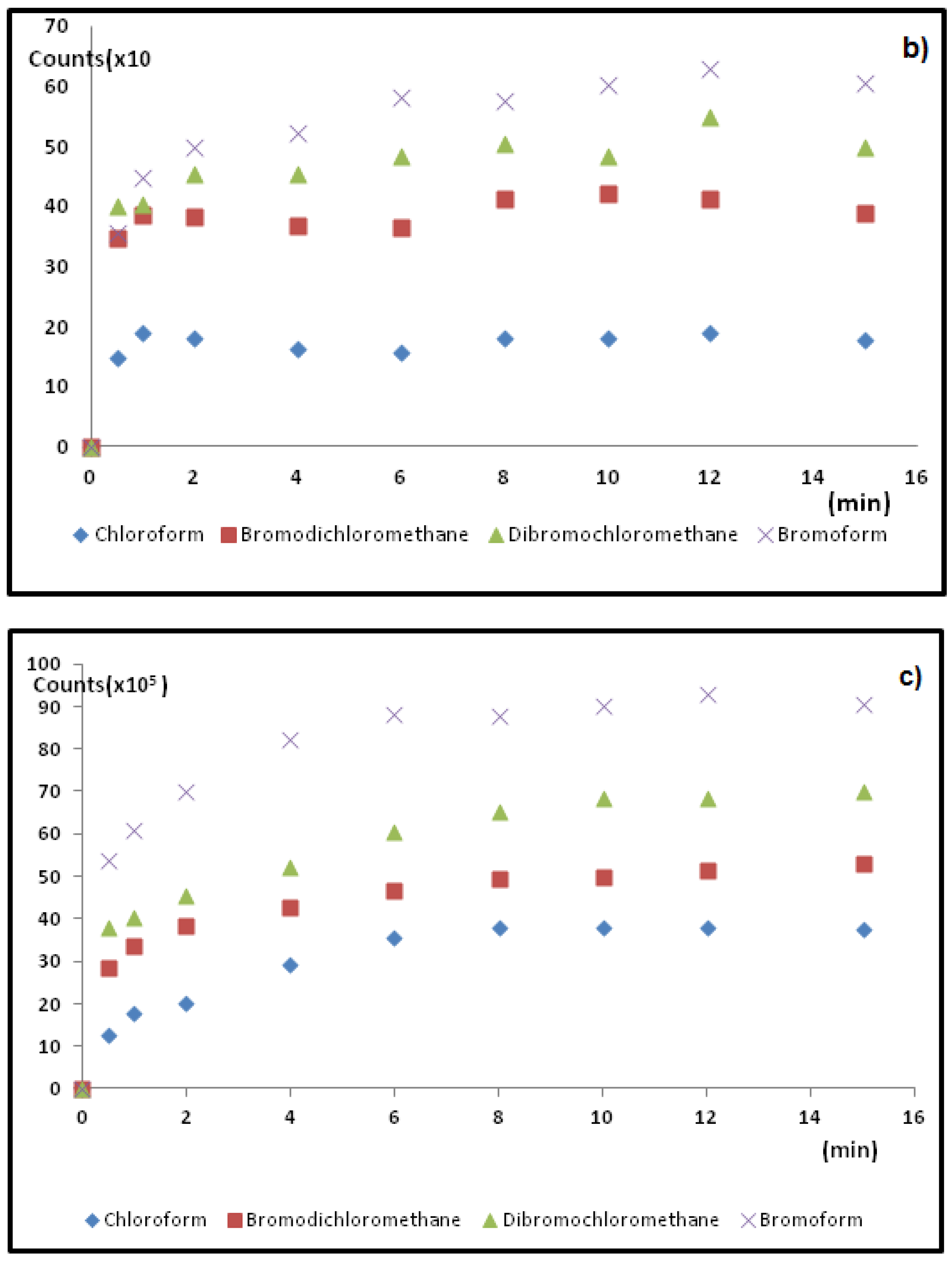
It is known that pH can change THM formation in natural waters [11]. Hence, it is important to maintain the same pH in the sampling vial. Ascorbic acid has been used [26] for slight pH adjustments, as it does not seem to change THM complexation. In order to re-adjust the pH to the water samples (pH~6, see Table 1) ascorbic acid was added to the vial after 10% wt./vol. of (NH4)2SO4 salt addition.
It has been noted that addition of (NH4)2SO4 salt can enhance THM extraction [16]. In sampling collected natural waters, a 10% wt./vol. of the salt was simply added to the water sample vials before extraction and analysis. For on-site sampling or for the determination of THMs under real water conditions this is not possible. Hence SPME sampling was carried out in three ways: (1) analysis by all methods from septum-sealed vials, were the salt and ascorbic acid was added; (2) TWA-SPME sampling in septum-sealed vials, with no salt or acid added; and (3) under on-site TWA-SPME sampling, when no salt or acid could be added. Under salt added conditions, for all methods, analysis of the six THM showed about a 10% GC peak area increase for all THM standards over similar non-salt added analysis. For on-site sampling, the natural water was found to have a conductivity of 60 µS, suggesting a low salt concentration (see Table 1). Hence a set of both non-salt (at 60 µS conditions) and a 10% salt-added calibration curves were created in order to relate on-site TWA and vial analyzed TWA conditions (see Table 2). The calibration curve obtained by HS-TWA vial sampling with no salt added was used for both HS-TWA sampling methods (by vial sampling and by on-site sampling) to determine the data (Table 1), for the two ponds under study.
3.3. GS/MS Analysis
A typical chromatogram of the six standard THMs (100 µg·L−1) by SPME vial sampling is shown in Figure 6 and illustrates that good peak shape and resolved components were obtained. Individual THM components in the water samples studied were identified by spiking the samples with standards and by MS identification. Quantification of the six THMs analytes were performed using the peak area ratios of the analyte relative to the calibration curve (with no salt added) from 3 to 800 µg·L−1. The sampling range was found to be linear and was chosen to conform to THM regulatory limits, for total THMs to be less than 100 µg·L−1 [15], and to provincial reports that have shown that total THMs have been found in natural waters up to 600 µg·L−1 [4]. Each sample and standard was analyzed in triplicate.
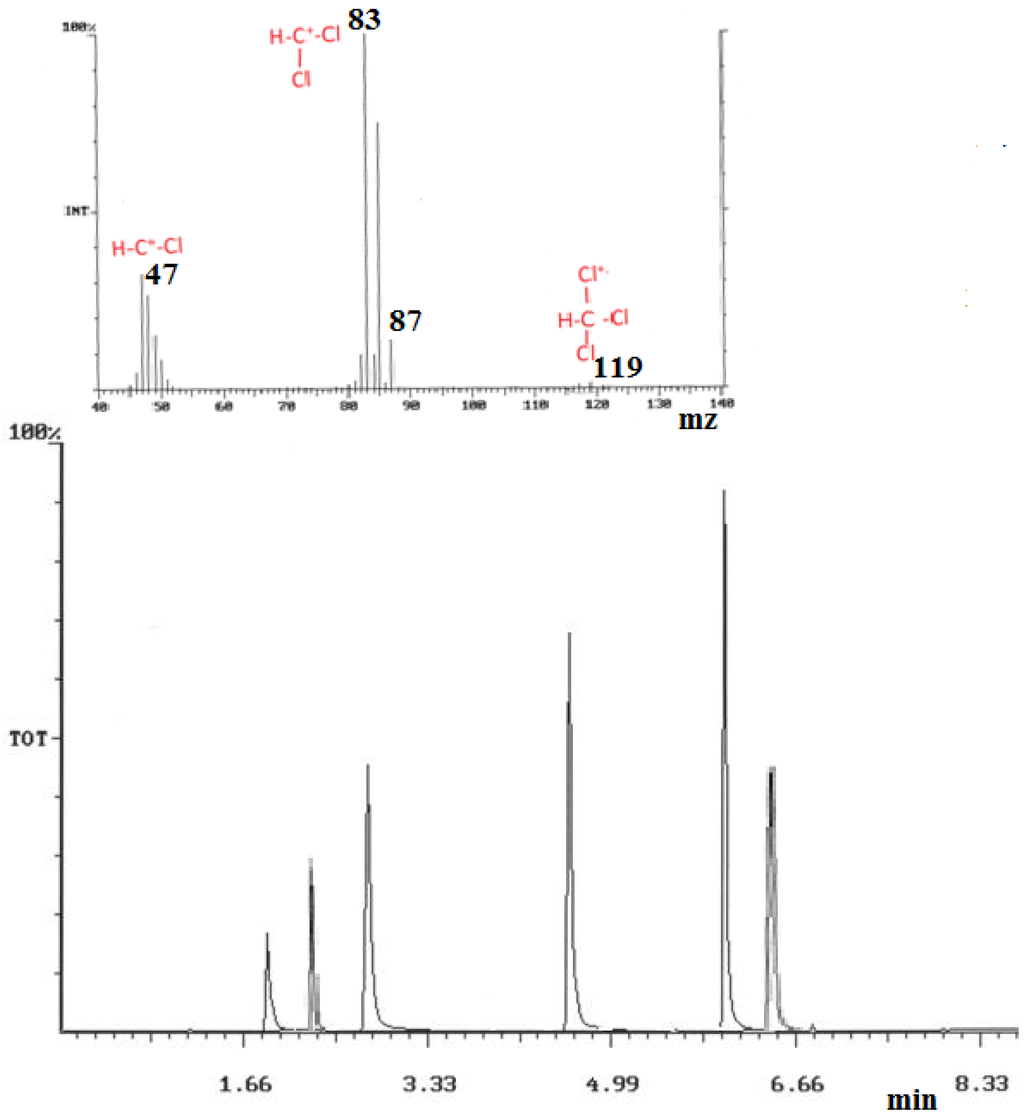
Figure 6.
(a) Top extracted MS of chloroform (1); (b) Bottom: GC/MS chromatogram of the 6 THM target components (and GC retention time): 1 = chloroform (1.88), 2 = 1,2-dichloroethane (2.21), 3 = bromodichloromethane (2.92), 4 = dibromochloromethane (4.59), 5 = bromoform (6.01), 6 = 1,2-dibromoethane (6.26).
Figure 6.
(a) Top extracted MS of chloroform (1); (b) Bottom: GC/MS chromatogram of the 6 THM target components (and GC retention time): 1 = chloroform (1.88), 2 = 1,2-dichloroethane (2.21), 3 = bromodichloromethane (2.92), 4 = dibromochloromethane (4.59), 5 = bromoform (6.01), 6 = 1,2-dibromoethane (6.26).
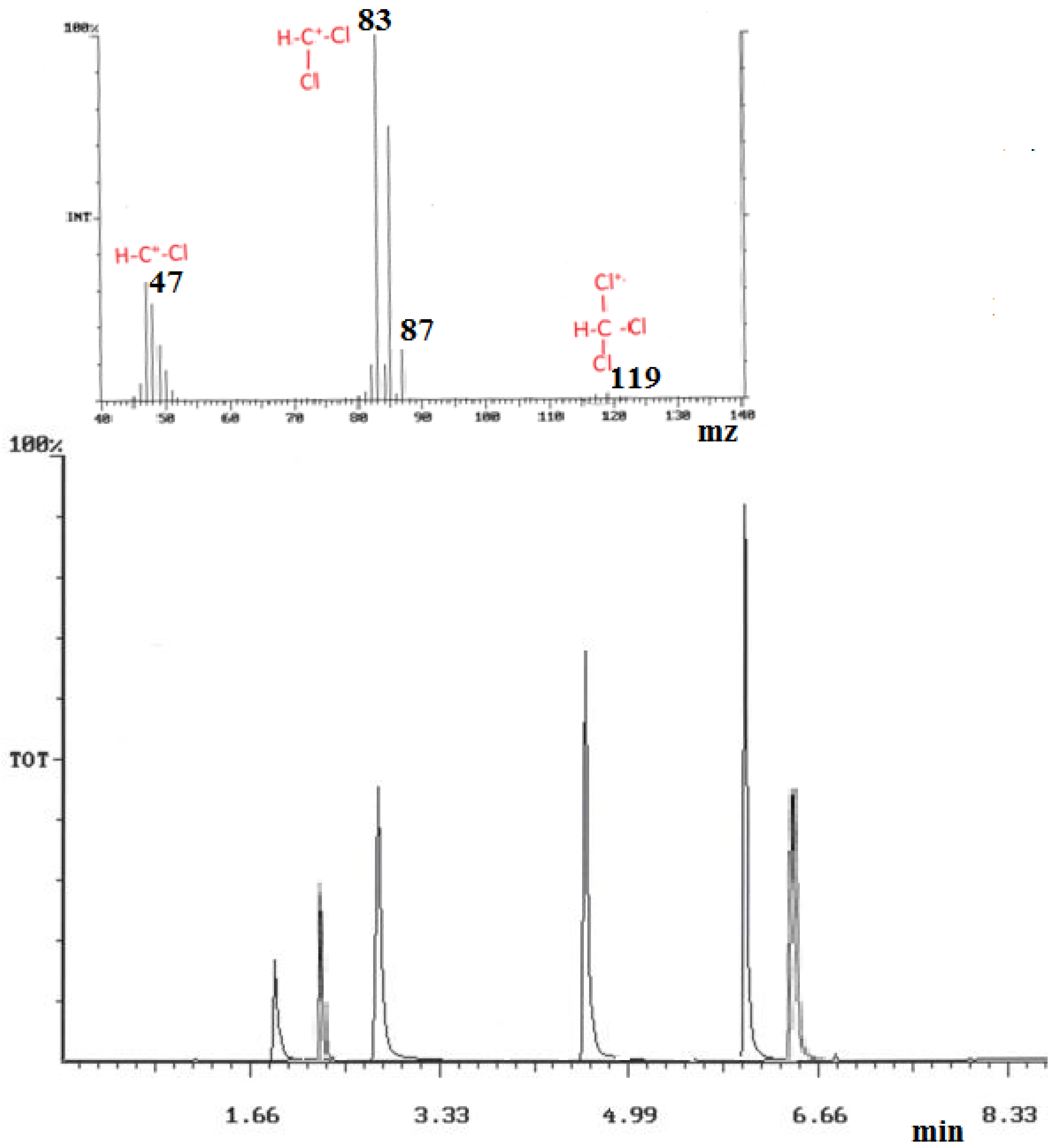
GC THM peaks areas were obtained and quantified by selected ion monitoring (SIM) mode for the optimized selected fragment ions given in the experimental section. Assessment of the largest GC peak area counts obtained for all six THM standards, was used to determine the optimized column conditions, which were found to be: 40 °C, held for 4 min, to 160 °C at a rate of 30 °C·min−1, and held for 4 min, then ramped to 210 °C at a rate of 50 °C·min−1, and held for 1 min, giving a total run time of 10 min. Under such conditions, all THM components, including those from real samples, were separated by the 6.8 min in the GC 10 min run time. To preclude any column contamination, the GC run included an extra time at 210 °C before the next run. Directly after injection, the SPME fiber was then desorbed into a heater port at 250 °C for a period of 2 min to negate sample carry over from the fiber. THM GC retention data tends to follow an increased retention time with increasing halo substitution, and with substitution of chlorine for bromine (chloroform elutes earlier than bromoform). Similar comparisons can be made with the compound Henry’s constant and to compound substitution, which suggests that there is a volatility decrease with increased halo substitution. For example, bromoform and dibromochloromethane give larger peak areas than do chloroform and bromodichloromethane (See Figure 6).
3.4. Comparisons of DI-, HS- and HS-TWA SPME Methods toward Water Analysis
Water sampling was carried out at the two municipal surface drinking water ponds near Corner Brook, NL, Canada: Trout Pond and 3 Mile Pond. Triplicate water samples were analyzed on-site for: conductivity, dissolved solids, pH and temperature (results are presented Table 1) and 1 L samples were collected for analysis for THMs. All methods used SPME fibers with 65 µm PDMS/DVB film. This fiber gave the best component extraction for all target THMs, based upon their GC peak areas under optimized conditions. Further, a zero sink test was performed to assess fiber compatibility for analysis of THMs. The results found that no analyte loss (<2%) from the fiber was seen during transfer periods from extraction to analysis. Such results suggest that the fiber is suitable for use in TWA studies for THM analysis.
Samples were studied for THMs in two scenarios. A portion of the collected water was sealed in vials with septum sealed tops. SPME sampling and extraction were achieved within 12 h from collection for DI- and HS-SPME methods. HS-TWA SPME method extracted analytes over a two days period with the SPME fiber retracted by 20 mm into the needle casing (optimized conditions). Otherwise similar optimized conditions were used in the HS-TWA method as for DI- and HS- sampling.
The second scenario was by on-site sampling, whereby the TWA-SPME fiber was left to sample for two days in the pond and then retrieved and analyzed. When on-site TWA water analysis data was compared to TWA-SPME vial sampling, results from on-site sampling showed consistently lower but reproducible results. Such differences were attributed to the change in solution conductivity, where salt had been added to the sampling vial method. Under natural on-site sampling conditions, the water conductivity was low (60 µS). With the addition of high salt (10% wt./vol. added) content, the relative volatility in the headspace is increased, because the increased salt concentration causes an increase in the gas/liquid equilibrium toward the fiber. Hence larger GC peaks areas would be obtained for smaller amounts of a THM component under salt enriched conditions. It has been reported that sodium sulfate provides a maximum extraction efficiency around 2.5 g of salt per 10 mL water for lower molecular weight THMs like (CHCl3 and CHCl2Br) [16,27]. The effect of adding salt to the sampling vial produced a ~10% signal enhancement in THM GC peak areas, found for all DI-, HS- and TWA- SPME analysis methods. Calibration curves constructed from this data also gave lower LODs for individual THM components (see Table 2). Hence a further calibration curve was necessary for TWA sampling from a vial where no salt was added. After comparing TWA vial sampling data, obtained under non-salted conditions, to on-site TWA sampling, the corrected results obtained were comparable to those obtained by HS- and DI- methods. The calibration curve produced under non-salt added conditions was used for quantification of TWA sampling of pond water by both on-site and by sampling vial methods, Data is given in Table 1. Under non-salt conditions, the method detection limits were usually higher for the individual THMs and generally smaller extracted peak areas were obtained for individual THM components for similar concentrations (see Table 2). This is perhaps a limitation to on-site sampling method development as careful attention has to be made to ensure the matching of ionic strength conditions for calibration samples to water samples analyzed. However, it may be the case, where the natural waters being sampled are briny or saline. Here the natural conductivity might closer mimic the artificial conditions to the “salt added” sample vial conditions and hence show minimal deviations.
All SPME methods were able to achieve low ppb detection limits and hence these methods were amenable to quantification of THMs in the analyzed waters. Further, DLs fall within a similar range of those reported elsewhere [21]. In comparison to the NL government monitored total THM values for these two ponds, the data obtained by all three methods (in Table 1) corresponds closely to their Total THM values, given that fluctuations with natural condition changes can occur. Further, there may be other THMs that have not been detected. In such cases, UV detection may be better and more universal for total THM and haloalkane concentration assessments. However, specific THM speciation cannot be determined by UV methods and such information is critical to gain knowledge about potential chemical reactivity and insight to individual reaction pathways, which SPME methods can provide.
In comparing HS- and DI- SPME sampling methods, optimized under 10% salt added salt conditions (in Table 2), the DI- method gives slightly lower LODs. However, when sample matrices are complex or contaminated, direct sampling methods, like DI-SPME, can be prone to contamination or fiber breakage. Under such conditions, alternate HS- or TWA- SPME sampling methods could be utilized. In this research, the drinking water pond samples and their calibration matrices were clean and free of debris.
HS- and TWA- methods gave similar results. However, TWA sampling is simpler to prepare. Further, it is much easier to carry multiple fibers away from a water body in the case of TWA-on-site sampling than to remove multiple volumes of water. This is an especially important consideration when contemplating measurements of water areas that are remote or where it is a physically difficult to carry multiple water samples out for analysis. Further TWA gives a more accurate assessment over time of a dynamic water system than does spot or grab sampling and hence TWA monitoring data would achieve more reliable background information about the water body.
4. Conclusions
Three SPME-GC/MS methods (HS-, DI- and HS-TWA-) are compared for the simultaneous analysis of six TMHs from raw aqueous solutions. SPME linking to GC/MS makes the method highly selective toward THM quantitation for water analysis, as demonstrated by the lack of significant signal interferences in both the standards and real sample matrices. Under optimized conditions, which included fiber selection, fiber position, sampling rate, extraction parameters and salt conditions, real samples were analyzed for natural THMs occurring in untreated aqueous waters. Of the seven SPME fibers tested, both the 65 µm DVB/PDMS and the 50-µm CAR/PDMS/DVB fiber performed best towards the six targeted THMs analytes. For our work, the 65 µm PDMS/DVB was selected because it was available and equally able to efficiently target all compounds over the range of molecular weights and volatilities required for this study.
Detection enhancement by addition of salt to the sampling matrix vial cannot be used in on-site TWA sampling conditions. The utility of salt modification in quantification studies on natural waters may very well depend upon the natural conditions. In low conductivity conditions, large deviations can occur if calibration solutions are not corrected for by matrix matching the ionic strength.
The SPME-GC/MS methods allow for simple, fast and simultaneous detection of THM compounds at low ppb levels and as such, may provide insight into the chemistry and the occurrence of halo-analytes in untreated waters. This is important as THMs have potential toxicity and impact to human health. SPME-TWA sampling is easy to perform and to analyze and with its ability to flatten episodic changes in dynamic aqueous systems, TWA methods may give better natural THM estimates and better representative data for determining natural background THM levels in surveys.
The utility of SPME techniques are most profound when the method is automated. This can be easily achieved with current samplers when water samples are collected and put into standard sampling vials. Under these circumstances, total optimization of SPME sampling, extraction and GC/MS injection parameters and analysis can be controlled under current software. However, in the case of on-site analysis, once the fibers have been retrieved, the development of an automated TWA SPME holder carousel would be useful to deliver samples for unattended automated analysis run. Further work to investigate such methodology is ongoing.
Acknowledgments
We acknowledge the Department of Environment, of the Province of Newfoundland and Labrador, Canada and for supplying provincial 2013 trihalomethanes (THMs) study results for various water bodies on the island of Newfoundland. We thank the Environmental Science Scholarship awarded to D. Barter and to R. Gaultois and for the Vice–President Research Award, Grenfell Campus, Memorial University of Newfoundland, Newfoundland and Labrador, Canada (MUN) for support of this work.
Author Contributions
Don-R. Parkinson conceived and designed the experiments, wrote the paper and provided oversight during data analysis. David Barter performed the initial experiments regarding the SPME-GC/MS and helped analyze the data. Robert Gaultois performed the initial experiments regarding the TWA-stop-flow and GC/MS analysis.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Vieira, R.F.; Berenguel, A.T.; Silva, M.A.; Vilaca, J.S.; Domingues, V.F.; Figueiredo, S.A. Natural organic matter fractionation along the treatment of water for human consumption. Glob. NEST J. 2012, 14, 399–406. [Google Scholar]
- Huber, S.G.; Kotte, K.; Scholer, H.F.; Williams, J. Natural abiotic formation of trihalomethanes in soil: Results from laboratory studies and field samples. Environ. Sci. Technol. 2009, 43. [Google Scholar] [CrossRef]
- Hassouna, M.E.M.; Badawy, M.I.; Gad-Allah, T.A.; Said, M.H. Fate of natural organic matter and THMs formation for raw and conventionally treated waters of El-Fayoum Governorate, Egypt. Int. J. Adv. Res. 2014, 2, 787–794. [Google Scholar]
- Department of Environment and Conservation. THMs Summary for Public Water Supplies in Newfoundland and Labrador; Department of Environment and Conservation: St. John’s, NL, Canada, 2013; pp. 1–13. [Google Scholar]
- Nollet, L.M.L. Handbook of Water Analysis, 2nd ed.; CRC Press: Boca Raton, FL, USA, 2007; p. 606. [Google Scholar]
- Dojlido, J.R.; Best, G.A. Chemistry of Water and Water Pollution; Ellis Hornwood Ltd.: New York, NY, USA, 1993; p. 34. [Google Scholar]
- Mullaugh, K.M.; Kieber, R.J.; Willey, J.D.; Avery, G.B. Long-term temporal variability in hydrogen peroxide concentrations in Wilmington, North Carolina USA rainwater. Environ. Sci. Technol. 2011, 45, 9538–9542. [Google Scholar] [PubMed]
- Weiner, E. Applications of Environmental Chemistry: A Practical Guide for Environmental Professionals; CRC Press: Boca Raton, FL, USA, 2000; p. 174. [Google Scholar]
- Larson, R.A.; Weber, E.J. Reaction Mechanisms in Environmental Organic Chemistry; CRC Press: Boca Raton, FL, USA, 1994; pp. 293–297. [Google Scholar]
- Hong, H.C.; Huang, F.Q.; Wang, F.Y.; Ding, L.X.; Lin, H.J.; Liang, Y. Properties of sediment NOM collected from drinking water reservoir in South China, and its association with THMs and HAAs formation. J. Hydrol. 2013, 476, 274–279. [Google Scholar] [CrossRef]
- Pedrot, M.; Dia, A.; Davranche, M. Dynamic structure of humic substances: Rare earth elements as a fingerprint. J. Colloid Interface. Sci. 2010, 345, 206–213. [Google Scholar] [PubMed]
- Hansen, K.M.S.; Willach, S.; Antoniou, M.G.; Mosboek, H.; Albrechtsen, H.; Andersen, H.R. Effect of pH on the formation of disinfection byproducts in swimming pool water. Water Res. 2012, 46, 6399–6409. [Google Scholar] [CrossRef] [PubMed]
- Liao, K.H.; Tan, Y.; Conolly, R.B.; Borghoff, S.J.; Gargas, M.L.; Andersen, M.E.; Clewell, H.J. Bayesian estimation of pharmacokinetic and pharmacodynamic parameters in a mode-of-action-based cancer risk assessment for chloroform. Risk Anal. 2007, 27, 1535–1531. [Google Scholar] [CrossRef] [PubMed]
- Health Canada. Guidelines for Canadian Drinking Water Quality—Summary Table. Water and Air Quality Bureau, Healthy Environments and Consumer Safety Branch, Health Canada, Ottawa, Ontario. Prepared by the Federal-Provincial-Territorial Committee on Drinking Water of the Federal-Provincial-Territorial Committee on Health and the Environment. October 2014. Available online: http://www.hc-sc.gc.ca/ewh-semt/pubs/water-eau/sum_guide-res_recom/index-eng.php (accessed on 5 August 2015). [Google Scholar]
- Government of the United States of America. Analysis of Trihalomethanes in Drinking Water. 1979; United States Environmental Protection Agency. Available online: http://www.epa.gov/region1/info/testmethods/index.html (accessed on 5 August 2015). [Google Scholar]
- Luo, Q.; Chen, X.; Wei, Z.; Xu, X.; Wang, D.; Wang, Z. Simultaneous and high-throughput analysis of iodo-trihalomethanes, haloacetonitriles, and halonitromethanes in drinking water using SPME/GC-MS: An optimization of sample preparation. J. Chromatogr. A 2014, 1365, 45–53. [Google Scholar] [PubMed]
- Maia, R.; Correia, M.; Pereira, I.M.; Bras, B.; Vitorino, M. Optimization of HS-SPME analytical conditions using factorial design for trihalomethanes determination in swimming pool water samples. Microchem. J. 2014, 112, 164–171. [Google Scholar]
- Wirth, T.C.; Later, J.B.; Oliphant, J.L.; Lee, E.D.; Later, D.W. Person-portable GC-MS for rapid on-site environmental screening of contaminants in drinking water. Am. Lab. 2012, 44, 34–35. [Google Scholar]
- Ouyang, G.; Zhao, W.; Alaee, M.; Pawliszyn, J. Time-weighted average water sampling with a diffusion-based solid-phase microextraction device. J. Chromatogr. A 2007, 1138, 42–46. [Google Scholar] [PubMed]
- Amdany, R.; Chimuka, L.; Cukrowska, E.; Kukucka, P.; Kohoutek, J.; Tolgyessy, P.; Vrana, B. Assessment of bioavailable fraction of POPS in surface water bodies in Johannesburg City, South Africa, using passive samplers: An initial assessment. Environ. Monit. Assess. 2014, 186, 5639–5653. [Google Scholar] [PubMed]
- Boyaci, E.; Rodriguez-Lafuente, A.; Gorynski, K.; Mirnaghi, F.; Souza-Silva, E.A.; Hein, D.; Pawliszyn, J. Sample preparation with solid phase microextraction and exhaustive extraction approaches: Comparison for challenging cases. Anal. Chim. Acta 2015, 873, 14–30. [Google Scholar] [PubMed]
- Stack, M.A.; Fitzgerald, G.; O’Connell, S.; James, K.J. Measurement of trihalomethanes in potable and recreational waters using solid phase micro extraction with gas chromatography-mass spectrometry. Chemosphere 2000, 41, 1821–1826. [Google Scholar] [CrossRef]
- Valencia, S.; Marin, J.; Restrepo, G. Method for trihalomethane analysis in drinking water by solid-phase microextraction with gas chromatography and mass spectrometry detection. Water Sci. Technol. Water Supply 2013, 13, 499–506. [Google Scholar]
- Pawliszyn, J. Handbook of Solid Phase Microextraction; Chemical Industry Press: Beijing, China, 2009; pp. 25–28, 47–48. [Google Scholar]
- Frazey, P.A.; Barkley, R.M.; Sievers, R.E. Solid-phase microextraction with temperature-programmed desorption for the analysis of iodination disinfection byproducts. Anal. Chem. 1998, 70, 638–644. [Google Scholar] [CrossRef] [PubMed]
- Bond, T.; Huang, J.; Templeton, M.R.; Graham, N. Occurrence and control of nitrogenous disinfection by-products in drinking water: A review. Water Res. 2011, 45, 4341–4354. [Google Scholar] [PubMed]
- Gough, R.; Holliman, P.J.; Willis, N.; Freeman, C. Dissolved organic carbon and trihalomethane precursor removal at a UK upland water treatment works. Sci. Total Environ. 2014, 468–469, 228–239. [Google Scholar] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).