

Extraction and Counter-Current Separation of Phenylpropanoid Glycosides from Pedicularis oederi Vahl by Deep Eutectic Solvent


Abstract
:1. Introduction
2. Materials and Methods
2.1. Apparatus
2.2. Reagents and Plant Material
2.3. UPLC Analysis
2.4. Preparation of DES
2.5. Optimization of Ultrasonic-Assisted Extraction Conditions
2.6. Ultrasonic-Assisted Extraction
2.7. Selection of Two-Phase Solvent System
2.8. Preparation of Two-Phase Solvent System and Sample Solution
2.9. High-Speed Counter-Current Chromatography Separation Procedure
3. Results and Discussion
3.1. Optimization of Ultrasonic-Assisted Extraction Conditions
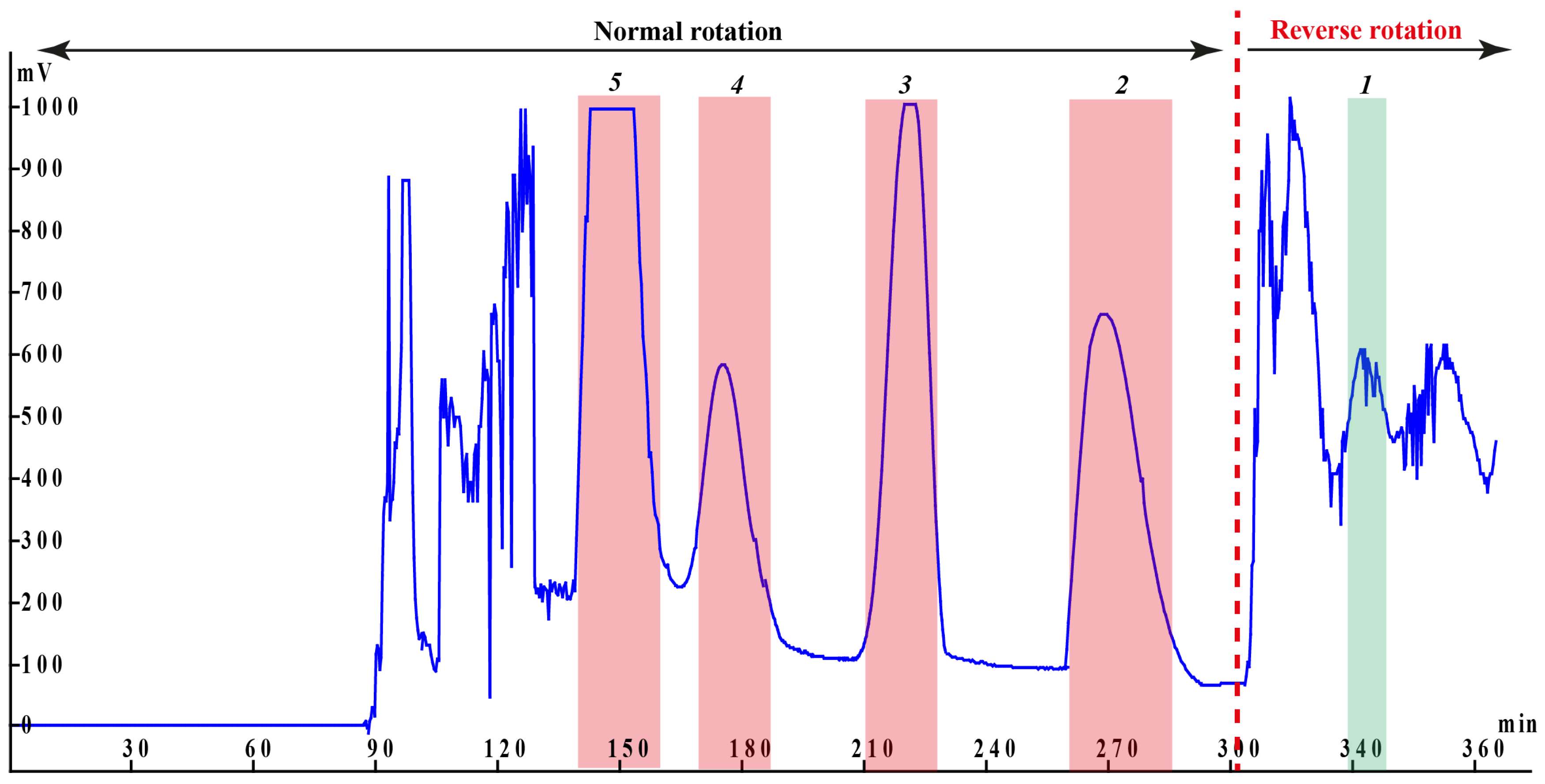
3.2. Selection of High-Speed Counter-Current Chromatography Experimental Conditions
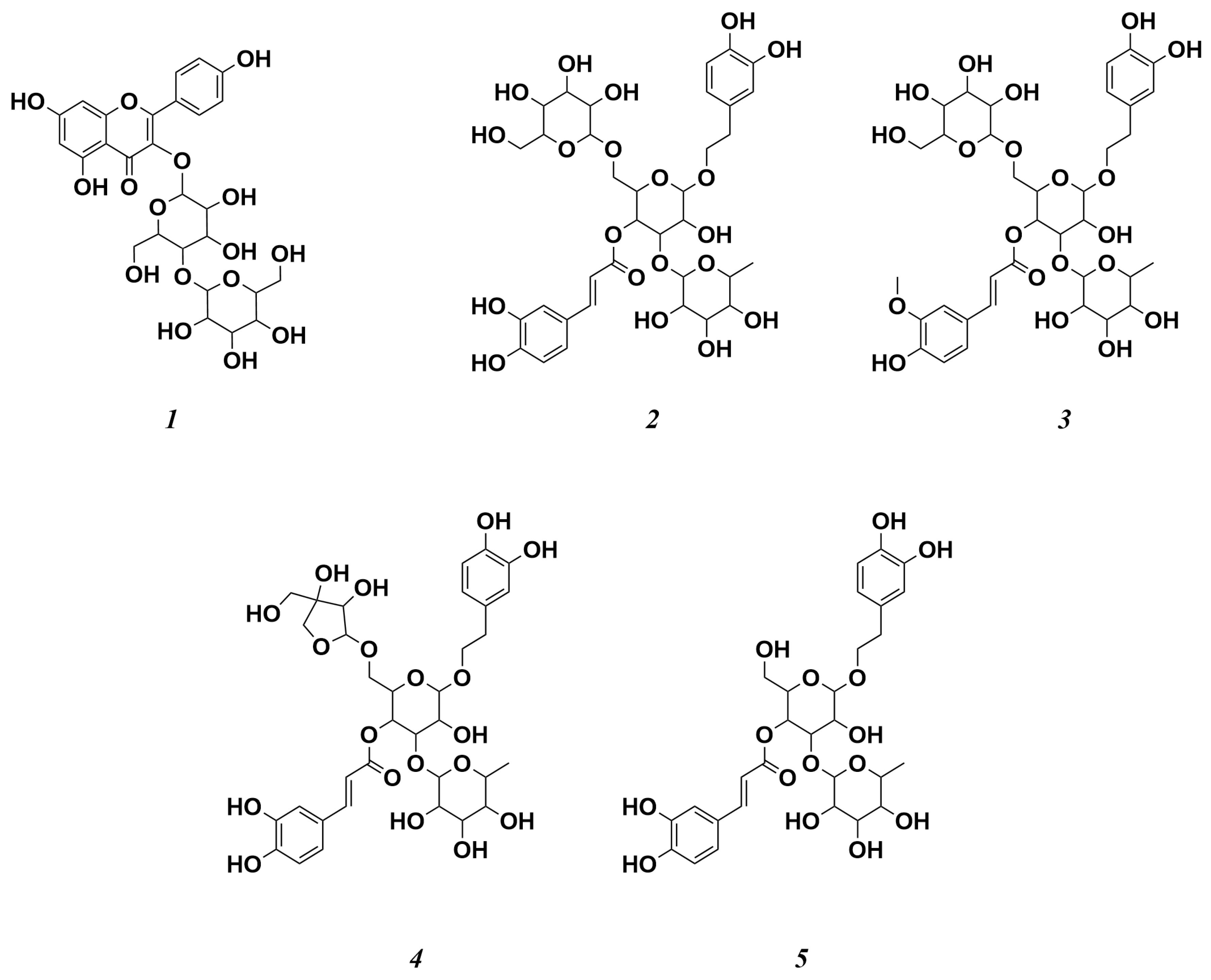
3.3. Structural Identification
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Tkach, N.; Ree, R.H.; Kuss, P.; Röser, M.; Hoffmann, M.H. High mountain origin, phylogenetics, evolution, and niche conservatism of arctic lineages in the hemiparasitic genus Pedicularis (Orobanchaceae). Mol. Phylogenetics Evol. 2014, 76, 75–92. [Google Scholar] [CrossRef]
- Yatoo, M.I.; Dimri, U.; Gopalakrishnan, A.; Karthik, K.; Gopi, M.; Khandia, R.; Saminathan, M.; Saxena, A.; Alagawany, M.; Farag, M.R.; et al. Beneficial health applications and medicinal values of Pedicularis plants: A review. Biomed. Pharmacother. 2017, 95, 1301–1313. [Google Scholar] [CrossRef]
- Chen, C.; Zhao, X.-H.; Yue, H.-L.; Li, Y.-L.; Chen, T. Separation of Phenylpropanoid Glycosides from a Chinese Herb by HSCCC. J. Chromatogr. Sci. 2013, 52, 395–399. [Google Scholar] [CrossRef]
- Jessop, P.G.; Phan, L.; Carrier, A.; Robinson, S.; Dürr, C.J.; Harjani, J.R. A solvent having switchable hydrophilicity. Green Chem. 2010, 12, 809–814. [Google Scholar] [CrossRef]
- Jessop, P.G. Searching for green solvents. Green Chem. 2011, 13, 1391–1398. [Google Scholar] [CrossRef]
- Cao, J.; Chen, L.; Li, M.; Cao, F.; Zhao, L.; Su, E. Two-phase systems developed with hydrophilic and hydrophobic deep eutectic solvents for simultaneously extracting various bioactive compounds with different polarities. Green Chem. 2018, 20, 1879–1886. [Google Scholar] [CrossRef]
- Erythropel, H.C.; Zimmerman, J.B.; de Winter, T.M.; Petitjean, L.; Melnikov, F.; Lam, C.H.; Lounsbury, A.W.; Mellor, K.E.; Janković, N.Z.; Tu, Q.; et al. The Green ChemisTREE: 20 years after taking root with the 12 principles. Green Chem. 2018, 20, 1929–1961. [Google Scholar] [CrossRef]
- Huang, J.; Guo, X.; Xu, T.; Fan, L.; Zhou, X.; Wu, S. Ionic deep eutectic solvents for the extraction and separation of natural products. J. Chromatogr. A 2019, 1598, 1–19. [Google Scholar] [CrossRef]
- Afonso, J.; Mezzetta, A.; Marrucho, I.M.; Guazzelli, L. History repeats itself again: Will the mistakes of the past for ILs be repeated for DESs? From being considered ionic liquids to becoming their alternative: The unbalanced turn of deep eutectic solvents. Green Chem. 2023, 25, 59–105. [Google Scholar] [CrossRef]
- Wang, T.; Wang, Q.; Li, P.; Yang, H. High-Speed Countercurrent Chromatography-Based Method for Simultaneous Recovery and Separation of Natural Products from Deep Eutectic Solvent Extracts. ACS Sustain. Chem. Eng. 2020, 8, 2073–2080. [Google Scholar] [CrossRef]
- Coskun, O. Separation techniques: Chromatography. North. Clin. Istanb. 2016, 3, 156. [Google Scholar]
- Zou, D.; Cui, Y.; Li, S.; Sang, D.; Liu, W.; Zhao, T.; Gu, X.; Chen, T.; Li, Y. The applicability of high-speed counter-current chromatography for preparative separation of biosynthesis products: Glycosylation products as example. J. Sep. Sci. 2021, 44, 4368–4375. [Google Scholar] [CrossRef]
- Chen, T.; Jia, J.; Zou, D.; Shen, C.; Wang, S.; Gao, X.; Yun, Z.; Ma, J.; Zhao, J.; Wang, H. Nuclear magnetic resonance-based solvent system selection for counter-current chromatography separation of compounds present in the same high-performance liquid chromatography peak: Flavonoids in barley seedlings as an example. J. Sep. Sci. 2023, 46, 2300406. [Google Scholar] [CrossRef]
- Zou, D.; Du, Y.; Kuang, J.; Sun, S.; Ma, J.; Jiang, R. PH-zone-refining counter-current chromatography with a hydrophilic organic/salt-containing two-phase solvent system for preparative separation of polar alkaloids from natural products. J. Chromatogr. A 2018, 1553, 1–6. [Google Scholar] [CrossRef]
- Wang, Q.; Chen, T.; Cui, Y.; Li, S.; Jiang, X.; Zhao, G.; Li, Y.; Zou, D. The applicability of pH-zone-refining counter-current chromatography for preparative separation of biosynthesis products: Glycosylation products as example. J. Chromatogr. A 2021, 1657, 462582. [Google Scholar] [CrossRef]
- Chen, T.; Wang, Q.; Wang, Q.; La, M.; Li, Y.; He, L.; Zou, D. Ab initio calculation based solvent system selection in silico for counter-current chromatography: Separation of resibufogenin glycosylation products. J. Chromatogr. A 2022, 1686, 463649. [Google Scholar] [CrossRef]
- Tanimura, T.; Pisano, J.J.; Ito, Y.; Bowman, R.L. Droplet Countercurrent Chromatography. Science 1970, 169, 54–56. [Google Scholar] [CrossRef]
- Ito, Y.; Conway, W.D. Development of countercurrent chromatography. Anal. Chem. 1984, 56, 534A–554A. [Google Scholar] [CrossRef]
- Conway, W.D.; Ito, Y. Resolution in Countercurrent Chromatography. J. Liq. Chromatogr. 1985, 8, 2195–2207. [Google Scholar] [CrossRef]
- Brent Friesen, J.; Pauli, G.F. GUESS—A Generally Useful Estimate of Solvent Systems for CCC. J. Liq. Chromatogr. Rel. Technol. 2005, 28, 2777–2806. [Google Scholar] [CrossRef]
- Ito, Y. Golden rules and pitfalls in selecting optimum conditions for high-speed counter-current chromatography. J. Chromatogr. A 2005, 1065, 145–168. [Google Scholar] [CrossRef]
- Zou, D.l.; Chen, T.; Li, H.m.; Chen, C.; Zhao, J.y.; Li, Y.l. Separation of three phenolic high-molecular-weight compounds from the crude extract of Terminalia Chebula Retz. by ultrasound-assisted extraction and high-speed counter-current chromatography. J. Sep. Sci. 2016, 39, 1278–1285. [Google Scholar] [CrossRef]
- Yu, P.; Ma, J.; Du, Y.; Wang, Q.; Chen, C.; Bai, X.; He, L.; Zou, D. An efficient strategy based on liquid-liquid extraction and pH-zone-refining counter-current chromatography for selective enrichment, separation, and purification of alkaloids and organic Acids from natural products. J. Sep. Sci. 2020, 43, 3607–3614. [Google Scholar] [CrossRef]
- Zou, D.; Zhu, X.; Zhang, F.; Du, Y.; Ma, J.; Jiang, R. An Efficient Strategy Based on Liquid–Liquid Extraction with Three-Phase Solvent System and High Speed Counter-Current Chromatography for Rapid Enrichment and Separation of Epimers of Minor Bufadienolide from Toad Meat. J. Agric. Food Chem. 2018, 66, 1008–1014. [Google Scholar] [CrossRef]
- Li, J.; Wang, Q.; Wang, Y.; La, M.; Mian, R.; He, L.; Suonan, J.; Zou, D. An efficient strategy for large-scale preparation of low polarity gingerols directly from ginger crude extract by high-speed countercurrent chromatography with different rotation mode. J. Sep. Sci. 2023, 46, 2300320. [Google Scholar] [CrossRef]
- Khuri, A.I.; Mukhopadhyay, S. Response surface methodology. Wiley Interdiscip. Rev. Comput. Stat. 2010, 2, 128–149. [Google Scholar] [CrossRef]
- Liu, G.; Feng, S.; Sui, M.; Chen, B.; Sun, P. Extraction and identification of steroidal saponins from Polygonatum cyrtonema Hua using natural deep eutectic solvent-synergistic quartz sand-assisted extraction method. J. Sep. Sci. 2023, 46, 2200823. [Google Scholar] [CrossRef]
- Vyviurska, O.; Koljančić, N.; Gomes, A.A.; Špánik, I. Optimization of enantiomer separation in flow-modulated comprehensive two-dimensional gas chromatography by response surface methodology coupled to artificial neural networks: Wine analysis case study. J. Chromatogr. A 2022, 1675, 463189. [Google Scholar] [CrossRef]
- He, Q.; Lei, Q.; Huang, S.; Zhou, Y.; Liu, Y.; Zhou, S.; Peng, D.; Deng, X.; Xue, J.; Li, X. Effective extraction of bioactive alkaloids from the roots of Stephania tetrandra by deep eutectic solvents-based ultrasound-assisted extraction. J. Chromatogr. A 2023, 1689, 463746. [Google Scholar] [CrossRef]
- Dean, A.; Voss, D.; Draguljić, D.; Dean, A.; Voss, D.; Draguljić, D. Response surface methodology. In Design and Analysis of Experiments; Springer: Berlin/Heidelberg, Germany, 2017; pp. 565–614. [Google Scholar]
- Oka, F.; Oka, H.; Ito, Y. Systematic search for suitable two-phase solvent systems for high-speed counter-current chromatography. J. Chromatogr. A 1991, 538, 99–108. [Google Scholar] [CrossRef]
- Sutherland, I.A. Recent progress on the industrial scale-up of counter-current chromatography. J. Chromatogr. A 2007, 1151, 6–13. [Google Scholar] [CrossRef]
- Conway, W.D. Counter-current chromatography: Simple process and confusing terminology. J. Chromatogr. A 2011, 1218, 6015–6023. [Google Scholar] [CrossRef]
- Hopmann, E.; Arlt, W.; Minceva, M. Solvent system selection in counter-current chromatography using conductor-like screening model for real solvents. J. Chromatogr. A 2011, 1218, 242–250. [Google Scholar] [CrossRef]
- Hopmann, E.; Frey, A.; Minceva, M. A priori selection of the mobile and stationary phase in centrifugal partition chromatography and counter-current chromatography. J. Chromatogr. A 2012, 1238, 68–76. [Google Scholar] [CrossRef]
- Hu, R.; Pan, Y. Recent trends in counter-current chromatography. TrAC Trends Anal. Chem. 2012, 40, 15–27. [Google Scholar] [CrossRef]
- Jang, Y.S.; Wang, Z.; Lee, J.-M.; Lee, J.-Y.; Lim, S.S. Screening of Korean Natural Products for Anti-Adipogenesis Properties and Isolation of Kaempferol-3-O-rutinoside as a Potent Anti-Adipogenetic Compound from Solidago virgaurea. Molecules 2016, 21, 226. [Google Scholar] [CrossRef]
- Zhang, L.; Yue, H.L.; Zhao, X.H.; Li, J.; Shao, Y. Separation of Four Phenylpropanoid Glycosides from a Chinese Herb by HSCCC. J. Chromatogr. Sci. 2014, 53, 860–865. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Std | ID | Run | Factor 1 | Factor 2 | Factor 3 | Response 1 |
|---|---|---|---|---|---|---|
| A: Ultrasonic Power | B: Water Content | C: Liquid/Solid Ratio | Content of Targets | |||
| W | % | 1 | % | |||
| 17 | 13 | 1 | 300 | 40 | 20 | 63.41 |
| 15 | 13 | 2 | 300 | 40 | 20 | 66.06 |
| 5 | 5 | 3 | 200 | 40 | 10 | 55.86 |
| 13 | 13 | 4 | 300 | 40 | 20 | 65.99 |
| 8 | 8 | 5 | 400 | 40 | 30 | 60.15 |
| 7 | 7 | 6 | 200 | 40 | 30 | 60.52 |
| 12 | 12 | 7 | 300 | 50 | 30 | 65.7 |
| 6 | 6 | 8 | 400 | 40 | 10 | 52.58 |
| 1 | 1 | 9 | 200 | 30 | 20 | 50.23 |
| 16 | 13 | 10 | 300 | 40 | 20 | 64.85 |
| 10 | 10 | 11 | 300 | 50 | 10 | 58.33 |
| 11 | 11 | 12 | 300 | 30 | 30 | 52.88 |
| 3 | 3 | 13 | 200 | 50 | 20 | 69.44 |
| 2 | 2 | 14 | 400 | 30 | 20 | 54.58 |
| 14 | 13 | 15 | 300 | 40 | 20 | 64.52 |
| 4 | 4 | 16 | 400 | 50 | 20 | 60.97 |
| 9 | 9 | 17 | 300 | 30 | 10 | 50.15 |
| Source | Sum of Squares | df | Mean Square | F-Value | p-Value | |
|---|---|---|---|---|---|---|
| Model | 589.59 | 9 | 65.51 | 56.48 | <0.0001 | significant |
| A—Ultrasonic power | 7.55 | 1 | 7.55 | 6.51 | 0.0381 | |
| B—Water content | 271.45 | 1 | 271.45 | 234.04 | <0.0001 | |
| C—Liquid/solid ratio | 62.33 | 1 | 62.33 | 53.74 | 0.0002 | |
| AB | 41.09 | 1 | 41.09 | 35.43 | 0.0006 | |
| AC | 2.12 | 1 | 2.12 | 1.83 | 0.2187 | |
| BC | 5.38 | 1 | 5.38 | 4.64 | 0.0682 | |
| A2 | 33.58 | 1 | 33.58 | 28.96 | 0.0010 | |
| B2 | 46.88 | 1 | 46.88 | 40.42 | 0.0004 | |
| C2 | 99.62 | 1 | 99.62 | 85.90 | <0.0001 | |
| Residual | 8.12 | 7 | 1.16 | |||
| Lack of fit | 3.24 | 3 | 1.08 | 0.8854 | 0.5207 | not significant |
| Pure error | 4.88 | 4 | 1.22 | |||
| Cor total | 597.71 | 16 |
| Solvent Systems | K2 | K3 | K4 | K5 |
|---|---|---|---|---|
| 5:9 (4:5) | 0.14 | 0.08 | 0.16 | 0.21 |
| 5:8 (3:5) | 0.20 | 0.19 | 0.26 | 0.32 |
| 5:7 (2:5) | 0.33 | 0.41 | 0.55 | 0.62 |
| 5:6 (1:5) | 0.42 | 0.69 | 0.78 | 0.93 |
| 6:6 (1:5) | 0.57 | 0.90 | 1.39 | 2.17 |
| 7:6 (1:5) | 0.94 | 1.12 | 1.49 | 2.66 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.; Dai, J.; Zhang, X.; Wang, Y.; He, F.; Liang, L.; Longzhu, D.; Zou, D. Extraction and Counter-Current Separation of Phenylpropanoid Glycosides from Pedicularis oederi Vahl by Deep Eutectic Solvent. Separations 2024, 11, 323. https://doi.org/10.3390/separations11110323
Wang Y, Dai J, Zhang X, Wang Y, He F, Liang L, Longzhu D, Zou D. Extraction and Counter-Current Separation of Phenylpropanoid Glycosides from Pedicularis oederi Vahl by Deep Eutectic Solvent. Separations. 2024; 11(11):323. https://doi.org/10.3390/separations11110323
Chicago/Turabian StyleWang, Yao, Jun Dai, Xiaoting Zhang, Yuhan Wang, Fangfang He, Lu Liang, Duojie Longzhu, and Denglang Zou. 2024. "Extraction and Counter-Current Separation of Phenylpropanoid Glycosides from Pedicularis oederi Vahl by Deep Eutectic Solvent" Separations 11, no. 11: 323. https://doi.org/10.3390/separations11110323
APA StyleWang, Y., Dai, J., Zhang, X., Wang, Y., He, F., Liang, L., Longzhu, D., & Zou, D. (2024). Extraction and Counter-Current Separation of Phenylpropanoid Glycosides from Pedicularis oederi Vahl by Deep Eutectic Solvent. Separations, 11(11), 323. https://doi.org/10.3390/separations11110323