
Synthesis and Characterization of Electrospun Sorbent for the Solid-Phase Extraction of Fluoroquinolones in Human Plasma and Their UHPLC-PDA Determination

 , ,
, ,  , , , and
, , , and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Chemicals and Materials
2.2. Apparatus and UHPLC Conditions
2.3. Preparation of Standard Solutions and Real Samples
2.4. Preparation of PAN Electrospun Nanofibers
2.5. Solid-Phase Extraction Procedure
2.6. Characterization
3. Results and Discussion
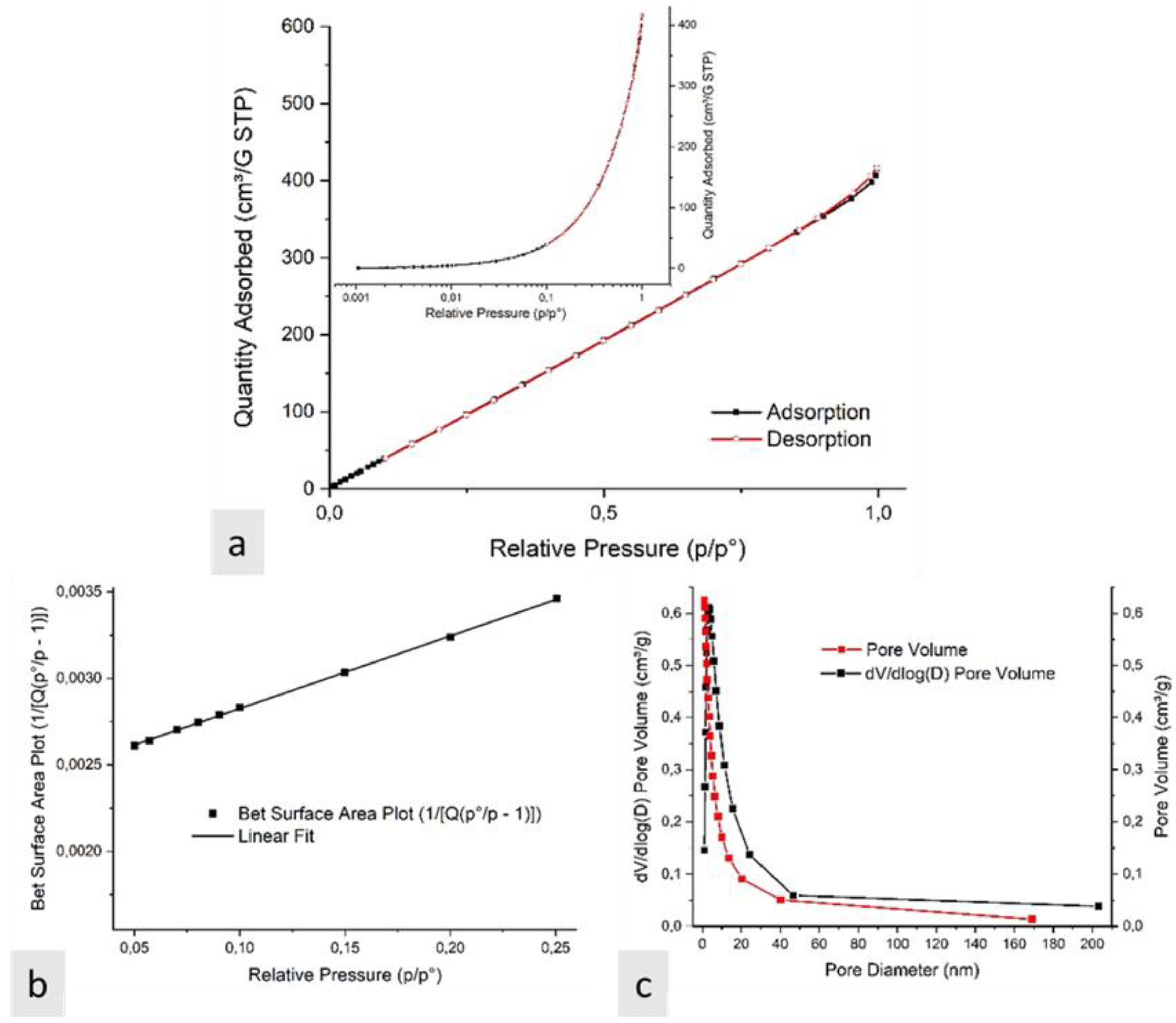
3.1. Characterization of Electrospun Nanofibers
3.2. Optimization of the Extraction Procedure
3.2.1. Effect of the pH
3.2.2. Effect of PAN-PMMA Nanofiber Amount and Sample Volume
3.2.3. Effect of Washing Solution
3.2.4. Effect of Elution Solvent Type and Volume
3.3. Analytical Performance of the Proposed SPE Method
3.4. Comparison with Existing Method in Literature
4. Conclusions
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Carlucci, G. Analysis of fluoroquinolones in biological fluids by high performance liquid chromatography: Review. J. Chromatogr. A 1998, 812, 343–367. [Google Scholar] [CrossRef]
- Lipsky, B.A.; Baker, C.A. Fluoroquinolone Toxicity Profiles: A Review Focusing on Newer Agents. Clin. Infect. Dis. 1999, 28, 352–364. [Google Scholar] [CrossRef] [PubMed]
- Kamberi, M. Pharmacokinetic/Pharmacodynamic Parameters: Rationale for Antibacterial Dosing of Mice and Men. Clin. Infect. Dis. 1998, 26, 1–12. [Google Scholar]
- Hooper, D.C. Mechanisms of action and resistance of older and newer fluoroquinolones. Clin. Infect. Dis. 2000, 31, S24–S28. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Guo, Z.; Wang, X.; Qiu, C. Sample preparation. J. Chromatogr. A 2008, 1184, 191–219. [Google Scholar] [CrossRef] [PubMed]
- Moldoveanu, S.C.; David, V. Sample Preparation in Chromatography; Elsevier: Amsterdam, The Netherlands, 2002. [Google Scholar]
- Buszewsky, B.; Szultka, M. Past, present and future of solid phase extraction: A review. Crit. Rev. Anal. Chem. 2012, 42, 198–213. [Google Scholar] [CrossRef]
- Ferrone, V.; Todaro, S.; Carlucci, M.; Fontana, A.; Ventrella, A.; Carlucci, G.; Milanetti, E. Optimization by response surface methodology of a dispersive magnetic solid phase extraction exploiting magnetic graphene nanocomposite coupled with UHPLC-PDA for simultaneous determination of new oral anticoagulants (NAOs) in human plasma. J. Pharm. Biomed. Anal. 2020, 179, 112992. [Google Scholar] [CrossRef]
- Ferrone, V.; Carlucci, M.; Palumbo, P.; Carlucci, G. Bioanalytical method development for quantification of ulifloxacin, fenbufen and felbinac in rat plasma by solid-phase extraction (SPE) and HPLC with PDA detection. J. Pharm. Biomed. Anal. 2016, 123, 205–212. [Google Scholar] [CrossRef]
- Pawliszyn, J.; Lord, H.L. Handbook of Sample Preparation; Wiley: New York, NY, USA, 2010. [Google Scholar]
- Milanetti, E.; Carlucci, G.; Olimpieri, P.P.; Palumbo, P.; Carlucci, M.; Ferrone, V. Correlation analysis based on the hydropathy properties of non-steroidal anti-inflammatory drugs in solid-phase extraction (SPE) and reversed-phase high performance liquid chromatography (HPLC) with photodiode array detection and their applications to biological samples. J. Chromatogr. A 2019, 1605, 3603351. [Google Scholar]
- Casado, N.; Gañán, J.; Zarcero, S.; Sierra, I. New Advanced Materials and Sorbent-Based Microextraction Techniques as Strategies in Sample Preparation to Improve the Determination of Natural Toxins in Food Samples. Molecules 2020, 25, 702. [Google Scholar] [CrossRef]
- Nayl, A.; Abd-Elhamid, A.; Awwad, N.; Abdelgawad, M.; Wu, J.; Mo, X.; Gomha, S.; Aly, A.; Bräse, S. Review of the Recent Advances in Electrospun Nanofibers Applications in Water Purification. Polymers 2022, 14, 1594. [Google Scholar] [CrossRef] [PubMed]
- Yoshimatsu, K.; YE, L.; Lindberg, J.; Chronakis, I. Selective molecular adsorption using electrospun nanofiber affinity membranes. Biosens. Bioelectron. 2008, 23, 1208–1215. [Google Scholar] [CrossRef] [PubMed]
- Xue, J.; Wu, T.; Dai, Y.; Xia, Y. Electrospinning and Electrospun Nanofibers: Methods, Materials, and Applications. Chem. Rev. 2019, 119, 5298–5415. [Google Scholar] [CrossRef] [PubMed]
- Luraghi, A.; Peri, F.; Moroni, P. Electrospinning for drug delivery applications: A review. J. Control. Release 2021, 334, 463–484. [Google Scholar] [CrossRef] [PubMed]
- Hong, C.; Yang, K.; Oh, S.; Ahn, J.; Cho, B.; Nah, C. Effect of blend composition on themorphology development of electrospunfibres based on PAN/PMMA blends. Polym. Int. 2008, 57, 1357–1362. [Google Scholar] [CrossRef]
- Wang, T.; Kumar, S. Electrospinning of polyacrylonitrile nanofibers. J. Appl. Polym. Sci. 2006, 102, 1023–1029. [Google Scholar] [CrossRef]
- Chinchillas, M.; Gaxiola, A.; Beltrán, C.; Orozco, V.; Cervantes, M.; Rodríguez, M.; Beltrán, A. A new application of recycled-PET/PAN composite nanofibers to cement–based materials. J. Clean. Prod. 2020, 252, 119827. [Google Scholar] [CrossRef]
- Babo, S.; Ferreira, J.L.; Ramos, A.M.; Micheluz, A.; Pamplona, M.; Casimiro, M.H.; Ferreira, L.; Melo, M. Characterization and Long-Term Stability of Historical PMMA: Impact of Additives and Acrylic Sheet Industrial Production Processes. Polymers 2020, 12, 2198. [Google Scholar] [CrossRef]
- Gopiraman, M.; Kim, I. Preparation, Characterization and Applications of Electrospun Carbon Nanofibers and Its Composites. In Electrospinning Electrospray–Techniques and Application; Intech Open: Rijeka, Croatia, 2019; Chapter 3. [Google Scholar]
- Kopec, M.; Lamson, M.; Yuan, R.; Tang, C.; Kruk, M.; Zhong, M.; Matyjaszewski, K.; Kowalewski, T. Polyacrylonitrile-derived nanostructured carbon materials. Prog. Polym. Sci. 2019, 92, 89–134. [Google Scholar] [CrossRef]
- Choi, S.; Han, E.; Park, K. Porosity Control of Electrospun PAN/PMMA Nanofiber Webs. Mol. Cryst. Liq. Cryst. 2019, 688, 68–74. [Google Scholar] [CrossRef]
- Abeykoon, N.; Bonso, J.; Ferraris, J. Supercapacitor performance of carbon nanofiber electrodes derived from immiscible PAN/PMMA polymer blends. RSC Adv. 2015, 5, 19865–19873. [Google Scholar] [CrossRef]
- Bazilevsky, A.; Yarin, A.; Megaridis, C. Co-electrospinning of Core-Shell Fibers Using a Single-Nozzle Technique. Langmuir 2007, 23, 2311–2314. [Google Scholar] [CrossRef] [PubMed]
- He, G.; Song, Y.; Chen, S.; Wang, L. Porous carbon nanofiber mats from electrospun polyacrylonitrile/polymethylmethacrylate composite nanofibers for supercapacitor electrode materials. J. Mater. Sci. 2018, 53, 9721–9730. [Google Scholar] [CrossRef]
- Khanlou, H.M.; Ang, B.C.; Talebian, S.; Afif, A.M.; Andriyana, A. Electrospinning of polymethyl methacrylate nanofibers: Optimization of processing parameters using the Taguchi design of experiments. Text. Res. J. 2015, 85, 356–368. [Google Scholar] [CrossRef]
- Xuan, D.; Liu, J.; Wang, D.; Lu, Z.; Liu, Q.; Liu, Y.; Li, S.; Zheng, Z. Facile Preparation of Low-Cost and Cross-Linked Carbon Nanofibers Derived from PAN/PMMA/Lignin as Supercapacitor Electrodes. Energy Fuels 2021, 35, 796–805. [Google Scholar] [CrossRef]
- Maroni, F.; Bruni, P.; Suzuki, N.; Aihara, Y.; Croce, F. Electrospun tin-carbon nanocomposite as anode material for all solid-state lithium-ion batteries. J. Solid State Electrochem. 2019, 23, 1697–1703. [Google Scholar] [CrossRef]
- Marinelli, L.; Cacciatore, I.; Eusepi, P.; Di Biase, G.; Morroni, G.; Cirioni, O.; Giacometti, A.; Di Stefano, A. Viscoelastic behaviour of hyaluronic acid formulations containing carvacrol prodrugs with antibacterial properties. Int. J. Pharm. 2020, 582, 119306. [Google Scholar] [CrossRef]
- Duan, G.; Zhang, C.; Li, A.; Yang, X.; Lu, L.; Wang, X. Preparation and Characterization of Mesoporous Zirconia Made by Using a Poly (methyl methacrylate) Template. Nanoscale Res. Lett. 2008, 3, 118–122. [Google Scholar] [CrossRef]
- Nataraj, S.K.; Yang, K.S.; Aminabhavi, T.M. Polyacrylonitrile-based nanofibers—A state-of-the-art review. Prog. Polym. Sci. 2012, 37, 487–513. [Google Scholar] [CrossRef]
- El-Deen, A.G.; Barakat, N.A.M.; Khalil, K.A.; Kim, H.Y. Development of multi-channel carbon nanofibers as effective electrosorptive electrodes for a capacitive deionization process. J. Mater. Chem. A 2013, 1, 11001–11010. [Google Scholar] [CrossRef]
- Kashiwagi, T.; Hirata, T.; Brown, J.E. Thermal and Oxidative Degradation of Poly(methylMethacrylate): Molecular Weight. Macromolecules 1985, 18, 131. [Google Scholar] [CrossRef]
- Rahaman, M.S.A.; Ismail, A.F.; Mustafa, A. A review of heat treatment on polyacrylonitrile fiber. Polym. Degrad. Stab. 2007, 92, 1421–1432. [Google Scholar] [CrossRef]
- ICH Guideline M10 on Bioanalytical Method Validation and Study Sample Analysis. European Medicines Agency. Guideline on Bioanalytical Method Validation 30/01/2022. Available online: https://www.ema.europa.eu/en/ich-m10-bioanalytical-method-validation-scientific-guideline (accessed on 27 January 2023).
- D’Angelo, V.; Tessari, F.; Bellagamba, G.; De Luca, E.; Cifelli, R.; Celia, C.; Primavera, R.; Di Francesco, M.; Paolino, D.; Di Marzio, L.; et al. Microextraction by packed sorbent and HPLC-PDA quantification of multiple anti-inflammatory drugs and fluoroquinolones in human plasma and urine. J. Enzym. Inhib. Med. Chem. 2016, 31, 110–116. [Google Scholar] [CrossRef] [PubMed]
- Esrafili, A.; Yamini, Y.; Ghambarian, M.; Shariati, S.; Moradi, M. Measurment of fluorochinolone antibiotics from human plasma using hollow fiber liquid-phase microextraction based on carried mediated transport. J. Liq. Chromatogr. Rel. Tech. 2012, 35, 343–354. [Google Scholar] [CrossRef]
- Zhang, J.; Chen, Z.; Tang, F.; Wu, F.; Luo, X.; Liu, G. Fabrication of highly fluorinated porphyrin-based organic frameworks decorated Fe3O4 nanospheres for magnetic solid phase extraction of fluoroquinolones. Microchim. Acta 2022, 189, 449. [Google Scholar] [CrossRef]
- Qiu, Q.; Wu, Y.; Yan, X.; Li, Y.; Li, J.; Chen, Y.; Wu, D. Porous electrospun microfbers for low fow-resistant solid phase extraction of fuoroquinolones in tap water, egg and milk samples. J. Chromatogr. A 2022, 1661, 462719. [Google Scholar] [CrossRef]
- He, H.; Dong, C.; Li, B.; Dong, J.P.; Bo, T.Y.; Wang, T.L.; Feng, Y.Q. Fabrication of enrofoxacin imprinted organic–inorganic hybrid mesoporous sorbent from nanomagnetic polyhedral oligomeric silsesquioxanes for the selective extraction of fuoroquinolones in milk samples. J. Chromatogr. A 2014, 1361, 23–33. [Google Scholar] [CrossRef]
- Wang, H.; Liu, Y.; Wei, S.; Yao, S.; Zhang, J.; Huang, H. Selective extraction and determination of fuoroquinolones in bovine milk samples with montmorillonite magnetic molecularly imprinted polymers and capillary electrophoresis. Anal. Bioanal. Chem. 2016, 408, 589–598. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Analyte | Concentration | Precision | Accuracy | ||
|---|---|---|---|---|---|
| Intra-Day | Inter-Day | Intra-Day | Inter-Day | ||
| Levofloxacin | LLOQ | 4.9 | 5.1 | +5.1 | +6.3 |
| QCL | 3.5 | 3.8 | +4.9 | +5.5 | |
| QCM | 4.7 | 5.0 | −2.0 | −4.7 | |
| QCH | 2.3 | 1.9 | −1.1 | −3.2 | |
| Ciprofloxacin | LLOQ | 4.9 | 5.0 | −1.9 | −3.2 |
| QCL | 3.1 | 3.9 | +1.5 | +1.9 | |
| QCM | 4.1 | 4.4 | −4.0 | −5.3 | |
| QCH | 4.4 | 3.9 | +5.3 | +6.0 | |
| Lomefloxacin | LLOQ | 5.0 | 4.9 | +7.0 | +7.4 |
| QCL | 1.2 | 3.0 | −6.4 | −6.8 | |
| QCM | 2.3 | 3.0 | −5.9 | −5.9 | |
| QCH | 2.2 | 2.2 | +3.1 | +3.2 | |
| Enrofloxacin | LLOQ | 4.8 | 5.0 | +5.9 | +6.9 |
| QCL | 2.2 | 2.5 | +0.7 | +2.1 | |
| QCM | 1.8 | 2.4 | +0.2 | +1.0 | |
| QCH | 3.1 | 2.3 | −4.2 | −5.5 | |
| Sparfloxacin | LLOQ | 5.0 | 4.9 | −0.9 | −1.5 |
| QCL | 2.6 | 2.7 | +3.1 | +3.6 | |
| QCM | 0.9 | 1.7 | +6.5 | +6.1 | |
| QCH | 1.1 | 2.0 | −3.2 | −3.3 | |
| Sarafloxacin | LLOQ | 4.7 | 3.3 | −4.4 | −5.9 |
| QCL | 3.1 | 3.9 | +3.2 | +3.6 | |
| QCM | 2.3 | 3.1 | +1.1 | +1.9 | |
| QCH | 1.5 | 4.0 | +4.2 | +4.0 | |
| Gatifloxacin | LLOQ | 4.0 | 5.1 | −3.1 | −3.4 |
| QCL | 2.2 | 4.0 | +2.7 | +3.6 | |
| QCM | 2.4 | 3.3 | +4.8 | +5.9 | |
| QCH | 1.4 | 3.0 | +6.5 | +6.0 | |
| Danofloxacin | LLOQ | 3.9 | 3.5 | −6.1 | −5.4 |
| QCL | 3.0 | 4,9 | −5.4 | −6.0 | |
| QCM | 1.5 | 3.1 | +2.6 | +3.1 | |
| QCH | 2.0 | 4.0 | +0.3 | +0.9 | |
| Analyte | Matrix | Sample Preparation | Instrumentation | Range (µg/mL) | Mean Recovery | LOQ (µg/mL) | Ref. |
|---|---|---|---|---|---|---|---|
| CIP, MOX a, LEV, ULI | Human plasma | MEPS b | HPLC-PDA | 0.1–10 | n.a. | 0.1 | [37] |
| OFL c, CIP | Human plasma | HF-LPME d | HPLC-UV | 0.01–5 | 30% | 0.01 | [38] |
| MAR e, LOM, DIF g, NOR h, CIP, ENR | Milk | dMSPE f | HPLC-PDA | 0.01–20 | 94.7% | 0.01 | [39] |
| CIP, ENR, SAR, DIF, DAN | Water, Egg, Milk | SPE | UHPLC-FLD | 0.005–10 | 101.9% | 0.005 | [40] |
| OFL, ENR, DAN | Milk | dMSPE | HPLC-UV | 0.05–1.0 | 80.1% | 0.05 | [41] |
| FLE i, GAT, LOM, NOR | Milk | dMSPE | CE j-UV | 0.03–1.0 | 102.1% | 0.03 | [42] |
| CIP, LEV, LOM, SPA, SAR, GAT, ENR, DAN | Human plasma | SPE | UHPLC-PDA | 0.005–10 | 98.9% | 0.005 | This work |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ferrone, V.; Carlucci, G.; Bruni, P.; Marinelli, L.; Avino, P.; Milanetti, E.; Pilato, S.; Sbrascini, L.; Di Profio, P.; Ferrari, S. Synthesis and Characterization of Electrospun Sorbent for the Solid-Phase Extraction of Fluoroquinolones in Human Plasma and Their UHPLC-PDA Determination. Separations 2023, 10, 104. https://doi.org/10.3390/separations10020104
Ferrone V, Carlucci G, Bruni P, Marinelli L, Avino P, Milanetti E, Pilato S, Sbrascini L, Di Profio P, Ferrari S. Synthesis and Characterization of Electrospun Sorbent for the Solid-Phase Extraction of Fluoroquinolones in Human Plasma and Their UHPLC-PDA Determination. Separations. 2023; 10(2):104. https://doi.org/10.3390/separations10020104
Chicago/Turabian StyleFerrone, Vincenzo, Giuseppe Carlucci, Pantaleone Bruni, Lisa Marinelli, Pasquale Avino, Edoardo Milanetti, Serena Pilato, Leonardo Sbrascini, Pietro Di Profio, and Stefania Ferrari. 2023. "Synthesis and Characterization of Electrospun Sorbent for the Solid-Phase Extraction of Fluoroquinolones in Human Plasma and Their UHPLC-PDA Determination" Separations 10, no. 2: 104. https://doi.org/10.3390/separations10020104
APA StyleFerrone, V., Carlucci, G., Bruni, P., Marinelli, L., Avino, P., Milanetti, E., Pilato, S., Sbrascini, L., Di Profio, P., & Ferrari, S. (2023). Synthesis and Characterization of Electrospun Sorbent for the Solid-Phase Extraction of Fluoroquinolones in Human Plasma and Their UHPLC-PDA Determination. Separations, 10(2), 104. https://doi.org/10.3390/separations10020104