Abstract
The primary cause of poor and ambiguous results obtained from the bioanalytical process is the sample pre-treatment, especially in clinical analysis because it involves dealing with complex sample matrices, such as whole blood, urine, saliva, serum, and plasma. So, the aim of this review is to focus attention on the classical and new techniques of pre-treatment for biological samples used in the bioanalytical process. We discussed the methods generally used for these types of complex samples. Undoubtedly, it is a daunting task to deal with biological samples because the analyst may encounter a substantial loss of the analytes of interest, or the overall analysis may be too time-consuming. Nowadays, we are inclined to use green solvents for the environment, but without sacrificing analytical performance and selectivity. All the characteristics mentioned above should be added to the difficulty of the withdrawal of samples like blood because it can be an invasive practice. For these reasons, now we can also find in the literature the use of saliva as alternative biological samples and new techniques that do not require substantial sample pre-treatment, such as fabric phase sorptive extraction (FPSE). The text has been divided into the following two distinct parts: firstly, we described clinical applications under different subsections, such as anticancer drugs, antibiotics, vitamins, antivirals, non-steroidal anti-inflammatory drugs, statin, imidazoles, and triazoles. The second part is dedicated to sample preparation techniques for diagnostic purposes and is divided into the following different sample preparation techniques: solid-phase microextraction (SPME), microextraction by packed sorbent (MEPS), dispersive liquid–liquid microextraction (DDLME), and fabric phase sorptive extraction (FPSE).
1. Introduction
The analysis of biological samples requires an adequate sample preparation procedure that has an inevitable role in the analytical process. In fact, the sample preparation step may influence the accuracy of results and consumes most of the time needed for the overall analytical workflow.
The traditional methods applied in many fields, including bioanalysis, such as solid-phase extraction (SPE) and liquid–liquid extraction (LLE), have shown some limitations due to their use of toxic organic solvents and long processing times; therefore, new methods based on microextraction techniques have been developed to achieve a high recovery of target analytes from simple to complex sample matrices and to comply with the concepts of Green Chemistry (GC) and Green Analytical Chemistry (GAC) [1].
Biological samples include whole blood, plasma, serum, urine, feces, saliva, bile, hair, sweat, breast milk, cerebrospinal fluid (CSF)), and tissues, and bio analytes (DNA, defined proteins, drugs, and specific metabolites or unknown molecules).
The stability of these samples is important. They are often rich in endogenous components, such as carbohydrates, proteins, lipids, and salts, which can interfere with the target analytes due to matrix effects. Moreover, analytes are often present at very low concentrations, and for this reason, the target analytes need to be preconcentrated prior to analysis. This often enhances the levels of interfering molecules (e.g., drugs, salts and metabolites, nucleic acids, proteins, and peptides) [2].
After collection from patients and appropriate storage, the sample clean-up and preparation step become crucial to avoid damage also caused by endogenous degrading properties (enzymes activities, cell death, etc.) and to perform an accurate and selective bioanalysis to obtain a real and correct “picture” of the studied system [3].
These sample preparation techniques include liquid phase microextraction (LPME) with its different modifications such as single-drop microextraction (SDME), dispersive liquid-liquid microextraction (DLLME), hollow fiber liquid-phase microextraction (HF-LPME) and solidified floating organic drop microextraction (SFO-DME). In LPME, the extraction phase is based on the action of different solvents (e.g., DESs, ILs, and ferrofluids) that, with their features and properties, play an important role for the entire process (in terms of recovery, enrichment factor, selectivity).
Other techniques include solid phase microextraction (SPME), microextraction by packed sorbent (MEPS), and fabric phase sorptive extraction (FPSE) [4,5]. These techniques manifest a rapid development in different fields, such as biological, environmental, food sciences, natural products, forensic medicine, and toxicology [1].
The clinical applications of (bio)analytical sample treatment procedures are very extensive in different fields, ranging from the search for biomarkers useful in the diagnosis of many diseases to those necessary for personalized therapeutic drug monitoring (TDM).
In this review, the attention is focused primarily on the clinical applications of these procedures, highlighting the major advantages and results that have been made in laboratory and clinical practices.
2. Clinical Applications
2.1. Anticancer Drugs
Zufía, Aldaz, and Giráldez (2004) reported the use of a mixture of ethyl acetate and acetonitrile (4:1, v/v) after sample acidification with orthophosphoric acid to simultaneously extract capecitabine, 5′-DFUR (5′-deoxy- 5-fluorouridine), 5-FU (5-fluorouracil), and 5-FUH2 (5,6-dihydro-5-fluorouracil) from plasma. A similar extraction method is also reported using LLE with a mixture of ethyl acetate and acetonitrile as the extractant (8:3, v/v) [6].
Piórkowska et al. (2014) have reported a similar extraction method using a mixture of ethyl acetate and acetonitrile (4:1, v/v) but without sample acidification. Acidification was not deemed necessary because the assay was developed to determine only capecitabine concentrations (no metabolites) [6].
Zufía, Aldaz, and Giráldez [7] used LLE, specifically using a mixture of ethyl acetate and acetonitrile (4:1, v/v) after acidification of samples to extract capecitabine, 5′-dFUR, 5-FU, and 5-FUH2 from human plasma. Piórkowska et al. [8] reported a similar LLE method using a mixture of ethyl acetate and acetonitrile (4:1, v/v), but without sample acidification, because they wanted to find only capecitabine and not its metabolites.
Table 1 provides a summary of sample-pretreatment procedures and the analytical figures of merit for capecitabine and its metabolites from different biological samples.

Table 1.
Pre-treatment of biological samples for capecitabine and its metabolites.
Many studies reported the use of protein precipitation-like sample treatment for bioanalytical analysis. Deng et al. [14] and Thorat et al. [15] used methanol as the solvent to precipitate the proteins from human plasma and human serum. Furthermore, Vainchtein et al. [17], with the aim of not losing the drug, added 10% v/v of trichloroacetic acid in water.
Another protocol used to extract anticancer drugs, specifically osimertinib, involved LLE. Fresnai et al. used tert-butyl methyl ether for simultaneous extraction and concentration of it. This method was developed to follow treatment for patients with non-small cell lung carcinoma with epidermal growth factor receptor activating mutation [20].
Llopis et al. studied nine kinase inhibitors, including cobimetinib, dasatinib, ibrutinib, imatinib, nilotinib, palbociclib, ruxolitinib, sorafenib, and vemurafenib; the following two active metabolites of them: N-desmethyl imatinib, N-oxide sorafenib; the following two anti-androgen drugs: abiraterone and enzalutamide. They used a single-step protein precipitation with 100 uL of aqueous Zinc Sulfate Monohydrate ZnSO4·H2O (10% w/v pH 5.4), adding this to 50 uL of human plasma followed by vortexing and centrifugation [21].
Locatelli et al. used whole blood, saliva, and urine samples. The difficulty, especially for whole blood, was to minimize the matrix interferents that can mess up the analysis. Usually, the first step is protein precipitation to convert whole blood into plasma. In this case, the authors directly used whole blood without converting it into plasma, thanks to the extraction protocol using fabric phase sorptive extraction (FPSE) to avoid the possible loss of analytical information related to the quantification of anastrozole, letrozole, and exemestane used in the treatment of metastatic breast cancer [22].
2.2. Antibiotics
Ferrari et al. wanted to highlight the matter of personal treatment of bacterial infections because different people may respond to the drugs in different ways. So, they followed pharmacokinetics studies on piperacillin, meropenem, linezolid, and teicoplanin in plasma samples from patients treated with the mentioned antibiotics. Pre-treatment involved a liquid–liquid extraction, vortexing for 10 s, and the addition of a precipitation buffer, and then vortexing followed by centrifugation. The supernatant was mixed with a dilution buffer and finally analyzed [23].
Wongchang et al. performed their sample preparation through protein precipitation. Plasma samples were aliquoted, and they added acetonitrile with cefotaxime as an internal standard. The samples were mixed and centrifuged. After this, the supernatant was loaded on the Phree phospholipid removal plate, and a vacuum was applied. Finally, the samples were diluted, mixed, and centrifuged [24].
Krnac et al. used human plasma as the sample for their analysis. The aim of this study was to monitor tazobactam, piperacillin, and meropenem in patients treated in the intensive care unit. The authors used a mixture of methanol–acetonitrile–water (6:2:2, v/v/v), specifically 200 uL for 10 uL of human plasma plus 10 uL of internal standard, to precipitate proteins. After the mixing, the samples were centrifuged, and the supernatant was used for the analyses [25].
2.3. Vitamin
Rola et al. used LLE to find the concentration of vitamin D and its metabolites in the serum. At the beginning of the procedure, the authors applied LLE followed by PP, but due to the low recovery of 24,25-dihydroxyvitamin D3 (24,25(OH)2 D3), they opted for only PP in the following two different steps: the first one for releasing the compound bound to the protein and the second step is to precipitate most proteins possible. In this case, the organic solvent was acetonitrile [26].
Moreover, Hotta et al. used PP to understand the pharmacokinetic profile of methylcobalamin, which is important in the reaction of Vitamin B12. In this case, the authors used methanol as a protein precipitation solvent because the extraction of methylcobalamin was higher with methanol than with acetonitrile [27].
2.4. Antiviral
Present HIV therapy appreciates the use of a combination of different drugs, such as dolutegravir, elvitegravir, reltegravir, nevirapine, and etravirine. Bollen et al. studied the therapeutic drug monitoring of these antiretroviral drugs to analyze the pharmacokinetics trials. The authors used plasma samples that pretreat with PP and used the supernatant as a liquid to analyze [28].
Elkady et al. analyzed the treatment of the hepatitis C viral infection. Specifically, the combination therapy of sofosbuvir and velpatasvir was found successful in the treatment. They used human plasma, which was pretreated with acetonitrile, then vortexed, and finally centrifuged [29].
2.5. Non-Steroidal Anti-Inflammatory Drugs (NSAIDs)
Raabova et al. used human serum taken from patients that were under treatment of intravenous infusion with 75 mg of sodium diclofenac. The blood samples were diluted 10 times with 20% aqueous acetonitrile, centrifuged, and the supernatant was analyzed [30].
Kabir et al. in 2018 focused attention on the analysis of inflammatory bowel disease (IBD). Among different treatment regimens used in IBD, therapeutic intervention by oral administration of cortisone, ciprofloxacin, and/or sulfasalazine, among others, are common practices. As such, the authors used a combination of cortisone, ciprofloxacin, and sulfasalazine as an oral treatment regimen. For pharmacokinetic studies, the authors used physiological samples such as plasma, serum, saliva, and urine. Whole blood was diluted with ultrapure water (1:5, v/v) and vortexed. Human plasma was directly vortexed after adding an internal standard, and the same protocol was used for urine. Finally, samples were ready for the FPSE procedure to analyze the supernatant [31].
Tartaglia et al. availed saliva samples from healthy volunteers in the absence of NSAIDs with the aim of analyzing furprofen, indoprofen, ketoprofen, fenbufen, flurbiprofen, and ibuprofen in the above samples. Thanks to FPSE, the authors did not need any pre-treatment, only the addition of the internal standard [32].
D’angelo et al. developed an analytical method based on MEPS and HPLC to simultaneously quantify FANS and fluoroquinolones. They collected plasma and urine samples from healthy volunteers 5 h after oral administration of FANS and FLQ. The addition of trichloroacetic acid aimed for the denaturation of proteins, and after a centrifugation step, the supernatant was used for the MEPS extraction [33].
2.6. Statin
Courlet et al. discussed about the risk of cardiovascular events in HIV patients. Atorvastatin, rosuvastatin, and pravastatin are the most prescribed lipid-lowering agents in this class of patients. In their study, blood samples from different patients for pharmacokinetics studies were collected. The pre-treatment of these samples was protein precipitation. Following centrifugation, the supernatant was then evaporated until dryness and then reconstituted in methanol as follows: H2O with 0.1% formic acid, vortexed, sonicated, and centrifuged. Finally, the superatant was ready to be analyzed [34].
2.7. Imidazoles and Triazoles
Campestre et al. studied imidazole as ketoconazole, bifonazole, clotrimazole, tioconazole, econazole, butoconazole, and miconazole and triazole as terconazole, voriconazole, posaconazole, ravuconazole, and itraconazole. Specifically, the research group was interested in the quantification of azole derivates in the human samples collected from healthy volunteers (plasma and urine). The samples were mixed with the working analyte solution as well as internal standard and vortexed. With the aim of denaturing proteins and reducing sample density, the samples were pre-treated with trichloroacetic acid and centrifuged, then the supernatant was directly analyzed [35].
The aim of this study was to simplify the clean-up procedures that precede the analysis in HPLC. These authors used plasma and urine samples from healthy volunteers, added analyte working solutions and internal standard f to them and applied or extraction procedure with FPSE membranes, and then analyzed the supernatant in HPLC [36].
3. Sample Preparation Techniques for Diagnostic Purposes
Particular attention is paid to novel extraction techniques, which significantly reduced the number of operations to be carried out during sample preparation. The aim was to develop extraction devices and technologies easily for operators to reduce time and minimize errors during operations [37]. As reported in Figure 1, sample preparation remain the principal source of error generated during chromatographic analysis. In the “other” section were reported all other points not specifically reported and labelled in Figure 1 (e.g. purity of chemical reagents, their "age" and expiration date, etc.).
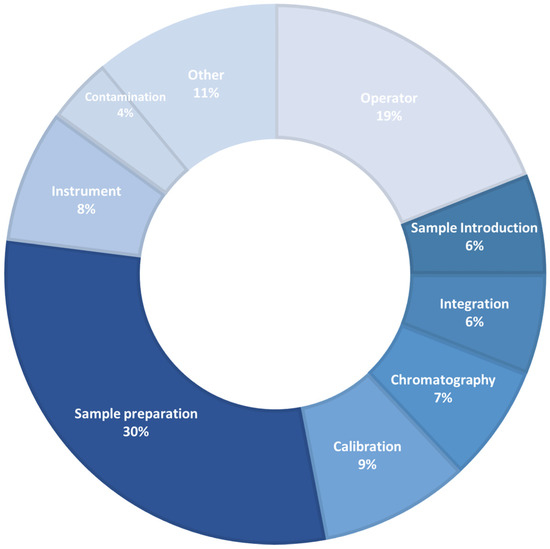
Figure 1.
Sources of error generated during chromatographic analysis.
3.1. Solidphase Microextraction (SPME)
Biological samples contain a variety of metabolites that can be used for clinical purposes in the diagnosis of various clinical conditions and many severe disorders.
Among sample preparation techniques, the most widely used procedure is solidphase microextraction (SPME), which includes sampling, extraction, and analyte pre-concentration into a single step. By contact with liquid bio-matrices, analytes are transferred to the adsorbent-coated solid surface thanks to the affinity of analytes to the coating material.
Direct immersion SPME (DI–SPME) and headspace SPME (HS–SPME) are the two modes of SPME operations. The first extraction technique is based on the sorption of analytes on the stationary phase immobilized on a fused silica glass rod that acts as the solid support. During the analytes’ extraction, the coated fiber is directly immersed into the sample solution, while in the HS–SPME, the fiber is exposed in the vapor phase above the sample. At equilibrium, the quantity of analyte extracted by the fiber is proportional to its concentration in the sample [38].
The main advantages of this method include simplicity and speed of operations, high sensitivity, and the volume of solvent reduction; for these reasons it has been applied to the investigation of metabolites and neurotransmitters for diagnostic purposes [2].
Many metabolites and neurotransmitters are useful for the diagnosis of neurological diseases. Neurotransmitters (NTs) are basic signaling molecules used for cell communications; they are involved in stress response mechanisms, the regulation of motor coordination, the control of psychomotor, gastrointestinal, and homeostatic function and neuronal communication.
Many psychomotor, psychiatric, neurodegenerative, and oncological disorders are characterized by an abnormal production, release, and/or metabolism of NTs; for this reason, NTs can be used as a potential biomarker for diagnoses of Alzheimer’s and Parkinson’s diseases, neuroendocrine cancers, and psychiatric illness such as schizophrenia.
The most common biological fluids in the NT analysis are urine and blood, but other alternative matrices include CSF and cerebral tissue, with the related difficulty due to the presence of interfering molecules and low levels of the target analytes. Therefore, the development of validated methods of sample preparation and analytical techniques for the quantitative analysis of NT in biological samples is needed [39].
Naccaro et al. developed a method for the determination of NTs such as dopamine (DOP), norepinephrine (NE), and serotonin (5-HT) in human urine by SPME coupled with gas chromatography–triple quadrupole mass spectrometry (GC-QqQ-MS).
The optimized SPME method provides a first step of extraction with polyacrylate fiber at room temperature for 45 min without the addition of NaCl; the second step was desorption at 300 °C, followed by a derivatization reaction carried out directly in a urine sample and SPME extraction in immersion mode in the same vial (without organic solvent or further sample treatment).
The developed method has shown good linearity with correlation coefficient values of 0.9995 for DOP, 0.9999 for 5-HT, and 0.9987 for NE; the accuracy values were between 92.8% and 103.0%, and RSD% ranged between 0.67% and 4.5%. LOD values were 0.587 µg/L for DOP, 0.381 µg/L for 5-HT, and 13.5 µg/L for NE, and LOQ values were 0.81 µg/L for DOP, 0.74 µg/L for 5-HT and 21.3 µg/L for NE.
The obtained satisfactory results indicate that the proposed method can be adapted for the analysis of urine samples, which contain a much lower concentration of DOP, 5-HT, and NE than those determined for healthy adults. Thanks to the rapidity and simplicity of this SPME method, with the advantages of minimal sample handling and high sensitivity, this procedure might be considered a valuable tool for the diagnosis of cancerous and neurological diseases correlated with the urinary levels of analyzed amines [40].
Among the markers proposed for the diagnosis of oncological diseases, the scientific literature proposes the analysis of volatile organic compounds (VOCs) as a valid alternative due to their importance in the identification and differentiation of cancer samples. In particular, the determination of urinary VOCs has been proposed as alternative cancer biomarkers for lung, bladder, and breast cancer.
Monteiro et al. validated a new HS-SPME method to obtain a urine volatile metabolomics profile useful for renal cell carcinoma (RCC). These compounds generally require a high efficiency of extraction that depends on the sample preparation method selected.
In this study, the authors validated an HS-SPME method after the optimization of different factors that may influence the results, such as the pH of the urine sample, fiber coating, time of incubation, and time and temperature of extraction. They tested different conditions (three pH levels, time, and temperatures of extraction, temperature of incubation) and fibers (five types of SPME fibers); amongst these, the best results were obtained with the exposition of 2 mL of a urine sample with optimal pH 2.0.
The use of an acidic pH value confirmed a better HS-SPME extraction in terms of the total of compounds and total chromatographic peak area, but strong acidic conditions may determine some errors due to the chemical degradation of some pH-sensitive compounds.
Moreover, the fiber with the highest and better performance for urinary volatile metabolites was divinylbenzene/polydimethylsiloxane (DVB/PDMS). The optimal analytical performance was also obtained with the addition of 0.59 g of NaCl, 9 min of incubation, and extraction at 68 °C in 24 min.
This method was simple, solvent-free, inexpensive, and fast, with a combination of extraction and preconcentration of a sample with minimal pre-treatment [41].
Deeva et al. validated an SPME method to obtain preconcentration of VOCs in the urine sample of patients with prostate cancer already confirmed by prostatic puncture biopsy for subsequent non-target GC-MS analysis.
In this procedure, 3 mL of urine with 0.3 g of sodium chloride was placed inside the headspace vial. The extraction equilibrium was obtained with stirring (250 rpm) on the hotplate. The 100-μm-thick polydimethylsiloxane fibers were used for SPME. These fibers were treated at 250 °C for 15 min to remove impurities before sample analysis. The fibers were placed in the headspace above the gas of the urine samples for 20 min at 50 °C and then were inserted into the hot GC injector with the adsorbed analytes, which were desorbed for 4 min in splitless modes. After this procedure, the samples were analyzed with GC-MS. This method showed a very high sensitivity, specificity, and accuracy, and might be a good approach in non-invasive prostate cancer screening [42].
Blood steroid levels are often required for the diagnosis of various endocrinological diseases involving the pituitary-adrenal axis, such as hypercortisolism and Cushing’s syndrome. In addition to blood, saliva also represents a biological sample of considerable interest for diagnostic purposes, due to the presence of these hormones in free form with concentration correlated to that present in the blood. Often the saliva concentration of endogenous steroids is very low, and it is necessary to employ a highly sensitive analytical technique.
Bessoneau et al. validated a method for the quantification of endogenous steroids (cortisol, testosterone, and progesterone, E1, E2, and E3) in saliva with in vivo SPME. The extraction method was developed using the biocompatible coated SPME blades that were first conditioned for 30 min with 1.5 mL of methanol/water (50:50, v: v) in 96-well-plates with agitation; all analytes required a time of 60 min for equilibrium extraction. To guarantee a complete immersion of the coating in the sample and to improve the method sensitivity, they have chosen a volume of saliva of 1.2 mL. After extraction, the method involves static washing with deionized water (1.5 mL) to remove matrix components from the surface of the coating and prevent subsequent desorption, no loss of analytes was detected after this step.
The optimal conditions of desorption for all analytes were 90 min in acetonitrile/water 80:20 (v: v). The LOQ was 178, 22, 105, 185, 29, and 16 pg mL−1 for cortisol, testosterone, progesterone, E3, E2, and E1, respectively. This method has shown advantages in terms of high sensitivity, reproducibility, and accuracy for the detection of endogenous steroids in saliva. These characteristics make the method better than the common immunoassays, which may lead to erroneous results due to the significant cross-reaction; the validated SPME method showed better results than liquid–liquid extraction (LLE) thanks to a cleaner extract that prevents matrix effects. Moreover, it was observed that in vivo sampling had a better efficiency than ex vivo sampling of saliva [43].
In recent years, a new special application of the SPME technique was observed that includes in vivo analysis. This method combines the advantages of the SPME technique for sample preparation that requires no, or small amount of, organic solvent and the advantages of the in vivo sampling that eliminates errors related to degradation or loss of short-life or unstable analytes; moreover, with this sampling method, it was possible to sample over time the same individual (e.g., drug monitoring).
SPME was coupled to GC-MS or LC-MS/MS to separate and detect analytes or directly to MS. Usually, this extraction technique depends on the fiber and matrix characteristics. The following different SPME coating fibers have been employed to determine numerous compounds with different volatility and polarity in complex matrices: CAR/PDMS (Carboxen/polydimethylsiloxane) have been used to determine VOCs, while PDMS-DVB fibers have been used to determine the non-polar or weakly polar VOCs with GC-MS analysis, drugs and metabolites with direct MS and endogenous hydrocarbons in human breath with GC-MS.
Different methods have been developed with this in vivo procedure for non-invasive and sensitive determination of different bio analytes in different types of samples in different fields of applications.
Amongst complex matrices used for in vivo SPME methods, the easiest to sample is the skin with direct contact of SPME fiber placed on the skin surface; moreover, breath and saliva can be used to obtain in vivo pathophysiological information.
For example, VOCs in human exhaled breath or emanating from human skin are potentially used as biomarkers for different disease diagnoses.
The methodology of in vivo SPME of VOCs in human skin was used to record the VOC profile in skin cancer (e.g., basal cell carcinoma), fibrotic skin disorders, or vascular lesions. In addition, VOCs in human breath are useful in the diagnosis of lung cancer, liver disease, myocardial infarction, or diabetes [44].
3.2. Microextraction by Packed Sorbent (MEPS)
Microextraction by packed sorbent (MEPS) is a miniaturized SPE technique with very interesting potential benefits, such as low solvent volume for analytes elution, small sample volume (10–250 µL), direct injection without further treatments, simple operation, and cost-effectiveness. In this procedure, about 1–2 mg of sorbent (such as C18, C8) is packed in the barrel of a gas-tight syringe positioned between the needle and the barrel as a cartridge. When the sample passed through the solid bed, the analytes were absorbed into the sorbent [1,38].
Ghimenti et al. validated an innovative method that combined MEPS with ultra-high-performance liquid chromatography coupled to electrospray ionization–triple quadrupole mass spectrometry (UHPLC-ESI-MS/MS) for the simultaneous determination of 8-isoprostaglandin F2α (8-isoPGF2α) and cortisol in saliva samples collected from patients with heart failure. During the optimization of this method, they maximized the extraction efficiency of 8-isoPGF2α and cortisol with the dilution of saliva samples with water (ratio 1:5, v/v) and filtration by syringe filter prior to MEPS extraction to avoid proteins, mucins, and other interferences that may cause deterioration of the sorbent and cartridge clogging, without loss of analytes. A LOD of 10 pg/mL was obtained for each target analyte; the coefficient of determination (R2) was 0.7, and the p-value was < 0.001.
Compared to other techniques, such as enzyme-linked immunosorbent assays (ELISA), which may lead to partially inaccurate results or solid-phase extraction (SPE) procedures, which required requires a larger volume of samples, a substantial problem for saliva analysis, the obtained results with MEPS showed that this is a good alternative with different advantages. MEPS procedure required a low volume of organic solvents (<100 μL/measurement) and a reusable sorbent (over 100 extractions with the same cartridge), which permits automating the extraction procedure with a reduction of the cost of analysis. In addition, this procedure showed a satisfactory recovery (95–110%) and an adequate limit of detection, which guarantees a reliable determination of low concentration levels of 8-isoPGF2α and cortisol in real saliva samples. The obtained results confirmed that the MEPS method allows reliable quantification of 8-isoPGF2α and cortisol as new potential non-invasive biomarkers for monitoring patients with heart failure and is useful for clinical purposes [45].
Berenguer et al. developed a highly sensitive MEPS/UHPLC method to detect and quantify lipid peroxidation biomarkers, such as leukotrienes E4 (LTE4), B4 (LTB4), and 11β-prostaglandin F2α (11βPGF2α) related to asthma, in the urine of child patients. The targeted analytes were isolated from urine using a semiautomatic miniaturization of solid-phase extraction, the MEPS; the LOD for LTB4 and 11βPGF2α was, respectively, 0.04 µg L−1 and 1.12 µg L−1 while LOQ values ranged between 0.10 µg L−1 for the LTB4 and 2.11 µg L−1 for 11βPGF2α. Compared to other methodologies, this technique showed more advantages, such as lower elution solvent volume consumption, high precision due to the semi-automatic procedure, excellent recoveries, and extraction efficiency. Moreover, the extraction procedure is fast, with a total analysis time of 31 min. The obtained values for the 11βPGF2α and LTB4 were about 1.8 times higher, and for the LTE4 were about 1.4 times higher in asthmatic patients than in healthy individuals suggesting the potential of these eicosanoids on asthma diagnosis. Consequently, MEPS/UHPLC-PDA represents a promising method for the simultaneous detection of these and other eicosanoids present in other biological matrices as plasma or saliva of patients with different inflammatory diseases [46].
3.3. Dispersive Liquid–Liquid Microextraction (DLLME)
Dispersive liquid–liquid microextraction (DLLME) is a type of sample preparation based on the liquid phase microextraction (LPME) technique in which the target analytes are extracted from an aqueous solution with small volumes of extraction solvent. In DLLME, the dispersion procedure increases the extraction kinetics by enlarging the exposure surface between the sample and the extractant, the emulsion was centrifuged, and the extractant was isolated. This technique showed several advantages, such as easy operation, low sample volume, and rapid processing with high recovery without the necessity of any appliance [2].
Zhou et al. validate an easy method of sample preparation with a high efficiency based on ultrasound-assisted ionic liquid dispersive liquid–liquid microextraction (UA-IL-DLLME) for simultaneous determination of neurotransmitters (NTs), such as γ-aminobutyric acid (GABA), acetylcholine (Ach), and glutamic acid (Glu) in mild cognitive impairment, mild dementia, and moderate dementia patients’ urine samples. The analysis of clinical samples showed that some NTs presented significant differences in different dementia stages. This method showed many advantages, such as selectivity, stability, sensitivity, and simple procedure of sample preparation. Thanks to these characteristics, the validated strategy of preparation and analysis might be used also with different types of biological samples [47]. Table 2 represents a summary of microextraction techniques used for diagnostic purposes.
Table 2.
Microextraction techniques used for diagnostic purposes.
3.4. Fabric Phase Sorptive Extraction (FPSE)
Fabric phase sorptive extraction (FPSE) is an innovative technique developed by Kabir and Furton [56] that offers a substantial simplification of the selective extraction of the target analytes from biological samples. Nowadays, thanks to the new analytical methods based on FPSE, it is possible to determine low concentrations of analytes with no/minimal sample pretreatment. In addition, the several steps operations in the treatment of complex biological matrices (for example, whole blood, urine, saliva, etc.), such as protein precipitation, sonication, elution of extract in an organic solvent, solvent evaporation and sample reconstitution, elution of extract, and more, are often the cause of most of the errors during the analytical process and can be eliminated from the sample preparation workflow when FPSE is used [57].
Briefly, when the FPSE membrane is immersed into the aqueous sample, the analyte(s) permeates through the sol-gel sorbent, such as an SPE disk, and establishes intermolecular interactions, such as an SPE disk. However, at the same time, the analyte(s) continue to accumulate on the FPSE membrane based on the partition coefficient between the sol-gel sorbent and the sample matrix. Therefore, this novel technique combines both the exhaustive extraction principle and the equilibrium-driven extraction principle by design [58].
Samanidou et al. used three FPSE media membranes, in particular, sol-gel poly (dimethyldiphenylsiloxane) (sol-gel PDMDPS), sol-gel poly (tetrahydrofuran) (sol-gel PTHF,) and sol-gel poly (ethylene glycol) (sol-gel PEG) to determinate benzodiazepines from human blood serum. The authors demonstrated the simplicity of this extraction in terms of the number of operations in comparison with other innovative techniques, such as SPE [57]. Figure 2 compares the steps involved in SPE and FPSE.
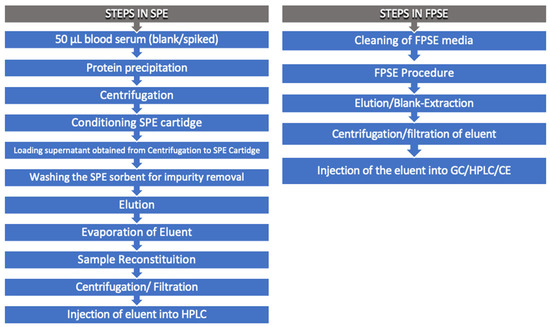
Figure 2.
Comparison between solid-phase extraction (SPE) and fabric phase sorptive extraction (FPSE).
As shown above, Locatelli M. et al. integrate the FPSE with HPLC-DAD for separation and quantification of anastrozole, letrozole, and exemestane from whole blood, plasma, and urine. The first step was the creation of a sol-gel sorbent coated membrane as follows: the selection of appropriate substrate and its pre-treatment, the design of the most appropriate sol solution to obtain maximum selectivity, the coat the pre-treated fabric substrate with sol-gel sorbent, and the condition and clean the FPSE membrane. Membranes were cut to have an identical surface area with an internal diameter of 11 cm. They were washed first with acetonitrile as follows: methanol (50:50, v/v) and then with milliQ water. Subsequently, the conditioned membranes were inserted into the sample (diluted whole blood, plasma, and urine) and were left for 30 min on the rotator. After this time, the analytes extracted were eluted from FSPE membranes using methanol, and the eluant sample was centrifugated. An aliquot from obtained supernatant was directly injected into the HPLC-DAD for the analysis [22].
Tartaglia et al. used eight different FPSE membranes for preliminary experiments to extract antidepressant drugs from conventional biological matrices (plasma and urine) and unconventional biological matrices (human saliva). For the optimization of the extraction process, all the parameters were gradually optimized to obtain the maximum extraction efficiency of the compounds. After the activation of the membranes, the following initial general conditions have been tested: each support was immersed in 500 µL of standard solution for 30 min under stirring, and then each membrane was immersed in 150 µL of MeOH for 30 min for analyte back-extraction. Subsequently, the eluant was tested and the following parameters were optimized: extraction time, elution solvents and mixtures, back extraction time, and volume. After the selection of optimal conditions using standard solutions, the optimization was performed on the biological complex matrices mentioned previously [59].
In the same way, Locatelli et al. have optimized all parameters to obtain the maximum extraction of twelve azole antimicrobial drug residues from human plasma and urine. They have investigated three different FPSE membranes and the different dimensions of these to study the optimum sample volume and the optimum extraction media dimension [36].
Kabir A. et al. have used this simple and powerful technique combined with HPLC-DAD for the simultaneous monitoring of three anti-inflammatory drugs (ciprofloxacin, sulfasalazine, and cortisone) administered in the treatment of inflammatory bowel disease. They have tested five different FPSE media membranes coated with different sorbents and different extraction parameters to develop an extraction procedure for molecules that are characterized by a wide range of LogP [31].
Gazioglu I. et al. have combined FPSE-extraction with HPLC-fluorimetric detection to simultaneously quantify Febuxostat and Montelukast in human blood [60].
Recently, FPSE extraction was also used by Tiris G. et al. to extract an anti-viral drug from breast milk [61] and was also applied for in vivo non-invasive exhaled breath aerosol sampling to monitor the exposure to xenobiotics by Locatelli M. et al. [62] and other xenobiotics [63,64].
The amazing benefits of FPSE as a new-generation sample preparation technique were further manifested in a study presented by G. Sidiropoulou et al. [65], where FPSE was combined with UPLC-ESI-MS/MS to monitor the presence of residual antiviral drugs, including amantadine, memantine, and rimantadine in human urine. Sol-gel poly(propylene glycol)-poly(ethylene glycol)-poly(propylene glycol)-coated FPSE membranes reached extraction equilibrium in just 20 min, thanks to the rapid mass transfer of the target analytes from the bulk of the sample to the FPSE membrane due to the engineered affinity of the sol-gel sorbent towards the target analytes via different intermolecular interactions and the sponge-like porous structure of the sol-gel sorbent. A matrix-matched calibration curve was developed in the range of 5–100 ng ml−1 to aid the quantitative analysis of the target analytes in the unknown clinical samples. The FPSE combined with UPLC-ESI-MS/MS method provides LOD values in the range of 0.2–0.8 ng/mL and LOQ value of 5 ng/mL for all target analytes. It is worth mentioning that, unlike other sample preparation techniques, FPSE did not require any kind of pretreatment of urine samples prior to the extraction of analytes on the FPSE membrane.
Another interesting study is presented by K. Mazaraki et al. [66], where the researchers investigated the presence of traces of six beta-blockers possessing widely varied logP values (from 0.1 to 3.1) in human serum and urine using FPSE as the sample preparation technique followed by UHPLC-ESI-MS/MS as the chromatographic technique. Sol-gel CW 20M sorbent coated on 100% cotton cellulose was identified as the most efficient FPSE membrane for the target analytes. After optimizing all the FPSE parameters, the FPSE-UHPLC-ESI-MS/MS composite method provided LOD values for the selected beta-blockers from 0.3 to 2 ng/mL, and the LLOQ values were calculated as 50 ng/mL in both serum and urine samples. The method was linear in the broad range of 50–5000 ng/mL.
G. Tris et al. [61] described an FPSE method followed by HPLC-UV analysis designed to monitor favipiravir, a promising antiviral agent for the treatment of coronavirus, in human plasma and breast milk samples. The overall chromatographic run was only 5 min. The LOQ values of the validated method were 0.2 µg/mL in plasma and 0.5 µg/mL.
By eliminating the protein precipitation and other sample pretreatment steps from the sample preparation workflow, FPSE has established itself as a rapid, simple, and green sample preparation technique for forensic toxicology. Recently, M. Locatelli et al. [67] proved the operational simplicity and performance superiority of FPSE in processing post-mortem samples, including whole blood and cerebrospinal liquor collected during autopsy to monitor seven common antidepressant drugs. A sol-gel CW20M-coated FPSE membrane was identified as the best FPSE sorbent for the selected drugs with an optimum extraction time of 20 min, back-extraction time of 5 min, and 150 µL of methanol as the eluent. The overall chromatographic run time was 20 min. The LOQ values were 0.1 µL/mL for all drug compounds except Venlafaxine, which was 0.2 µL/mL.
The performance superiority, simplicity, and easiness in the application of this novel technique have been demonstrated in many articles and reviews, in particular, in extractions from complex sample matrices [32,63,64].
4. Conclusions
As can be seen from the manuscript, the last several years were important to develop new strategies and techniques to apply in bioanalytical studies. The principal aim was to maximize the performance with these samples in terms of efficiency and operational steps and speed, complying with green chemistry principles and the possibilities to use them for different applications. It is important to emphasize the diversification of the samples, from conventional samples such as whole blood or urine to unconventional such as saliva or breast milk, but at the same time duration of the sample preparation workflow, efficiency, reproducibility, and the potential reuse of the sample preparation device to reduce the overall analytical cost. Moreover, the wide range of samples can be applied in different clinical analyses, useful for the research’s progress. The clinical analysis includes different studies such as pharmacokinetics studies or studies that may give help in a specific treatment regimen or studies that diagnose some pathologies. Therefore, the employment of different matrices means different approaches in terms of preparation and pre-pre-treatment, and it follows the possibilities to differ specific studies. For complex sample matrices, the principal issue can be the pre-treatment and extraction of the sample. In fact, pre-treatment is often the principal cause of the error. This is the primary rationale as to why we discussed new techniques, easy for operators, simple to execute, and capable of so they can be able to reduce overall sample preparation time and the number of steps involved in operations.
Author Contributions
All Authors contributed equally in terms of data curation, writing, and editing. M.L., A.G., F.S., U.d.G., A.K., H.I.U., C.D. and I.A.: conceptualization; M.L., A.G., F.S., U.d.G., A.K., H.I.U., C.D. and I.A.: supervision and project administration; V.G., O.M., E.R., M.P., L.C., M.L., A.G., F.S., U.d.G., A.K., H.I.U., C.D. and I.A.: writing—original draft preparation and writing—review and editing. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data and information are available on request to the authors.
Acknowledgments
Authors would like to thank the University “G. d’Annunzio” for the support in the literature survey.
Conflicts of Interest
The authors declare no conflict of interest.
References
- D’Ovidio, C.; Bonelli, M.; Rosato, E.; Tartaglia, A.; Ulusoy, H.I.; Samanidou, V.; Furton, K.G.; Kabir, A.; Ali, I.; Savini, F.; et al. Novel Applications of Microextraction Techniques Focused on Biological and Forensic Analyses. Separations 2022, 9, 18. [Google Scholar] [CrossRef]
- Ingle, R.G.; Zeng, S.; Jiang, H.; Fang, W.J. Current developments of bioanalytical sample preparation techniques in pharmaceuticals. J. Pharm. Anal. 2022, 12, 517–529. [Google Scholar] [CrossRef]
- Pandey, K.; Mishra, O.P. Advancement in Analytical and Bioanalytical Techniques as a Boon to Medical Sciences. In Biochemical Testing; Bobbarala, V., Zaman, G.S., Desa, M.N.M., Akim, A.M., Eds.; IntechOpen: London, UK, 2020; Volume 12, pp. 29–46. [Google Scholar]
- Pourshamsi, T.; Amri, F.; Abniki, M. A comprehensive review on application of the syringe in liquidand solid phase microextraction methods. J. Iran. Chem. Soc. 2021, 18, 245–264. [Google Scholar] [CrossRef]
- Hansen, F.A.; Pedersen-Bjergaard, S. Emerging Extraction Strategies in Analytical Chemistry. Anal. Chem. 2020, 92, 2–15. [Google Scholar] [CrossRef]
- Knikma, J.E.; Rosing, H.; Guchelaar, H.J.; Cats, A.; Beijnen, J.H. A review of the bioanalytical methods for the quantitative determination of capecitabine and its metabolites in biological matrices. Biomed. Chromatogr. 2020, 34, e4732. [Google Scholar]
- Zufia, L.; Aldaz, A.; Giraldez, J. Simple determination of capecitabine and its metabolites by liquid chromatography with ultraviolet detection in a single injection. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2004, 809, 51–58. [Google Scholar] [CrossRef]
- Piórkowska, E.; Kaza, M.; Fitatiuk, J.; Szlaska, I.; Pawinski, T.; Rudzki, P.J. Rapid and simplified HPLC-UV method with on-line wavelengths switching for determination of capecitabine in human plasma. Pharmazie 2014, 69, 500–505. [Google Scholar]
- Licea-Perez, H.; Wang, S.; Bowen, C. Development of a sensitive and selective LC-MS/MS method for the determination of á-fluoroâ-alanine, 5-fluorouracil and capecitabine in human plasma. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2009, 877, 1040–1046. [Google Scholar] [CrossRef]
- Salvador, A.; Millerioux, L.; Renou, A. Simultaneous LC-MS-MS Analysis of Capecitabine and its Metabolites (50 -deoxy5-fluorocytidine, 50 -deoxy-5-fluorouridine, 5-fluorouracil) After OffLine SPE from Human Plasma. Chroma 2006, 63, 609–615. [Google Scholar] [CrossRef]
- Buchner, P.; Mihola, E.; Sahmanovic, A.; Steininger, T.; Dittrich, C.; Czejka, M. Validation of a Simple Assay for the Quantification of the Capecitabine Metabolites 5′–DFCR and 5′–DFUR for Drug Monitoring in Patients Receiving Outpatient Chemotherapy. Anticancer Res. 2013, 33, 881–886. [Google Scholar]
- Farkouh, A.; Ettlinger, D.; Schueller, J.; Georgopoulos, A.; Scheithauer, W.; Czejka, M. A rapid and simple HPLC assay for quantification of capecitabine for drug monitoring purposes. Anticancer Res. 2010, 30, 5207–5211. [Google Scholar]
- Xu, Y.; Grem, J.L. Liquid chromatography-mass spectrometry method for the analysis of the anti-cancer agent capecitabine and its nucleoside metabolites in human plasma. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2003, 783, 273–285. [Google Scholar] [CrossRef] [PubMed]
- Deng, P.; Ji, C.; Dai, X.; Zhong, D.; Ding, L.; Chen, X. Simultaneous determination of capecitabine and its three nucleoside metabolites in human plasma by high performance liquid chromatography-tandem mass spectrometry. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2015, 989, 71–79. [Google Scholar] [CrossRef] [PubMed]
- Thorat, S.G.; Chikhale, R.V.; Tajne, M.R. A rapid and simple HPTLC assay for therapeutic drug monitoring of capecitabine in colorectal cancer patients. Biomed. Chromatogr. 2018, 32, e4100. [Google Scholar] [CrossRef] [PubMed]
- Guichard, S.M.; Mayer, I.; Jodrell, D.I. Simultaneous determination of capecitabine and its metabolites by HPLC and mass spectrometry for preclinical and clinical studies. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2005, 826, 232–237. [Google Scholar] [CrossRef]
- Vainchtein, L.D.; Rosing, H.; Schellens, J.H.M.; Beijnen, J.H. A new, validated HPLC-MS/MS method for the simultaneous determination of the anti-cancer agent capecitabine and its metabolites: 50 -deoxy-5- fluorocytidine, 50-deoxy-5-fluorouridine, 5-fluorouracil and 5-fluorodihydrouracil, in human plasma. Biomed. Chromatogr. 2010, 24, 374–386. [Google Scholar] [CrossRef]
- Wang, Z.; Li, X.; Yang, Y.; Zhang, F.; Li, M.; Chen, W.; Gao, S.; Chen, W. A Sensitive and Efficient Method for Determination of Capecitabine and Its Five Metabolites in Human Plasma Based on One-Step LiquidLiquid Extraction. J. Anal. Methods Chem. 2019, 2019, 9371790. [Google Scholar] [CrossRef]
- Deenen, M.J.; Rosing, H.; Hillebrand, M.J.; Schellens, J.H.M.; Beijnen, J.H. Quantitative determination of capecitabine and its six metabolites in human plasma using liquid chromatography coupled to electrospray tandem mass spectrometry. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2013, 913–914, 30–40. [Google Scholar] [CrossRef]
- Fresnai, M.; Roth, A.; Foerster, K.I.; Jäger, D.; Pfister, S.M.; Haefeli, W.E.; Burhenne, J.; Longuespée, R. Rapid and Sensitive Quantification of Osimertinib in Human Plasma Using a Fully Validated MALDI–IM–MS/MS Assay. Cancers 2020, 17, 1897. [Google Scholar] [CrossRef]
- Llopis, B.; Robidou, P.; Tissot, N.; Pinna, B.; Gougis, P.; Aubart, F.C.; Campedel, L.; Abbar, B.; Weil, D.R.; Uzunov, M.; et al. Development and clinical validation of a simple and fast UPLC-ESI-MS/MS method for simultaneous quantification of nine kinase inhibitors and two antiandrogen drugs in human plasma: Interest for their therapeutic drug monitoring. J. Pharm. Biomed. Anal. 2021, 197, 113968. [Google Scholar] [CrossRef]
- Locatelli, M.; Tinari, N.; Grassadonia, A.; Tartaglia, A.; Macerola, D.; Piccolantonio, S.; Sperandio, E.; D’Ovidio, C.; Carradori, S.; Ulusoy, H.I.; et al. FPSE-HPLC-DAD method for the quantification of anticancer drugs in human whole blood, plasma, and urine. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2018, 1095, 204–213. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, D.; Ripa, M.; Premaschi, S.; Banfi, G.; Castagna, A.; Locatelli, M. LC-MS/MS method for simultaneous determination of linezolid, meropenem, piperacillin and teicoplanin in human plasma samples. Journal of Pharmaceutical and biomedical analysis. J Pharm Biomed Anal. 2019, 169, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Wongchang, T.; Winterberg, M.; Tarning, J.; Sriboonvorakul, N.; Muangnoicharoen, S.; Blessborn, D. Determination of ceftriaxone in human plasma using liquid chromatography–tandem mass spectrometry. Wellcome Open Res. 2021, 4, 47. [Google Scholar] [CrossRef]
- Krnac, D.; Reiffova, K.; Rolinski, B. A new HPLC-MS/MS analytical method for quantification of tazobactam, piperacillin, and meropenem in human plasma. J. Sep. Sci. 2021, 44, 2744–2753. [Google Scholar] [CrossRef] [PubMed]
- Rola, R.; Kowalski, K.; Bienkowski, T.; Studzinska, S. Improved sample preparation method for fast LC-MS/MS analysis of vitamin D metabolites in serum. J. Pharm. Biomed. Anal. 2020, 190, 113529. [Google Scholar] [CrossRef]
- Hotta, K.; Wang, Y.; Mano, Y. A sensitive bioanalytical assay for methylcobalamin, an endogenous and light-labile substance, in human plasma by liquid chromatography with tandem mass spectrometry and its application to a pharmacokinetic study. J. Pharm. Biomed. Anal. 2020, 191, 113621. [Google Scholar] [CrossRef] [PubMed]
- Bollen, P.D.J.; de Graaff-Teulen, M.J.A.; Schalkwijk, S.; van Erp, N.P.; Burger, D.M. Development and validation of an UPLC-MS/MS bioanalytical method for simultaneous quantification of the antiretroviral drugs dolutegravir, elvitegravir, raltegravir, nevirapine and etravirine in human plasma. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2019, 1105, 76–84. [Google Scholar] [CrossRef]
- Elkadya, E.F.; Aboelwaf, A.A. Rapid bioanalytical LC-MS/MS method for the simultaneous determination of sofosbuvir and velpatasvir in human plasma-application to a pharmacokinetic study in Egyptian volunteers. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2018, 1102–1103, 116–124. [Google Scholar] [CrossRef]
- Raabovà, H.; Havlíková, L.C.; Erben, J.; Chvojka, J.; Švec, F.; Šatínský, D. Polycaprolactone Composite Micro/Nanofibrous Material as an Alternative to Restricted Access Media for Direct Extraction and Separation of Non-Steroidal Anti-Inflammatory Drugs from Human Serum Using Column-Switching Chromatography. Nanomaterials 2021, 11, 2669. [Google Scholar] [CrossRef]
- Kabir, A.; Furton, K.G.; Tinari, N.; Grossi, L.; Innosad, D.; Macerola, D.; Tartaglia, A.; Di Donato, V.; D’Ovidio, C.; Locatelli, M. Fabric phase sorptive extraction-high performance liquid chromatography photo diode array detection method for simultaneous monitoring of three inflammatory bowel disease treatment drugs in whole blood, plasma and urine. J. Chromatogr. B 2018, 1084, 53–63. [Google Scholar] [CrossRef]
- Tartaglia, A.; Kabir, A.; D’Ambrosio, F.; Ramundo, P.; Ulusoy, S.; Ulusoy, H.I.; Merone, G.M.; Savini, F.; D’Ovidio, C.; De Grazia, U.; et al. Fast off-line FPSE-HPLC-PDA determination of six NSAIDs in saliva samples. J. Chromatogr. B 2020, 1144, 9. [Google Scholar] [CrossRef] [PubMed]
- D’angelo, V.; Tessari, F.; Bellagamba, G.; De Luca, E.; Cifelli, R.; Celia, C.; Primavera, R.; Di Francesco, M.; Paolino, D.; Di Marzio, L.; et al. Microextraction by packed sorbent and HPLC–PDA quantification of multiple anti-inflammatory drugs and fluoroquinolones in human plasma and urine. J. Enzym. Inhib. Med. Chem. 2016, 31 (Suppl. S3), 110–116. [Google Scholar] [CrossRef] [PubMed]
- Courlet, P.; Spaggiari, D.; Desfontaine, V.; Cavassini, M.; Saldanha, S.A.; Buclin, T.; Marzolini, C.; Csajka, C.; Decoster, L.A. UHPLC-MS/MS assay for simultaneous determination of amlodipine, metoprolol, pravastatin, rosuvastatin, atorvastatin with its active metabolites in human plasma, for population-scale drug-drug interactions studies in people living with HIV. J. Chromatogr. B. 2019, 1125, 11. [Google Scholar] [CrossRef] [PubMed]
- Campestre, C.; Locatelli, M.; Guglielmi, P.; De Luca, E.; Bellagamba, G.; Menta, S.; Zengin, G.; Celia, C.; Di Marzio, L.; Carradori, S. Analysis of imidazoles and triazoles in biological samples after MicroExtraction by packed sorbent. J. Enzym. Inhib. Med. Chem. 2017, 32, 1053–1063. [Google Scholar] [CrossRef]
- Locatelli, M.; Kabir, A.; Innosa, D.; Lopatriello, T.; Furton, K.G. A fabric phase sorptive extraction-High performance liquid chromatography-Photo diode array detection method for the determination of twelve azole antimicrobial drug residues in human plasma and urine. J. Chromatogr. B 2017, 1040, 192–198. [Google Scholar] [CrossRef]
- Kabir, A.; Locatelli, M.; Ulusoy, H.I. Recent Trends in Microextraction Techniques Employed in Analytical and Bioanalytical Sample Preparation. Separations 2017, 4, 36. [Google Scholar] [CrossRef]
- Locatelli, M.; Tartaglia, A.; Piccolantonio, S.; Di Iorio, L.A.; Sperandio, E.; Ulusoy, H.I.; Furton, K.G.; Kabir, A. Innovative configurations of sample preparation Techniques Applied in bioanalytical chemistry: A review. Curr. Anal. Chem. 2019, 15, 731–744. [Google Scholar] [CrossRef]
- Matys, J.; Gieroba, B.; Jó’zwiak, K. Recent developments of bioanalytical methods in determination of neurotransmitters in vivo. J. Pharm. Biomed. Anal. 2020, 180, 12. [Google Scholar] [CrossRef]
- Naccarato, A.; Gionfriddo, E.; Sindona, G.; Tagarelli, A. Development of a simple and rapid solid phase microextraction-gas chromatography–triple quadrupole mass spectrometry method for the analysis of dopamine, serotonin and norepinephrine in human urine. Anal. Chim. Acta 2014, 810, 17–24. [Google Scholar] [CrossRef]
- Monteiro, M.; Carvalho, M.; Henrique, R.; Jeronimo, C.; Moreira, N.; de Lourdes Bastos, M.; de Pinho, P.G. Analysis of volatile human urinary metabolome by solid-phase microextraction in combination with gas chromatography–mass spectrometry for biomarker discovery: Application in a pilot study to discriminate patients with renal cell carcinoma. Eur. J. Cancer 2014, 50, 1993–2002. [Google Scholar] [CrossRef]
- Deev, V.; Solovieva, S.; Andreev, E.; Protoshchak, V.; Karpushchenko, E.; Sleptsov, A.; Kartsova, L.; Bessonova, E.; Legin, A.; Kirsanov, D. Prostate cancer screening using chemometric processing of GC–MS profiles obtained in the headspace above urine samples. J. Chromatogr. B 2020, 1155, 8. [Google Scholar] [CrossRef] [PubMed]
- Bessonneau, V.; Boyaci, E.; Maciazek-Jurczyk, M.; Pawliszyn, J. In vivo solid phase microextraction sampling of human saliva for non-invasive and on-site monitoring. Anal. Chim. Acta 2015, 856, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Costa Queiroz, M.E.; Donizeti de Souza, I.; de Oliveira, I.G.; Grecco, C.F. In vivo solid phase microextraction for bioanalysis. Trends Anal. Chem. 2022, 153, 18. [Google Scholar]
- Ghimenti, S.; Lomonaco, T.; Bellagambi, F.G.; Biagini, D.; Salvo, P.; Trivella, M.G.; Scali, M.C.; Barletta, V.; Marzilli, M.; Di Francesco, F.; et al. Salivary lactate and 8-isoprostaglandin F2á as potential non-invasive biomarkers for monitoring heart failure: A pilot study. Sci. Rep. 2020, 10, 7441. [Google Scholar] [CrossRef]
- Berenguer, P.H.; Camacho, I.C.; Câmara, R.; Oliveira, S.; Câmara, J.S. Determination of potential childhood asthma biomarkers using a powerful methodology based on microextraction by packed sorbent combined with ultra-high pressure liquid chromatography. Eicosanoids as case study. J. Chromatogr. A 2019, 1584, 42–56. [Google Scholar] [CrossRef] [PubMed]
- Zhou, G.-S.; Yuan, Y.-C.; Yin, Y.; Tang, Y.P.; Xu, R.-J.; Liu, Y.; Chen, P.-D.; Yin, L.; Duan, J.-A. Hydrophilic interaction chromatography combined with ultrasound-assisted ionic liquid dispersive liquid-liquid microextraction for determination of underivatized neurotransmitters in dementia patients’ urine samples. Anal. Chim. Acta 2020, 1107, 74–84. [Google Scholar] [CrossRef]
- Monteleone, M.; Naccarato, A.; Sindona, G.; Tagarelli, A. A reliable and simple method for the assay of neuroendocrine tumor markers in human urine by solid-phase microextraction–gas chromatography-triple quadrupole mass spectrometry. Anal. Chim. Acta 2013, 759, 66–73. [Google Scholar] [CrossRef]
- Lord, H.; Yu, Y.; Segal, A.; Pawliszyn, J. Breath analysis and monitoring by membrane extraction with sorbent interface. Anal. Chem. 2002, 74, 5650–5657. [Google Scholar] [CrossRef]
- Lee, C.Y.J.; Jenner, A.M.; Halliwell, B. Rapid preparation of human urine and plasma samples for analysis of F 2-isoprostanes bygas chromatography-mass spectrometry. Biochem. Biophys. Res. Commun. 2004, 320, 696–702. [Google Scholar] [CrossRef]
- Perestrelo, R.; Silva, C.L.; Câmara, J.S. Determination of urinary levels of leukotriene B4 using ad highly specific and sensitive methodology based on automatic MEPS combined with UHPLC-PDA analysis. Talanta 2015, 144, 382–389. [Google Scholar] [CrossRef]
- Xiong, X.; Zhang, Y.; Zhao, R. Quantitative Measurement of Plasma Free Metanephrines by a Simple and Cost-Effective Microextraction Packed Sorbent with Porous Graphitic Carbon and Liquid Chromatography-Tandem Mass Spectrometr. J. Anal. Met. Chem. 2021, 2021, 8821276. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.-R.; Lew, B.-L.; Sim, W.-Y.; Hong, J.; Chung, B.-C. Serial Hydrolysis for the Simultaneous Analysis of Catecholamines and Steroids in the Urine of Patients with Alopecia Areata. Molecules 2021, 26, 2734. [Google Scholar] [CrossRef] [PubMed]
- Castillo-Aguirre, A.; Maldonado, M. Preparation of Methacrylate-Based Polymers Modified with Chiral Resorcinarenes and Their Evaluation as Sorbents in Norepinephrine Microextraction. Polymers 2019, 11, 1428. [Google Scholar] [CrossRef] [PubMed]
- Xiong, X.; Zhang, Y. Simple, rapid, and cost-effective microextraction by the packed sorbent method for quantifying of urinary free catecholamines and metanephrines using liquid chromatography-tandem mass spectrometry and its application in clinical analysis. Anal. Bioanal. Chem. 2020, 412, 2763–2775. [Google Scholar] [CrossRef] [PubMed]
- Kabir, A.; Furton, K.G. Fabric Phase Sorptive Extractors. U.S. Patents 9557252, 31 January 2017. [Google Scholar]
- Samanidou, V.; Kaltzi, I.; Kabir, A.; Furton, K.G. Simplifying sample preparation using fabric phase sorptive extraction technique for the determination of benzodiazepines in blood serum by high-performance liquid chromatography. Biomed. Chromatogr. 2016, 30, 829–836. [Google Scholar] [CrossRef]
- Tartaglia, A.; Locatelli, M.; Kabir, A.; Furton, K.G.; Macerola, D.; Sperandio, E.; Piccolantonio, S.; Ulusoy, H.I.; Maroni, F.; Bruni, P.; et al. Comparison between Exhaustive and Equilibrium Extraction Using Different SPE Sorbents and Sol-Gel Carbowax 20M Coated FPSE Media. Molecules 2019, 24, 382. [Google Scholar] [CrossRef]
- Tartaglia, A.; Covone, S.; Rosato, E.; Bonelli, M.; Savini, F.; Furton, K.G.; Gazioglu, I.; D’Ovidio, C.; Kabir, A.; Locatelli, M. Fabric phase sorptive extraction (FPSE) as an efficient sample preparation platform for the extraction of antidepressant drugs from biological fluids. Adv. Sample Prep. 2022, 3, 100022. [Google Scholar] [CrossRef]
- Gazioglu, I.; Tekkeli, S.E.K.; Tartaglia, A.; Aslan, C.; Locatelli, M.; Kabir, A. Simultaneous determination of febuxostat and montelukast in human plasma using fabric phase sorptive extraction and high-performance liquid chromatography-fluorimetric detection. J. Chromatogr. B 2022, 1188, 123070. [Google Scholar] [CrossRef]
- Tiris, G.; Gazioglu, I.; Furton, K.G.; Kabir, A.; Locatelli, M. Fabric phase sorptive extraction combined with high performance liquid chromatography for the determination of favipiravir in human plasma and breast milk. J. Pharm. Biomed. Anal. 2023, 223, 115131. [Google Scholar] [CrossRef]
- Locatelli, M.; Tartaglia, A.; Ulusoy, H.I.; Ulusoy, S.; Savini, F.; Rossi, S.; Santavenere, F.; Merone, G.M.; Bassotti, E.; D’Ovidio, C.; et al. Fabric-Phase sorptive membrane array as a noninvasive in vivo sampling device for human exposure to different compounds. Anal. Chem. 2021, 93, 1957–1961. [Google Scholar] [CrossRef]
- Tartaglia, A.; Kabir, A.; Ulusoy, S.; Sperandio, E.; Piccolantonio, S.; Ulusoy, H.I.; Furton, K.G.; Locatelli, M. FPSE-HPLC-PDA analysis of seven paraben residues in human whole blood, plasma, and urine. J. Chromatogr. B 2019, 1125, 10. [Google Scholar] [CrossRef] [PubMed]
- Locatelli, M.; Furton, K.G.; Tartaglia, A.; Sperandio, E.; Ulusoy, H.I.; Kabir, A. An FPSE-HPLC-PDA method for rapid determination of solar UV filters in human whole blood, plasma, and urine. J. Chromatogr. B 2019, 1118, 40–50. [Google Scholar] [CrossRef] [PubMed]
- Sidiropoulou, G.; Kabir, A.; Furton, K.G.; Kika, F.S.; Fytianos, K.; Tzanavaras, P.D.; Zacharis, C.K. Combination of fabric phase sorptive extraction with UHPLC-ESI-MS/MS for the determination of adamantine analogues in human urine. Microchem. J. 2022, 176, 107250. [Google Scholar] [CrossRef]
- Mazaraki, K.; Kabir, A.; Furton, K.; Fytianos, K.G.; Samanidou, V.F.; Zacharis, C.K. Fast fabric phase sorptive extraction of selected β-blockers from human serum and urine followed by UHPLC-ESI-MS/MS analysis. J. Pharm. Biomed. Anal. 2021, 199, 114053. [Google Scholar] [CrossRef]
- Locatelli, M.; Covone, S.; Rosato, E.; Bonelli, M.; Savini, F.; Furton, K.G.; Gazioglu, I.; D’Ovidio, C.; Kabir, A.; Tartaglia, A. Analysis of seven selected antidepressant drugs in post–mortem samples using fabric phase sorptive extraction followed by high performance liquid chromatography-photodiode array detection. Forensic. Chem. 2022, 31, 100460. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).