Abstract
The paper is dedicated to the research output presented at the jubilee 50th edition of the international scientific conference Synthesis and Analysis of Drugs 2022 (SAL), which was organized and hosted by the Faculty of Pharmacy of Masaryk University in Brno. Since its launch in 1971, when Jozef Čižmárik from the Faculty of Pharmacy of the Comenius University in Bratislava started organizing SAL, significant progress in medicinal chemistry has been documented every year through the experimental works of researchers from different scientific institutions, both universities and academies of sciences as well as companies. We must point out long-term guest of SAL, Miroslav Protiva, co-founder of the antihistamine dosulepin and antiemetic moxastine-teoclas. As already mentioned, SAL 2022 was held at the Faculty of Pharmacy in Brno, which has been part of the University of Veterinary and Pharmaceutical Sciences for 25 years but became part of Masaryk University in 2021. Hence, the year 2022 was unique not only because of the 50th anniversary of this meeting but also for this year’s conference was the first within this institution. What topics were discussed among SAL conference participants? The meeting addressed all issues related to drug research—their design, preparation, analysis, and detection methods, as well as their biological activity studies, ADME studies, etc. The multidisciplinary character of the research, including topics on pharmaceutical technology and innovative dosage form investigation, was an additional value of the conference and its outcomes. We would like to thank the organization team and all participants for both high-quality research presentations resulting in the output of this paper and for the informal and friendly atmosphere.
Keywords:
drug designing; antibacterials; cancerostatics; antivirotics; theranostics; inhibitors; mass spectrometry; UV/VIS detection; high-performance liquid chromatography; capillary electrophoresis; separation techniques; pharmaceutical and biomedical analysis; lipophilicity; biological activity testing; biodegradable polymers; innovative dosage forms; drug carrier systems; nanoparticles 1. Computational Modeling of 3′-Phosphoadenosine 5′-Phosphosulfate Synthase Papss
1
College of Biomedical Sciences, Larkin University, 18301 North Miami Avenue, Miami, FL 33169, USA
2
Center for Nanobiology and Structural Biology, Institute of Microbiology Academy of Sciences of the Czech Republic, Zamek 136, CZ-37333 Nove Hrady, Czech Republic
3
College of Allophatic Medicine, NOVA Southeastern University, Fort Lauderdale, FL 33314, USA
The sulfur nucleotide PAPS (3′-phosphoadenosine 5′-phosphosulfate) is the universal sulfuryl donor of the cell. In mammals, 3′-phosphoadenosine 5′-phosphosulfate synthase (PAPSS), using ATP, biochemically converts inert inorganic sulfate to the metabolically active PAPS. It is a bi-functional enzyme and catalyzes the formation of PAPS in two sequential steps [1]. In the first step, inorganic sulfate reacts with ATP to form APS and pyrophosphate. The resulting phosphoric-sulfuric anhydride bond has high energy, which is the chemical basis of sulfate activation. The second step is catalyzed by the kinase domain of PAPSS and involves the reaction of APS with ATP to form PAPS and ADP. The proper function of PAPSS is essential for normal physiology in the human being. As the ubiquitous sulfate donor in most biological systems, the product of the enzyme PAPS plays an essential role in ECM formation, embryonic development, and biomolecule secretion. PAPSS has also been shown to be involved with the pathophysiology of a number of diseases, and deficiency in humans results in osteochondrodysplasias or defective cartilage and bone metabolism as evidenced in the clinical condition of the recessively inherited, spondyloepimetaphyseal dysplasia (SEMD). Using a combination of molecular docking, homology modeling, and molecular dynamics simulations in combination with experimental work, we try to understand how the three-dimensional structure of PAPSS determines the enzyme function, focusing on the roles of specific amino acid residues [2], the overall structures on the dynamics of the enzyme in an aqueous solution, and the related quaternary arrangements of the enzyme. Results are discussed that give a realistic picture of the enzyme activity.
References:
- Venkatachalam, K.V. IUBMB Life 2003, 55, 1–11
- K.V. Venkatachalam K.V., Ettrich R.H. Biochemistry and Biophysics Reports 2021, 28, 101155
2. Sustainable Approaches to the Preparation of Isotope-Labelled Pharmaceuticals
Faculty of Chemistry and Chemical Technology, University of Ljubljana, Večna pot 113, Ljubljana 1000, Slovenia
The preparation of isotope-labelled organic compounds, particularly deuterated compounds, has recently become a research area of significant interest for drug development [1,2]. This surge in interest was partly triggered by the FDA approval of deutetrabenazine in 2017, the first for a deuterated pharmaceutical [3].
Two strategies are generally used for the synthesis of deuterium-labeled compounds: (i) a total-synthetic approach starting with commercially available deuterium-labeled precursors and (ii) late-stage functionalization via hydrogen–deuterium (H–D) exchange. The latter approach can potentially produce deuterated compounds more rapidly and cost-effectively [4].
H–D exchange is typically conducted using disposable iridium catalysts due to their regio- and stereo-selectivity, use of relatively mild reaction conditions, and treatment of a broad scope of substrates [5]. However, iridium is the ninth least abundant metal [6] and is currently experiencing a drastic price hike due to a surge in demand for its use in other applications, such as the electrodes in fuel cells for electric cars. Thus, alternative approaches to iridium catalysis are being explored, and these will be expounded during my presentation, with a particular focus on embedding iridium within recyclable macromolecular architectures either as homogeneous or heterogeneous catalysts. I will also briefly introduce my research which centers on the development of recyclable dendrimer catalysts.
The research was supported by the European Union’s Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement No 945380.
References:
- Pirali, T.; Serafini, M.; Cargnin, S.; Genazzani, A. A. J. Med. Chem. 2019, 62(11), 5276–5297.
- Prakash, G.; Paul, N.; Oliver, G. A.; Werz, D. B.; Maiti, D. Chem. Soc. Rev., 2022, 51, 3123–3163.
- Mullard, A. Nat Rev Drug Discov. 2017, 16, 305.
- Kopf, S.; Bourriquen, F.; Li, W.; Neumann, H.; Junge, K.; Beller, M. Chem. Rev. 2022, 122(6), 6634–6718.
- Di Guiseppe, A.; Castarlenas, R.; Oro, L. A. Compt. Rend. Chim. 2015, 18, 713–741.
- Payne, D. 2016. Nature Chem., 8, 392.
3. Design and Synthesis of Aspartyl(Asparaginyl)-Beta-Hydroxylase Inhibitors for the Treatment of Cancer
1
Department of Pharmaceutical Sciences, College of Pharmacy, Midwestern University, 19555 N. 59th Ave., Glendale, AZ, USA, 85308
2
Pharmacometrics Center of Excellence, Midwestern University, 555 31st Street, Downers Grove, IL 60515, USA
Aspartyl (Asparaginyl)-Beta-Hydroxylase (ASPH) is a 2-oxoglutarate (2OG) utilizing iron-dependent dioxygenase and is closely related to epigenetic enzymes, such as KDM, TET1-3, and FTO [1]. ASPH catalyzes post-translational hydroxylation of critically positioned aspartic acids and asparagines in specific calcium-binding epidermal growth factor (cbEGF) domains. Biologically, ASPH is involved in trophoblast invasion of the uterine wall and is expressed in the endoderm of developing embryos, although expression in healthy adult tissue is extremely limited. Mutations in ASPH are linked with Traboulsi syndrome in humans [2]. Experimentally confirmed cbEGF substrates of ASPH include LDLR, C1R, JAGGED1, and FX, and computationally predicted substrates include NOTCH1-4, JAGGED1&2, DLL1&4, DNER, and DLK1&2 among others. A crystal structure of ASPH is available [3]. Hepatocellular carcinoma and pancreatic cancer are known to significantly over-express ASPH on the cell surface, conferring an aggressive, invasive phenotype. Other cancers, such as mammary carcinoma, may also over-express ASPH. ASPH has been demonstrated to aberrantly activate the NOTCH signaling pathway [4]. ASPH inhibitors have been rationally designed and synthesized and demonstrate predicted activities in vitro, including the suppression of migration, invasion, and NOTCH pathway-related proteins [5]. In vivo proof-of-principle experiments demonstrate significant suppression of tumor growth of aggressive cancers, including cholangiocarcinoma [6]. Current ASPH inhibitors are orally bioavailable, are not genotoxic, have no identified in vitro safety liabilities, and have not demonstrated intestinal toxicity, unlike Gamma-secretase inhibitors. These compounds have demonstrated context-dependent NOTCH pathway inhibition in vivo.
The study was supported by Midwestern University.
References:
- Kanwal, M.; Smahel, M.; Olsen, M.; et al. J Exp Clin Cancer Res. 2020, 39(1), 163.
- Patel, N.; Khan, A.O.; Mansour, A.; et al. Am J Hum Genet. 2014, 94(5), 755.
- Pfeffer, I.; Brewitz, L.; Krojer, T.; et al. Nat Commun. 2019, 10(1), 4910.
- Lahousse, S.A.; Carter, J.J.; Xu, X.J.; et al. BMC Cell Biol., 2006 7, 41.
- Aihara, A.; Huang, C.K.; Olsen, M.J.; et al. Heptatology, 2014, 60(4), 1302.
- Nagaoka, K.; Ogawa, K.; Ji, C.; et al. Dig Dis Sci, 2021, 66(4), 1080.
4. Antimicrobial Activity of Ruta chalepensis: A Resazurin Assay and Scanning Electron Microscopy-Based Approach
1
Centre for Natural Products Discovery (CNPD), School of Pharmacy and Biomolecular Sciences, Liverpool John Moores University, James Parsons Building, Byrom Street, Liverpool L3 3AF, UK
2
Laboratory of Growth Regulators, Institute of Experimental Botany ASCR & Palacký University, Šlechtitelů 27, 78371 Olomouc, Czech Republic
Iraq is surrounded by Turkey in the north, Saudi Arabia in the south, Iran in the east and Syria in the west. It has a rich diversity in its flora; there are ca. 360 medicinal plants from 270 genera and 98 families found in Iraq. However, about 3300 plant species belonging to 908 genera and 136 families grow in Iraq. Iraqi medicinal plants are the building blocks of Iraqi traditional medicinal heritage and practice, which still thrive in Iraq. Iraqi traditional medicine can be traced back to the Sumerian period (3000-1970 BCE) and then the Assyrian period (1970 to 589 BCE). Iraqi medicinal plants and their products have long been used for the treatment of various human diseases, including infections.
As a part of our studies involving evidence-based phytotherapy, as well as plant-based drug discovery, which is the core research activity within the Centre for Natural Products Discovery at Liverpool John Moores University, different plant parts of Ruta chalepensis L., collected from Iraq, were studied for their antimicrobial activity, using a combination of the resazurin assay and scanning electron microscopy (SEM) to validate its traditional medicinal uses as an antimicrobial agent. Several antimicrobial compounds, predominantly coumarins and isoquinoline alkaloids, were isolated from R. chalepensis by preparative-reversed-phase-HPLC, and the structures were elucidated by spectroscopic means. Some of these isolated compounds exhibited anti-MRSA activity against clinical isolates of methicillin-resistant Staphylococcus aureus.
5. Natural Products from Seagrasses (Alismatales): Chemical Diversity, Bioactivity, and Ecological Function
Pharmazeutisches Institut, Universität zu Kiel, Germany
Seagrasses are the only higher plants living in marine environments; they play a significant role in coastal ecosystems. Seagrasses inhabit the coastal shelves of all continents except Antarctica and can grow in depths up to 90 m. Many studies have been dedicated to seagrasses and their ecology because of their eminent ecological importance. However, the phytochemistry has not been equally well investigated yet, and many of the existing studies in chemical ecology are only investigating the chemistry at the level of compound classes, e.g., phenolics, and not at the level of chemically defined metabolites. Until 2016 [1], a total of 154 chemically defined natural products have been reported from about 70 seagrass species known worldwide. Compounds reported include simple phenols derivatives (four compounds), phenylmethane derivatives (14 compounds), phenylethane derivatives (four compounds), phenylpropane derivatives including their esters and dimers (20 compounds), chalkones (four compounds), flavonoids including catechins (57 compounds), phenylheptanoids (four compounds), one monoterpene derivative, one sesquiterpene, diterpenoids (13 compounds), steroids (31 compounds), and one alkaloid. Recent studies from our group have been focused on apolar diarylheptanoids and the seasonal variation of natural products from Zostera marina from the Baltic Sea [2,3,4,5,6]
References:
- Zidorn, C., 2016. Secondary metabolites of seagrasses (Alismatales and Potamogetonales; Alismatidae): Chemical diversity, bioactivity, and ecological function. Phytochemistry 124, 5–28. doi:10.1016/j.phytochem.2016.02.004
- Grauso, L., Li, Y., Scarpato, S., Shulha, O., Rárová, L., Strnad, M., Teta, R., Mangoni, A., Zidorn, C., 2020. Structure and conformation of zosteraphenols, tetracyclic diarylheptanoids from the seagrass Zostera marina: an NMR and DFT study. Org. Lett. 22, 78–82. doi:10.1021/acs.orglett.9b03964
- Li, Y., Grauso, L., Scarpato, S., Cacciola, N.A., Borrelli, F., Zidorn C., Mangoni, A., 2021. Stable catechol keto tautomers in cytotoxic heterodimeric cyclic diarylheptanoids from the seagrass Zostera marina. Org. Lett. 23, 7134–7138. doi:10.1021/acs.orglett.1c02537
- Li, Y., Mangoni, A., Shulha, O., Çiçek, S.S., Zidorn, C., 2019. Cyclic diarylheptanoids deoxycymodienol and isotedarene A from Zostera marina (Zosteraceae). Tetrahedron Lett. 60, 150930. doi:10.1016/j.tetlet.2019.07.021
- Li, Y., Mangoni, A., Zidorn, C., 2022a. Seasonal variation of diarylheptanoids in Zostera marina (Zosteraceae) from the Baltic Sea, impact of drying on diarylheptanoids and phenolics in Zostera marina, and first report 3-keto-steroids from Zostera marina. Biochem. Syst. Ecol. 103, 104446. doi:10.1016/j.bse.2022.104446
- Li, Y., Rárová, L., Scarpato, S., Çiçek, S.S., Jordheim, M., Štenclová, T., Strnad, M., Mangoni, A., Zidorn, C., 2022b. Seasonal variation of phenolic compounds in Zostera marina (Zosteraceae) from the Baltic Sea. Phytochemistry 196, 113099. doi:10.1016/j.phytochem.2022.113099
6. Polymer Particles as Contrast Agents for Photoacoustic Tomography
1
Institute of Macromolecular Chemistry, Czech Academy of Sciences, Heyrovského nám. 2, 162 06 Prague 6, Czech Republic
2
Charles University, First Faculty of Medicine, Center for Advanced Preclinical Imaging, Prague, Czech Republic
3
Institute of Experimental Medicine, Czech Academy of Sciences, Vídeňská 1083, 142 20 Prague 4, Czech Republic
Photoacoustic (PA) imaging detects an acoustic signal induced by light. This unique method simultaneously provides ultrasound anatomical information with a high resolution along with a functional photoacoustic signal, which is created by the transformation of a laser pulse to a mechanical wave by some light-absorbing chromophore. PA imaging shows great potential for various clinical procedures from diagnosis to therapy guidance, which arises from its ability to gather functional and molecular information in a real-time regime with a high spatial resolution at clinically relevant depths, together with the absence of ionizing beaming.
To maximize the contrast effect of the exogenous contrast agents (CA) in the living organism, the optical absorption of the CA should optimally be in the near-infrared (NIR) regions ~700–1100 nm and 1200–2000. We developed new heterogenous syntheses of polypyrrole (PPY) particles with PA contrast properties in NIR, which allow for good control of size (10 nm step within the range from 80–300 nm) (Figure 1). Besides widely used linear water-soluble polymer stabilizers of the dispersion polymerizations, classical emulsifiers were also successfully employed in their synthesis, which broadens possibilities to employ less hydrophilic comonomers in the aqueous polymerization.
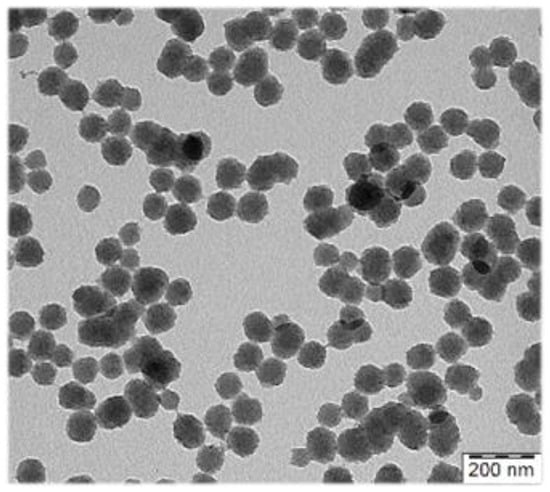
Figure 1.
Transmission electron micrograph of polypyrrole particles prepared for photoacoustic imaging.
This work was supported by the Ministry of Education, Youth and Sport of the Czech Republic (project no. LTAUSA18173 and Czech-BioImaging LM2018129).
7. G-Quadruplexes Recognition by P53 Family and Modulation by Ligands
1
Department of Molecular Pharmacy, Faculty of Pharmacy, Masaryk University, Palackého tř. 1946/1, 612 00 Brno, Czech Republic
2
Institute of Biophysics of the AS CR, v.v.i., Královopolská 135, 612 65 Brno, Czech Republic
Putative G-quadruplex (G4) motifs are frequently found in promoters 5’UTRs and 3ʹUTRs. Recent evidence suggests G4s are involved in key genome functions such as transcription, translation, and epigenetic regulation. To date, different compounds have been developed that aim to target G4 structures (G4 ligands) and modulate or stabilize G4 structure formation. p63 and p73, members of the p53 family of transcription factors, can induce or repress the transcription of an extensive network of common and unique genes by direct binding to a number of regulatory sites. We and others observed a strong correlation between DNA topology and the transcription of p53 target genes. We also established p53 and G4 interactions [1]. G4 from the c-myc promoter was strongly recognized by the p53 and p53 binding leads to the c-myc promoter repression in vivo. Furthermore, NMM stabilized G4 conformation of telomeric G4 was the G4 form of human telomere quadruplex favored by p53, suggesting conformational selectivity. We have also shown that R273H oncogenic mutant p53 frequently interacts with PQSs around transcription sites. The interaction of mutp53 with PQSs is presented as one of the mechanisms by which mutp53 actively promotes cancer. The main goal of our work is to prove G-quadruplexes as regulatory elements in p53 family biology. Several potential G-quadruplex forming regions in promoter, intronic or 5´UTR regions were selected based on our bioinformatics analysis. The project started with a biophysical G4 structure analysis of potential G4 forming motifs derived from G-rich regulatory regions of selected p63 target genes (e.g., KRT14, BTG2, EGFR) and p73 target genes. The G4 structure of these sequences was examined by CD spectroscopy. The analysis of NA binding properties of p63 isoforms was compared with p53. Different G4 ligands were used to probe protein-G4 interaction and target gene regulations in p53/p63/p73 biology [2,3].
The study was supported GAČR (grant No. 19-15168S).
References:
- Petr, M.; Helma, R.; Polaskova, A.; et al. Bioscience Rep 2016, 36.
- Adamik, M.; Kejnovska, I.; Bazantova, P.; et al. Biochimie 2016, 128, 83–91.
- Quante, T.; Otto, B.; Brazdova, M.; et al. Cell Cycle 2012, 11 (17), 3290–303.
8. The Effect of the “Catalytic Triangle” on the Lipase Activity
Department of Medicinal Chemistry, Collegium Medicum in Bydgoszcz, Faculty of Pharmacy, Nicolaus Copernicus University in Torun, Dr. A. Jurasza 2, 85-089 Bydgoszcz, Poland,
Lipases in free or immobilized forms belong to the most applicable catalysts pharmaceutical industry. The immobilization of lipases onto supports allows for obtaining higher catalytic activity. The intramolecular interactions between lipase, support, and substrate exist as a “catalytic triangle”. Due to the evaluation of the enzyme activity of lipase in a broad substrate spectrum, the optimal catalytic system can be gained [1,2,3]. The polyunsaturated (PUFAs) and monounsaturated (MUFAs) fatty acids, commonly named omega fatty acids, are characterized by multidirectional activity in human metabolism (e.g. cardioprotective, antiatherosclerotic, neuroprotective) [1,2]. Therefore, they were used as the substrates of fish and vegetable oils to evaluate lipase lipolytic activity [2]. The obtained results in our study showed high lipolytic activity of Amano lipase PS from Burkholderia cepacia (APS-BCL). Despite immobilized lipase activity decrease, compared with the free form [1,2], the “cut-off limit” to oils with a higher ω6/ω9 ratio (2.33 or higher) above the lipolytic activity was higher has been confirmed. The interactions mentioned above in the developed “catalytic triangle” (diffusion of substrate info support, dispersion of lipase to substrate, lipase hyperactivation, the reaction medium, and the effect of immobilization) could influence lipase behavior. Therefore, it is suggested that an optimized catalytic system can be helpful in the future in vitro digestion of fats.
References:
- J. Dulęba, T. Siódmiak, M.P. Marszałł, Amano Lipase PS from Burkholderia cepacia—Evaluation of the Effect of Substrates and Reaction Media on the Catalytic Activity, Current Organic Chemistry, 24 (2020) 798–807.
- J. Dulęba, T. Siódmiak, M.P. Marszałł, The influence of substrate systems on the enantioselective and lipolytic activity of immobilized Amano PS from Burkholderia cepacia lipase (APS-BCL), Process Biochemistry, 130 (2022), 126–137.
- T. Siódmiak, G.G. Haraldsson, J. Dulęba, M. Ziegler-Borowska, J. Siódmiak, M.P. Marszałł, Evaluation of Designed Immobilized Catalytic Systems: Activity Enhancement of Lipase B from Candida antarctica, Catalysts, 10 (2020) 21
9. Aromatic Schiff Bases as Aminopeptidase N Inhibitors
1
Department of Chemical Drugs, Faculty of Pharmacy, Masaryk University, Palackého tř. 1946/1, 612 00 Brno, Czech Republic;
2
Department of Pharmacology and Toxicology, Faculty of Pharmacy, Masaryk University, Palackého tř 1946/1, 612 00 Brno, Czech Republic
Aminopeptidase N (APN) is a broad specificity zinc metallopeptidase involved in many physiological and pathological processes. Some of its inhibitors, such as ubenimex or tosedostat were used as anticancer drugs. In the frame of this study, a series of thiosemicarbazones and semicarbazones derived from 2-, 3- or 4-(2-aminocetamido)acetophenones, where the amino group is a part of either a dialkylamino group or a saturated 5–7 membered nitrogenous heterocycle, which can further be substituted, was prepared. Target compounds were isolated either as free bases, hydrochlorides, or perchlorates. The compounds were tested for inhibition activity against APN either by a standardized VIS-spectrophotometry method using a 96-well plate reader or by an improved RP-HPLC procedure based on the method of Xiong et al. [1]. The activity results were processed into a QSAR study where the dependence of APN inhibition on lipophilicity expressed as log P and on electronic properties represented by the isoelectric point pI was demonstrated. Furthermore, five compounds with the best values of IC50 for APN (ranging between 13–23.5 μmol/L) underwent testing for the inhibition of cell proliferation on the three different cell lines, which differ one from each other on the level of APN expression [2,3,4]. All five compounds triggered a significant antiproliferative effect in the cell lines expressing APN, THP-1, and MCF-7, while in the cell line DU-145 with no APN expression, four of these five compounds did not affect proliferation at all. The remaining compound also inhibited DU-145 cell proliferation but less than in APN-positive, THP-1, or MCF-7 lines. These results could suggest that the antiproliferative activity is linked with APN inhibition, although other mechanisms can also participate in it.
The study was supported by MASARYK UNIVERSITY, grant numbers MUNI/IGA/0932/2021 and MUNI/A/1682/2020.
References:
- Xiong, X., Barathi, A., Beuerman, R.W., Tan, D.T.H.; J. Chromatogr. B. 2003, 796, 63.
- Licona-Limón, I., Garay-Canales, C.A., Muños-Paleta, O., Ortega E.; J. Leucoc. Biol. 2015, 98(1), 85.
- Yongkang, G., Yaqun, J.; et al. Mol. Pharmaceutics 2020, 17(1), 358.
- Joshi, S., Chen, L.; et al. Sci. Rep. 2017, 7(1), 1424.
10. Novel 1,3,5-Triazinyl Aminobenzenesulfonamides as Potent Anti-Vre Agents
1
Department of Chemical Drugs, Faculty of Pharmacy, Masaryk University, Palackého tř. 1-3, 612 42 Brno, CZ
2
Department of Chemical Biology, Faculty of Science, Palacky University, Slechtitelu 27, 783 71 Olomouc, CZ
3
Department of Pharmaceutical Chemistry, Faculty of Pharmacy, Comenius University, Odbojarov 10, 832 32 Bratislava, SK
4
Section of Farmaceutical and Neutraceutical Sciences, Neurofarba Department, University of Florence, 50019 Sesto Fiorentino, IT
Carbonic anhydrases (CA, EC 4.2.1.1) are metalloenzymes catalyzing the reversible hydration of CO2, thereby affecting the pH and affecting related physiological processes in various organisms [1].
In pathogenic bacteria, CAs play an important role in survival and growth [2]. The inhibition of bacterial CAs leads to growth retardation and defects and makes bacteria vulnerable to host defense mechanisms. Bacterial Cas are, therefore, a very promising target in the search for new antibiotics.
A series of 1,3,5-triazinyl aminobenzenesulfonamides, substituted by aminoalcohol, aminostilbene, and aminochalcone structural motifs, were synthesized as potential bacterial Cas inhibitors. The compounds were tested against vancomycin-resistant Enterococcus faecalis (VRE) isolates. A great number of the tested compounds exhibit significant inhibitory activity against VRE.
To evaluate the selectivity of the compounds against bacterial Cas towards human Cas (hCA), the inhibitory activity of compounds against tumor-associated hCA IX and hCA XII, hCA VII isoenzyme present in the brain, and the physiologically important hCA I and hCA II were determined. Tested compounds had only a negligible effect on physiologically important isoenzymes.
In conclusion, newly prepared compounds have great potential as antibacterial agents with high activity and, at the same time, with high selectivity for bacterial CA in comparison with metabolically important hCA isoenzymes (e.g., hCA I, hCA II) found in the human body.
The study was supported by INGA MU (MUNI/A/1202/2020).
References:
- Angeli A.; Carta F.; Nocentini A.; Winum J.-Y.; et al. Metabolites. 2020, 10(10), 412.
- Flaherty P.; Seleem D. M. N.; Supuran C. T. Future Med. Chem. 2021, 13(19), 1619.
11. Synthetic Polymers as Carriers of Biologically Active Compounds
Institute of Macromolecular Chemistry, Czech Academy of Sciences, Heyrovského nám. 2, 162 06, Prague 6, Czech Republic, Czech Republic
The use of nanomedicines consisting of polymer-carriers as drug delivery systems is a promising strategy to reduce adverse effects and enhance the therapeutic efficacy of several disorders, namely, cancer. Polymer carriers can protect numerous bioactive compounds, improve their pharmacokinetic profile, and prolong their circulation time in the body. These compounds can be attached to the carrier via suitable biodegradable spacers responsive to various tumor-associated stimuli. Also, polymer-based nanomedicines are passively accumulated in solid tumors to a higher extent due to the “enhanced permeability and retention" (EPR) effect. Biologically active compounds can be released in the target tissue or cells after their specific target, followed by their interaction with suitable receptors, thus improving their action [1,2,3].
Synthetic polymer-carriers based on N-(2-hydroxypropyl)methacrylamide (HPMA) copolymers are especially attractive for the construction of in vivo applicable nanomedicines since they are fully biocompatible, water-soluble, and lack any toxicity or immunogenicity. Their favorable pharmacokinetics enable a higher uptake in solid tumors or inflammation sites, therefore reducing dose-related adverse side effects on healthy organs with an enhanced therapeutic outcome. The therapeutic specificity and potential of biocompatible nanomedicines can also be improved by attaching specific targeting moieties along the polymer backbone; thus, their selective interaction with overexpressed tumor cell receptors can trigger numerous biological activities, which makes them potential nanomedicines for anticancer therapy.
Supported by the Ministry of Health of the Czech Republic, grant nr. NU21-08-00510. All rights reserved.
References:
- Ulbrich, K.; Holá, K.; Šubr, V.; et al. Chem. Rev. 2016, 116, 5338–5431.
- Chytil, P.; Koziolová, E.; Etrych, T.; Ulbrich, K. Macromol. Biosci. 2018, 18, 1–15.
- Kopeček, J.; Yang, J. Adv. Drug Deliv. Rev. 2020, 156, 40–64.
12. Imaging of Elements and Proteins in Biological Tissues: Medical and Pharmaceutical Applications
1
Department of Chemical Drugs, Masaryk University in Brno, Palackého třída 1946/1, 612 00 Brno
2
Department of Chemistry, Faculty of Science, Masaryk University, Kotlářská 2, 611 37 Brno, Czech Republic
3
Department of Pathological Physiology, Faculty of Medicine, Masaryk University, Kamenice 5, 625 00, Brno, Czech Republic
4
Department of Pharmacology and Toxicology, Masaryk University, Palackého třída 1946/1, 612 00 Brno
5
Mendel Univ Brno, Dept Chem & Biochem, Zemedelska 1, CZ-61300 Brno, Czech Republic
In the last decades, there has been increased interest in imaging of element, metal, and protein distribution, mainly in pharmaceuticals, biomedicine research, or life sciences with direct application of research to specific problems or diseases [1,2,3]. The main emphasis of this contribution is to demonstrate a unique method of laser ablation with inductively coupled plasma and mass spectrometry (LA-ICP-MS). It is a well-established method for multi-elemental analysis of elements at trace and ultra-trace. Nowadays, it is also starting to be used as a technique for the simultaneous specific determination of a protein of interest, which opens the possibility of achieving the so-called multiplex analysis.
Proteins are imaged using an immunohistochemical method (binding nanoparticle-labeled antibodies to a specific protein) and LA-ICP-MS. The main advantages of utilizing LA-ICP-MS are the acquisition of comprehensive (metallo)proteomic information about the tissue of a given disease (in this case cancer or stroke) and the low detection limits compared with other conventionally employed protein imaging techniques in combination with multiplex analysis of one sample.
The study was supported by Czech Science Foundation No. 20-20203S, Grant Agency of Masaryk University—Career Restart, 109576.
Supported by Ministry of Health of the Czech Republic, grant nr. NU21-08-00510. All rights reserved.
References:
- A. Lores-Padin, B. Fernandez, L. Alvarez, H. Gonzalez-Iglesias, I. Lengyel, R. Pereiro, Multiplex bioimaging of proteins-related to neurodegenerative diseases in eye sections by laser ablation—Inductively coupled plasma—Mass spectrometry using metal nanoclusters as labels, Talanta, 221 (2021) 10.
- A. Matusch, C. Depboylu, C. Palm, B. Wu, G.U. Hoglinger, M.K.H. Schafer, J.S. Becker, Cerebral Bioimaging of Cu, Fe, Zn, and Mn in the MPTP Mouse Model of Parkinson’s Disease Using Laser Ablation Inductively Coupled Plasma Mass Spectrometry (LA-ICP-MS), Journal of the American Society for Mass Spectrometry, 21 (2010) 161–171.
- C. Giesen, T. Mairinger, L. Khoury, L. Waentig, N. Jakubowski, U. Panne, Multiplexed Immunohistochemical Detection of Tumor Markers in Breast Cancer Tissue Using Laser Ablation Inductively Coupled Plasma Mass Spectrometry, Analytical Chemistry, 83 (2011) 8177–8183.
13. A Rapid Hplc-Elsd Method for Separation of Sugars and Sugar Alcohols in Fruits of Czech Sorbus Species
1
Department of Natural Drugs, Faculty of Pharmacy, Masaryk University, Palackého tř. 1946/1, 612 00 Brno, Czech Republic
2
Department of Chemical Drugs, Faculty of Pharmacy, Masaryk University, Palackého tř. 1946/1, 612 00 Brno, Czech Republic
Plants of the genus Sorbus are spread all over the world, and their fruits are used in folk medicine, particularly rowanberries are known for their diuretic effect, and S. domestica fruits are traditionally consumed as an antidiabetic agent. However, the landscape of the Czech Republic provides a unique environment that enables the creation of several endemic microspecies due to the ability of apomixis of the Sorbus species. Unfortunately, these endemic species are overlooked, and we know nothing about their phytochemical profile so far. Therefore, this is the first study dealing with Czech Sorbus species focused on the content of sugars and sugar alcohols. For this purpose, a rapid HPLC-ELSD method has been developed in order to simultaneously detect sugars and sugar alcohols that commonly occur in various berries. All tested components were completely separated within 12 min. Unsurprisingly, the analysis revealed the fruits of the Sorbus species to be rich in glucose and sorbitol. However, based on the content of the dominant sugars and based on the presence of minor ones, it can be pretty easily recognized which subgenera they belong to. Therefore, this separation method can be used for both analytical and chemophenetic purposes.
14. Synthesis and In Vitro Antimycobacterial Activity of Hybrid Compounds Containing An 2-/3-/4-Alkoxyphenylcarbamic Acid Structural Motif
Department of Pharmaceutical Chemistry, Faculty of Pharmacy, Comenius University Bratislava, Odbojárov 10, 832 32 Bratislava, Slovak Republic
This research focused on a synthesis, in silico evaluation, and in vitro antimycobacterial screening of the molecules Ia–Vd consisting of a 2-/3-/4-alkoxyphenylcarbamoyloxy moiety (alkoxy = methoxy to butoxy), connecting 2-hydroxypropan-1,3-diyl chain and 4-[(substituted)phenyl-/pyrimi-din-2-yl]piperazin-1-yl privileged structures (Figure 1). The lipophilicity, polarity, molecular flexibility, saturation, drug-likeness, and pharmacokinetic descriptors for the basic forms IaB–VdB of the final derivatives Ia–Vd were predicted by employing several online predictor tools and software packages. The molecules Ia–Vd were tested against M. tuberculosis CNCTC My 331/88 (identical with H37Rv and ATCC 2794), M. kansasii CNCTC My 235/80 (identical with ATCC 12478), M. avium CNCTC My 330/88 (identical with ATCC 25291), and M. kansasii 6509/96 clinical isolate, respectively, by standardized microdilution methods using isoniazid, rifampicin, and ethambutol reference drugs [1,2,3]. The insight into the structure–antimycobacterial activity relationships, considering the in silico and estimated biological descriptors, was provided as well.
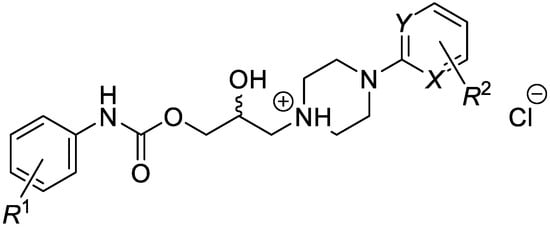
Figure 1.
Structure of synthesized 1-[3-(2-/3-/4-alkoxyphenylcarbamoyloxy)-2-hydroxypropyl]-4-[(substituted)phenyl/pyrimidin-2-yl]piperazin-1-ium chlorides.
(Ia–Vd; alkoxy = methoxy to butoxy), which were in vitro screened against the M. tuberculosis CNCTC My 331/88 (identical with H37Rv and ATCC 2794), M. kansasii CNCTC My 235/80 (identical with ATCC 12478), M. avium CNCTC My 330/88 (identical with ATCC 25291) and M. kansasii 6509/96 strain, respectively.
References:
- Waisser, K.; Doležal, R.; Čižmárik, J.; et al. Folia Pharm. Univ. Carol. 2007, 35–36(1), 45–48.
- Waisser, K.; Doležal, R.; Čižmárik, J.; et al. Sci. Pharm. 2007, 75(2), 55–61.
- Malik, I.; Lukac, M.; Galba, J.; et al. Fresenius Environ. Bull. 2016, 25(6), 2052–2062.
15. Preliminary Investigation of the Structure–Activity Relationships for Several Groups of Janus Kinase Inhibitors Using a Chemometric Principal Component Analysis
Department of Pharmaceutical Chemistry, Faculty of Pharmacy, Comenius University Bratislava, Odbojárov 10, 832 32 Bratislava, Slovak Republic
This research focused on the structure–activity relationships (SARs) for several groups of Janus kinase isoenzyme (JAK1–JAK3) inhibitors (n = 74) containing a purinone, pyrazolopyrimidine, or benzimid-azole scaffold (Figure 1). The SAR analyses inspected possible connections between in silico descriptors of those derivatives and their ability to inhibit JAK1–JAK3 [1] in vitro employing a principal component analysis (PCA) tool for dimensionality reduction, data compression, feature extraction as well as data visualization. All JAK1–JAK3 inhibitory activity constants (IC50 values) were adopted [2,3,4]. The calculated physicochemical descriptors were molecular weight (MW), number of carbon atoms in sp3 hybridization (csp3), molecular volume (MV), molar refractivity (MR), or lipophilicity parameters (log p values were predicted by several methods), among others, using several interactive online predictor tools as well as software packages. Positive correlation defined by a Pearson’s correlation coefficient (r) was found between predicted lipophilicity (LOGP (SILICOS-IT) method) and JAK1 inhibitory activity of the ligands (r = 0.694); the given conclusion mainly concerned the purinone derivatives (n = 29, r = 0.765). The csp3 descriptor for purinone compounds inversely correlated with both JAK1 (r = −0.670) and JAK2 (r = −0.706) inhibitory efficiency. The current study hypothesized that analyzed molecules might inhibit both JAK1 and JAK2 in a similar manner.
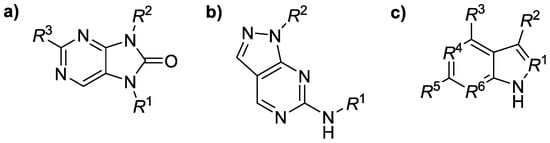
Figure 1.
General structure of Janus kinase inhibitors containing a variously substituted (a) purinone, (b) pyrazolopyrimidine or (c) benzimidazole scaffold.
References:
- Xu, P.; Shen, P.; Yu, B.; et al. Eur. J. Med. Chem. 2020, 192, art. no. 112155 (28p).
- Bach, J.; Eastwood, P.; González, J.; G.; et al. J. Med. Chem. 2019, 62(20), 9045–9060.
- Yang, T.; Hu, M.; Qi, W.; et al. J. Med. Chem. 2019, 62(22), 10305–10320.
- Jones, P.; Storer, R. I.; Sabnis Y. A.; et al. J. Med. Chem. 2017, 60(2), 767–786.
16. Pharmacokinetic Profiling via Dried Blood Spot Sampling Method
1
Department of Biochemistry, Faculty of Science, Masaryk University, Kamenice 5, 625 00 Brno, Czech Republic
2
Department of Pharmacology, Faculty of Medicine, Masaryk University, Kamenice 5, 625 00 Brno, Czech Republic
Pharmacokinetic (PK) profiles are still an inevitable part of preclinical safety studies. Generally, PK studies require the collection of a series of blood samples in sufficient volumes (usually about 200 µL). Typically, animal protocols limit the total blood draw between 10 and 15% of the total blood volume allowed within a 2-week period. Further, animal models are subjected to carefully monitored ethical standards, and the principles of the 3Rs are required: replacement, reduction, and refinement [1].
Blood microsampling can be defined as a blood sample collection that is no more than 50 µL total volume. Blood microsampling techniques cover capillary blood or plasma microsampling, dried blood spots (DBS), and accurate volume dried blood sampling. The usage of DBS in preclinical PK studies enables serial sampling from a single animal, which results in data quality improvement, significantly reduces the number of animals required, and offers significant ethical and cost benefits supporting the overall objectives of the 3Rs. Further, a recent scientific paper suggests a very good correlation between PK parameters obtained from whole blood in the form of DBS and plasma samples [2].
Our pilot PK study of clozapine (CLO) was conducted in a single Wistar albino rat. The study was approved by the local and national animal welfare committee (No. 58013/2017-MZE-17214). CLO was subcutaneously administrated in a single dose bolus (20 mg/kg), and blood samples were taken from a retroorbital plexus by a calibrated glass capillary and spotted onto the card (Whatman 903 Protein Saver Card). CLO concentrations were acquired by the LC-MS method, and corresponding PK parameters were calculated by Kinetica 4.4 software. The obtained pharmacokinetic parameters are in agreement with those previously reported [3].
The study was supported by the Grant Agency of Masaryk University, project MUNI/G/1464/2018 and MUNI/A/1440/2021.
References:
- Russel, W.M.S.; Burch, L.R. The principles of Humane Experimental Technique, Mathuen, London 1959.
- Baldo, M.N.; Angeli, E.; Gareis, N.C.; et al. Lab. Anim. 2018, 52(2), 125–134.
- Sun, L.; Lau C.E. Eur. J. Pharmacol. 2000, 398(2), 225–238.
17. Prenylated Phenolics as Pleiotropic Bioactive Substances
1
Department of Natural Drugs, Faculty of Pharmacy, Masaryk University, Palackého tř. 1946/1, 61200 Brno, Czech Republic
2
Department of Pharmacology and Toxicology, Veterinary Research Institute, Hudcova 296/70, 62100 Brno, Czech Republic
3
Department of Molecular Pharmacy, Faculty of Pharmacy, Masaryk University, Palackého tř. 1946/1, 61200 Brno, Czech Republic
Natural substances often have a pleiotropic effect and can affect several cellular processes in parallel. They can have parallel anti-inflammatory and antibacterial effects, together with the current antiviral effect. Their mechanism of action is complex. However, the problem with natural substances is often their limited solubility and, consequently, also problematic bioavailability [1]. A series of prenylated phenols were isolated from Paulowniaceae, Moraceae, and Euphorbiaceae plants [2,3,4,5]. As part of the lecture, we will introduce the isolation and identification of prenylated phenols with potential antiviral and anti-inflammatory effects; we will describe their bioactivity and formulations to increase solubility and will describe the possibilities of their further development. We describe the effects of phenolics in vitro in cellular or biochemical systems on the production and release of inflammation-related cytokines, their effects on the inhibition of cyclooxygenases and lipoxygenases, and also some in vivo experiments confirming the activity. In the end, an improvement of solubility by incorporating tested substances into liposomes is presented.
The work was supported by the Czech Science Foundation, project no. 21-38204L Complexes of selected transition metals with plant-derived compounds with anti-NF-kappa B and pro-PPAR dual activities.
References:
- Brezani V, Smejkal K, Hosek J et al. (2018) Curr. Med. Chem. 25: 1094–159.
- Lelakova V, Smejkal K, Jakubczyk K, et al. (2019). Food Chem. 285:431–40.
- Hanakova Z, Hosek J, Kutil Z, et al. (2017). J. Nat. Prod. 2017; 80(4):999–1006.
- Malaník M, Treml J, Leláková V, et al. (2020) Bioorganic Chemistry, 104, art. no. 104298.
- Čulenová M, Sychrová A, Hassan STS, et al. (2020) J. Ethnopharmacol. 248, art. no. 112296.
18. Perhaloacetaldehydes in the Enantioselective Organocatalyzed Fridel-Crafts Alkylation: Reaction Conditions Development
Department of Chemical Drugs, Faculty of Pharmacy, Masaryk University, Palackého 1946/1, 612 00 Brno, Czech Republic
Trihaloacetaldehydes represent useful electrophiles in asymmetric processes that can grant access to many biologically active compounds containing CX3 groups. One of the possibilities for obtaining aromatic trihaloethanols is the asymmetric organocatalyzed Friedel-Crafts reaction between phenol and trihaloacetaldehyde, which represents the subject of our current research (Figure 1). As such, we started with the extensive three-phase catalyst screening. Cinchona alkaloid-based amide derivatives showed the best enantioselectivity in the initial stage of catalyst testing. Improvement of the catalyst structure revealed 3,5-dinitrobenzamide of 9-aminoepicinchonidine as the lead catalytic molecule. Next, a series of optimizations were performed to establish the most suitable reaction conditions. Having the optimal parameters in hand, the reaction between electron-rich phenols and trihaloacetaldehydes or their hemiacetals conveniently provided enantioenriched adducts with good to excellent enantiomeric ratios (up to 99:1) within 12–24 h at 25 °C. The substrate scope included 29 derivatives containing –CF3, –CCl3, –CF2Cl, and –CF2Br groups. Additionally, several stereoretentive downstream transformations of products were identified. This work constitutes the first organocatalyzed method for the synthesis of chiral non-racemic 2,2,2-trihalo-1-hydroxyalkylphenols [1].
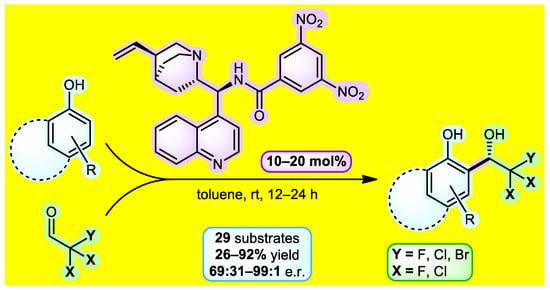
Figure 1.
Reaction scheme for obtaining trihaloethanols.
The study was supported by the projects MUNI/A/1510/2020 and MUNI/A/1682/2020.
References:
- Švestka, D.; Otevřel, J.; Bobáľ, P. Adv. Synth. Catal. 2022, 364(13), 2174.
19. Introduction to Theranostics
1
NEXARS, The Science Campus Park, Palachovo nám. 2, 62500 Brno, Czech Republic
2
Department of Immunology, Faculty of Medicine and Dentistry, Hněvotínská 3, University of Palacky, 77515 Olomouc, Czech Republic;
Drug delivery systems (DDS) are extensively studied and developed in commercial products to improve the efficacy and administration of active pharmaceutical compounds, such as low molecular drugs, recombinant proteins and enzymes, peptides, vaccines, antibodies, various contrast agents etc. There are also some limitations for the application of nanosystems to medicine. Nanotoxicity and interactions with the immune system are the most important ones from a pharmacological point of view, e.g., first-pass effect, instability in body fluids, intolerance and induction of immune unwanted and harmful response, fluctuations in plasma drug levels resulting in limited effectiveness, lack of selectivity, poor bioavailability, etc. A plethora of controlled DDS has been developed to achieve the goal of targeted and selective delivery.
The DDS has become an indispensable tool also for the development of commercial products for preclinical and clinical theranostics, combining both diagnostic and therapeutic properties to improve medical intervention in various diseases. The diagnosis and treatment of cancer are first in line for the application of theranostics, but also brain diseases and inflammation are of great importance.
Some aspects of modern theranostics and imaging systems will be presented and discussed.
20. The Combined Treatment of Methotrexate with Carnosic Acid Influences Hepatic mRNA Expression of Inflammatory and Antioxidant Genes in Model of Adjuvant Arthritis in Rat
1
Department of Cellular and Molecular Biology of Drugs, Pharmaceutical Faculty of Comenius University, SK-83232 Bratislava, Slovakia,
2
Centre of Experimental Medicine, Slovak Academy of Sciences, SK-84104 Bratislava, Slovakia
Rheumatoid arthritis (RA) is a chronic autoimmune inflammatory disease characterized by polyarthritis and systemic low-grade inflammation. The inflammation is also accompanied by oxidative stress and changes in the levels of antioxidant enzymes, such as catalase (CAT), paraoxonase (PON-1), superoxide dismutase 1 (SOD-1), or heme oxygenase 1 (HO-1) [1]. A combination of standard therapy with natural compounds with anti-inflammatory or antioxidant activities may improve treatment effectiveness and allow for reduced dosing and thus decrease the side effects. The aim of our study was to test the therapeutic potential of carnosic acid (CA), a natural compound with anti-inflammatory and antioxidant activities [2], as a monotherapy or in combination with methotrexate (MTX) for the treatment of adjuvant arthritis (AA). AA, which has a pathology similar to RA, was induced in rats by intradermal administration of heat-inactivated Mycobacterium butyricum in an incomplete Freund´s adjuvant. The expression of antioxidant and inflammatory genes was measured in the liver after the isolation of RNA and analysis by quantitative PCR. In our experimental settings, the mRNA expression of SOD-1 was unchanged under inflammatory conditions. Although in monotherapy CA was not effective, combination treatment with MTX was in arthritic rats associated with a significant reduction of hind paw volume and arthritic score and also with reduced gene expression of IL-1β, HO-1 and upregulated gene expression of CAT, PON-1. Since PON1 is also part of the HDL, its upregulation may improve HDL functionality and thus improve cardiovascular health in patients. These results suggest a promising therapeutic potential of CA in combination with MTX.
The study was supported by VEGA 1/0429/21 and FAF/15/2022.
References:
- Smallwood, M.J.; Nissim, A.; Knight, A.R.; Whiteman, M.; Haigh, R.; Winyard, P.G. Oxidative stress in autoimmune rheumatic diseases. Free Radic Biol Med 2018, 125, 3–14
- Birtić, S., et al. Carnosic acid. Phytochemistry, 2015/07/01/ 2015, 115, 9–19.
21. Liberation of Ibuprofen from Polysaccharide Hydrogels
1
Department of Galenic Pharmacy, Faculty of Pharmacy, Comenius University Bratislava, Odbojárov 10, 83232 Bratislava
2
Department of Pharmaceutical Analysis and Nuclear Pharmacy, Faculty of Pharmacy, Comenius University Bratislava, Odbojárov 10, 83232 Bratislava
3
Toxicological and Antidoping Center, Faculty of Pharmacy, Comenius University Bratislava, Odbojárov 10, 83232 Bratislava
In this comparative study, the rheological and texture properties of 11 polysaccharide-based hydrogels were comprehensively investigated, along with the in vitro release of the model drug ibuprofen (IBU). At first, the rheological properties of the hydrogels as dependencies of shear stress on the shear rate were evaluated to recognize the type of flow. Based on viscosity values, three categories of hydrogels were selected, i.e., low-, medium-, and high-viscous, and two later categories were further studied for texture properties. In the second step, hardness, adhesiveness, and minimal retracting force were evaluated by texture profile analysis.
Drug release kinetics were compared through the coefficient of determination expressed for the regression line of five kinetic models. The values indicated that the drug release of the hydrogels during in vitro diffusion studies follows mostly the Higuchi model, whereas drug release from CS hydrogels follows the zero-order kinetic. Significant differences in IBU diffusion were statistically confirmed among the groups of hydrogels composed of positively charged, neutral, or negatively charged polysaccharides. Here, differences in provided interactions (mainly attractive–repulsive Coulomb forces) of the ionic polymers with the formulated anionic drug played a key role in different IBU release profiles, as supported by statistical analysis (Student’s t-test) and illustrated by Figure 1 [1]. The knowledge of the provided interactions can be utilized when formulating one or more drugs with their different character and release kinetics.
The knowledge of rheological and texture properties, as well as drug release kinetic profiles of individual polysaccharide hydrogels, as comprehensively presented in this work, can also be advantageously utilized in the creation of various hydrogel preparations with modified drug release profiles suitable for particular drug delivery purposes.
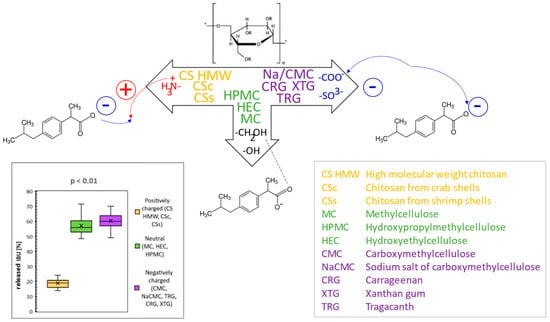
Figure 1.
General scheme of interactions between different polysaccharides as hydrogel constituents and ibuprofen as model drug and resulting released amounts of ibuprofen from different polysaccharide hydrogels.
This work was supported by the projects VEGA 1/0514/22, KEGA 027UK-4/2020, APVV-15-0585, FaF/21/2022, and UK/73/2022.
References:
- Mikušová, V.; Ferková, J.; Žigrayová, D.; Krchňák, D.; Mikuš, P. Gels. 2022, 8, 168.
22. Determination of the Critical Micellar Concentration of Heptacainium Chloride in Aqueous and Alcohol Solutions Using the Optical Density Method
Department of Pharmaceutical Chemistry, Faculty of Pharmacy, Comenius University, Odbojárov 10, 832 32, Bratislava, Slovak Republic
Micelles are self-assembled aggregates that are formed, in many different structures, from surfactants and have a wide range of applications. The critical micellar concentration (CMC) is the concentration of surfactant in a bulk phase, above which aggregates of surfactant molecules start to form. The CMC is an important characteristic of surfactants.
The CMC value of heptacainium chloride [1] in an aqueous medium at laboratory temperature was determined by measuring the optical density on a microtiter plate [2].
Subsequently, the CMC value of heptacainium chloride in solutions of monohydric alcohols—methanol, ethanol, and propanol—in concentrations of 10%, 20%, 30%, and 40% was determined by this method. In the case of all three monohydric alcohols, a gradual increase in CMC was observed, together with an increasing concentration of alcohol in the solutions, and when a certain point was reached, the CMC value began to decrease again.
The study was supported by the Grant FaF/19/2022
References:
- Čižmárik, J.; Borovanský, A. Acta Facult. Pharm. Univ. Comenianae. 1976, 29,53–80.
- Gališinová, J.; Andriamainty, F.; Čižmárik, J.; et al. J. Bioequiv. Availab. 2013, 5, 161–164.
23. Schiff Bases as Potential Inhibitors of Aminopeptidase N as Anticancer Agents
1
Department of Chemical Drugs, Faculty of Pharmacy, Masaryk University, Palackého tř. 1946/1, 612 00 Brno, Czech Republic
2
Department of Pharmacology and Toxicology, Faculty of Pharmacy, Masaryk University, Palackého tř 1946/1, 612 00 Brno, Czech Republic
Many Schiff bases appear to be important intermediates in a number of enzymatic reactions. The presence of the azomethine group, as well as their interesting physical and chemical properties, cause the widespread application of their metal complexes [1]. Synthesized groups of basic thiosemicarbazone and semicarbazone derivatives of acetophenone have these properties as well.
Aminopeptidase-N (AP-N), or membrane alanyl aminopeptidase (m-AAP), is a neutral zinc-binding metallopeptidase that cleaves N-terminal residues from protein and peptides. This aminopeptidase turns out to be identical to the human cluster differentiation antigen CD13 expressed on the surface of myeloid progenitors and myeloid leukemia cells [2]. Potential inhibitors of AP-N may offer effective and broad-spectrum therapy.
Determining AP-N inhibition by using high-performance liquid chromatography has become the analytical method of choice. L-Leucine-p-nitroanilide was used as a substrate, and its hydrolytic product, p-nitroaniline, was monitored at 405 nm absorption wavelengths. The compounds with the best inhibitory activity on the APN enzyme underwent testing for inhibition of cell proliferation on the three different cell lines. A simple QSAR model describing the dependence between the inhibitory activity expressed as IC50 and the descriptors derived from the chemical structure was constructed.
The study was supported by the project CZ.02.2.69/0.0/0.0/19_073/0016943 Internal grant agency of Masaryk University (MUNI/IGA/0916/2021).
References:
- Hossain, A.M.S., Méndez-Arriaga, J.M., Xia, C., Xie, J., Gómez-Ruiz, S., 2022. Metal complexes with ONS donor Schiff bases. A review. Polyhedron, 2022, 217, 115692.
- Turner, A.J., 2013. In: Rawlings, ND, Salvesen, G (Eds.), Handb. Proteolytic Enzym. Third Ed., Academic Press, pp. 397–403.
24. Synthesis of Biologically Active Heterocycles Derived from Isatin
Department of Pharmaceutical Chemistry, Faculty of Pharmacy, Comenius University, Odbojárov 10, 832 32, Bratislava, Slovak Republic
Isatin has been widely used in drug discovery. The ring is part of many naturally occurring compounds and is used to design compounds with diverse biological activities. Structurally, isatin is a fusion of a six-membered benzene ring and a five-membered ring containing nitrogen. Isatin derivatives possess several biological properties, such as anticancer effect in different types of cancer [1], antibiotics and antidepressants [2], anxiogenic, sedative, anticonvulsant [3], antibacterial, antifungal and antidiabetic [4], etc.
Diabetes mellitus is a group of metabolic disorders of carbohydrate metabolism characterized by high blood glucose levels (hyperglycemia) resulting from defects in insulin secretion, insulin action, or both [5]. A set of four novel isatin-thiazole derivatives with potential antidiabetic activity [6] were synthesized by three-step synthesis from commercially available isatines (Figure 1). Final isatin-thiazole-derived compounds were prepared in an overall yield of 48–66%. The individual intermediates and the final products were confirmed by 1H NMR and 13C NMR spectroscopy.
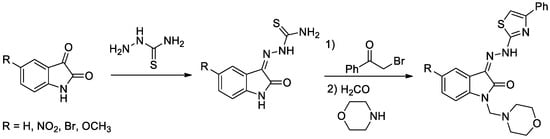
Figure 1.
Reaction scheme for obtaining isatin-thiazole-derived compounds.
The study was supported by FaF/8/2022
References:
- Liang, C.; Xia, J.; Lei, D.; et al. Eur. J. Med. Chem. 2014, 74, 742–750.
- Pakravan, P.; Kashanian, S.; Khodaei, M. M.; et al. Pharma Rep. 2013, 65, 313–335
- Nath Pandeya, S.; Smitha, S.; Jyoti, M.; et al. Acta Pharm. 2005, 65, 27–46.
- Varun, Ch.; Sonam, M.; Kakkar, R. Med. Chem. Comm. 2019, 10, 351–368.
- DeFronzo, R.A. Med. Clin. N. Am. 2004, 88, 787–835.
- Xie, Z.; Wang, G.; Wang, J.; et al. Molecules 2018, 28(2), 113–118.
25. Determination of Sugar Profile in Milk and Special Infant’s Formulas by Hplc
Department of Chemical Drugs, Faculty of Pharmacy, Masaryk University
Lactose is a disaccharide, typically called milk sugar, composed of galactose and glucose. This disaccharide is present in a higher amount of milk carbohydrate content. For babies, it is the most important saccharide, and it is cleaved by an enzyme called lactase (hydrolase) to monosaccharides, which are utilized. The aim of this paper is primarily to develop and apply a method for the determination and quantification of lactose, but in the case of sample contents of other sugars, such as glucose, galactose, sucrose, and maltodextrins, we can determine and quantify. In commercially available samples of whole cow´s milk and selected neonatal dairy nutrition, lactose was determined and quantified. In the case that milk was treated at a high temperature (UHT, ultra-high temperature, special types of milk), it could be present in lactulose, a synthetic isomer of lactose.
HPLC (high-performance liquid chromatography) in the mode HILIC (hydrophilic interaction chromatography) could be quite often used in pharmacy. HILIC has some benefits, including the speed of analyses (typically high flow grade) and type of analytes (in our case, carbohydrates, which cannot be easily determined by normal reversed phase chromatography). With a suitable combination of polar and non-polar components in the mobile phase, we can quickly separate carbohydrates. However, since carbohydrates do not contain any chromophore in their structure, a conventional UV detector is unusable, and thus an evaporative light-scattering detector (ELSD) can be advantageously used. The stacionary phase used in HILIC mode is silica, which is, in most cases, modified with polar groups (hydroxy, amine, amide, etc.). The mobile phase is usually a mixture of water (polar) and acetonitrile (nonpolar).
The method was developed on a Dionex UltiMate 3000 HPLC kit (ThermoFisher Scientific, Waltham, MA, USA) with a HALO Penta–HILIC 4.6 × 150 mm column (AMT, Wilmington, DE, USA) with a stationary phase of modified silica particles with a maximum size of 2.7 µm and an ELS detector Varian 380–LC (Varian, Palo Alto, CA, USA). Chromeleon and MS Excel were used to evaluate the obtained data. After optimizing the conditions of the method, the temperature was determined to be 10 °C, the flow rate of the mobile phase 2 mL per minute, and the use of a mixture of acetonitrile and 30 mmol/L ammonium formate buffer as the mobile phase. Gradient elution was used. Detection conditions were determined at a nitrogen flow of 1 slm, a temperature of a nebulizer, and an evaporator of 40 °C.
Under our optimized conditions, the method can determine lactose in 12.60 min, lactose–lactulose resolution 1.70; repeatability (peak area) 2.73%; repeatability (lactose retention time) 2.26%; LOD 60.26 mg/mL and LOQ 109.01 mg/mL, recovery (method of externally standard addition) 96.7–107.1%.
Acknowledgments: IGA project MUNI/A/1236/2021 of Masaryk University Brno, Czech Republic
26. Application of Thin Layer Chromatography Analysis for Evaluation of the Lipophilicity of Selected Antiandrogen Drugs
1
Doctoral School of the Medical University of Silesia in Katowice, Faculty of Pharmaceutical Sciences in Sosnowiec, 41-200 Sosnowiec, Poland
2
Department of Analytical Chemistry Faculty of Pharmaceutical Sciences in Sosnowiec, Medical University of Silesia in Katowice, 41-200 Sosnowiec, Poland
Lipophilicity is an important physicochemical parameter of a drug. It affects its activity and toxicity [1]. This value determines the bioavailability of the drug as well as its solubility in body fluids. Lipophilicity is a widely used parameter in the process of designing substances as candidates for new drugs. The most frequently used methods for determining lipophilicity parameters are reverse-phase thin-layer chromatography (RP-TLC) and high-performance chromatography (RP-HPLC). We also distinguish computational methods, i.e., computer programs [2]. The subjects of the research were drugs belonging to the group of antiandrogens such as abiraterone, bicalutamide, flutamide, nilutamide, leflunomide, teriflunomide and ailantoine. They are used in the treatment of prostate cancer and belong to different, including a newer generation of antiandrogens. In this work, the usefulness of the TLC method and selected computational algorithms for the determination of the lipophilicity parameter of these drugs were assessed. In the conducted lipophilicity studies, the following chromatographic plates were used as stationary phases—RP2F254, RP18F254, and RP18WF254—and the mixture of ethanol-water, propanol-water, acetonitrile-water in various volume ratios as the mobile phases. Compounds were analyzed at λ = 254 nm. The chromatographic parameter of lipophilicity of the tested compounds was compared with the theoretical values estimated with the use of a computer program. The obtained results indicate the usefulness of the TLC method for determining the experimental lipophilicity parameter of the tested compounds. It was stated that cluster analysis allowed the estimation of the similarity between the compounds studied on the basis of obtained results of both chromatographic and theoretical parameters of lipophilicity.
The research was carried out as part of a project financed by the Medical University of Silesia in 2021
Katowice No. PCN-1-045-N/1/F.
References:
- Jóźwiak, K.; Szumiło, H.; Soczewiński, E. Lipophilicity, methods of determination and its role in biological effect of chemical substances. Wiad Chem. 2001, 55, 1047–1074.
- www.vcclab.org/lab/alogps (accessed on 15 May 2022).
27. Biologically Active Xanthones from Maclura pomifera
Department of Natural Drugs, Faculty of Pharmacy, Masaryk University, Palackého tř. 1946/1, 612 00 Brno, Czech Republic
Xanthones are secondary metabolites occurring only in a few plant families and are known for their in vitro cytotoxic, antitumor, and antimalarial activity [1].
Maclura pomifera is an interesting source of biologically active xanthones, often with side prenylated modifications. Based on the literature data and our previous research [2,3], an ethanolic extract of maclura stem bark was prepared using maceration and ultrasonication; the liquid/liquid extraction followed. Column chromatography of ethyl acetate fraction led to the isolation of three xanthones: toxyloxanthone C, mesuaxanthone A, and osajaxanthone. Isolated compounds were identified using HPLC, MS and 1D, and 2D NMR experiments.
Subsequent semi-preparative HPLC chromatography of selected subfraction enabled the isolation of another two xanthones, which are currently being identified.
Isolated compounds will be screened for biological activity, especially anti-inflammatory and antioxidant effects, to broaden the spectrum of potential therapeutical applications of these xanthones. Based on further studies, they could find application in the therapy of some skin diseases demanding the combination of dual biological activity. Moreover, our isolation of xanthones with different substituent modifications can enable us to better describe the structure-activity relationship.
The study was supported by Masaryk University (Specific research—support for student projects), grant number MUNI/A/1688/2020.
References:
- da Costa, C. T.; Margolis, S. A.; Benner B. A.; et al. J. Chromatogr. A 1999, 831(2), 167–178.
- Khan, M. T. H; Orhan, I.; Senol, F. S.; et al. Chem. Biol. Interact. 2009, 181(3), 383–389.
- Orhan, I.; Senol, F. S.; Kartal, M.; et al. Food Chem. Toxicol. 2009, 47(8), 1747–1751.
28. Study of Ibuprofen Liberation from Hydrogels Containing Combined Gelling Agents
1
Department of Galenic Pharmacy, Faculty of Pharmacy, Comenius University Bratislava, Odbojárov 10, 83232 Bratislava, Slovakia
2
Department of Pharmaceutical Analysis and Nuclear Pharmacy, Faculty of Pharmacy, Comenius University Bratislava, Odbojárov 10, 83232 Bratislava, Slovakia
3
Toxicological and Antidoping Centre, Faculty of Pharmacy, Comenius University Bratislava, Odbojárov 10, SK-832 32 Bratislava, Slovakia
In pharmaceutical preparation, polysaccharide-based excipients are substances capable of influencing and controlling the release of the drug and, therefore, its bioavailability. By their combination, it is possible to additionally modify and adjust the drug release. In this work, combinations of cationic hydrogels with neutral or anionic ones were performed to illustrate the possibilities of providing modified ibuprofen (IBU) release profiles. Three combinations of polysaccharide hydrogels were studied for rheological and texture properties and ibuprofen diffusion: (i) cationic hydrogel with ionic (anionic) one, (ii) cationic hydrogel with ionizable (anionic) one, and (iii) cationic hydrogel with a neutral one. For this, 3% high molecular weight chitosan CS HMW was combined with 0.5% and 1% sodium salt of carboxymethylcellulose NaCMC (i), carboxymethylcellulose CMC (ii), and methylcellulose MC (iii). In this context, chitosan was presented as an effective modifier of diffusion profiles for negatively charged drugs formulated into combined polymeric systems, providing their prolonged release.
A combination of cationic hydrogel (CS) with the neutral one (MC) showed the slowest IBU release. It can be simply explained by an increased number of free functional groups of the polymers offering additional interactions or binding (mainly H-bonds) with IBU. On the other hand, a combination of cationic hydrogel (CS) with anionic ones (NaCMC and CMC) speeds up the IBU release. The IBU diffusion can be influenced by several mechanisms, including ionic crosslink of the polymers via their oppositely charged groups, the attraction of IBU via free (non-crosslinked) amino groups of CS, the repulsion of IBU via free (non-crosslinked) carboxylate groups of NaCMC or CMC, and, obviously, binding via several electroneutral functional groups of NaCMC or CMC (mainly through H-bonds).
Hence, for future studies, when considering the formulation of a group of hydrophobic drugs possessing a negatively charged functional group (as the model drug ibuprofen), CS can be advantageously utilized as a basis for designing new (e.g., combined polymer) systems for controlled drug release that is mainly prolonged or sustained.
This work was supported by the projects FaF/21/2022, UK/73/2022, VEGA 1/0514/22, KEGA 027UK-4/2020, and APVV-15-0585.
This poster was created with thanks to the support of the Operational Program Integrated Infrastructure for the project: National infrastructure for supporting technology transfer in Slovakia II—NITT SK II, co-financed by the European Regional Development Fund.
29. Isolation and Identification of Diterpenes from Coleus forsteri ‘Marginatus’ and Plectranthus ciliatus
1
Department of Natural Drugs, Faculty of Pharmacy, Masaryk University, Palackého tř. 1946/1, 612 00 Brno, Czech Republic
2
Institute of Pharmacognosy, Faculty of Pharmacy, University of Szeged, Eötvös u. 6, 6720 Szeged, Hungary
Coleus forsteri “Marginatus” (Benth.) A.J.Paton and Plectranthus ciliatus E.Mey. are ornamental plants belonging to the Lamiaceae family [1]. Although only a little information regarding their phytochemistry has been published [2,3,4], many species of the medicinally important genus Plectranthus s.l. have been shown to produce phenolic compounds and terpenes. Within the group of terpenes, diterpenes have become the most studied subgroup because of their structural diversity and various biological activities such as antimicrobial, antiprotozoal, cardiotonic, gastroprotective, and cytotoxic activity [1,4].
In our present work, we aimed to investigate the methanolic extracts of the aerial parts of C. forsteri “Marginatus” and P. ciliatus, which led to the isolation of four abietane and two kaurane diterpenes, respectively. Five of them have never been reported in these species. The substances were isolated using flash chromatography and RP-HPLC, and their identification was conducted by high-resolution mass spectrometry, one-dimensional and two-dimensional nuclear magnetic resonance spectroscopy, as well as circular dichroism spectroscopy.
The study was supported by the Grant Agency of Masaryk University (grant number MUNI/A/1688/2020), the Centre for International Cooperation of Masaryk University (Exchange program ERASMUS+), and the NKFIH, Hungary (K-134704).
References:
- Lukhoba, C.W.; Simmonds, M.S.J.; Paton, A.J. J. Ethnopharmacol. 2006, 103(1), 1–24.
- Jordheim, M.; Calcott, K.; Gould, K.S.; et al. Phytochemistry 2016, 128, 27–34.
- Viljoen, A.M.; Demirci, B.; Başer, K.H.C.; et al. S. Afr. J. Bot. 2006, 72(1), 99–104.
- Gáborová, M.; Šmejkal, K.; Kubínová, R. Molecules 2022, 27(1), 166.
30. 2-Hydroxynaphthalene-1-Carboxamides Possessing Antimycobacterial Activity
1
Department of Chemical Drugs, Faculty of Pharmacy, Masaryk University, Palackého tř. 1946/1, 612 00 Brno, Czech Republic
2
Department of Infectious Diseases and Microbiology, Faculty of Veterinary Medicine, VETUNI, Palackého tř 1946/1, 612 42 Brno, Czech Republic
3
Department of Analytical Chemistry, Faculty of Natural Sciences, Comenius University Bratislava, Ilkovičova 6, 842 15 Bratislava, Slovakia
Despite the approval of some new antituberculosis drugs in recent years (such as bedaquillin, delamanid, and pretomanid), tuberculosis (TB) now ranks alongside human immunodeficiency virus as a leading death-causing infectious disease worldwide. Based on the recent World Health Organization tuberculosis report, TB killed 1.5 million people in 2020 worldwide; 9.9 million people are estimated to have fallen ill with TB in 2020. Almost 70% of new TB cases were caused by rifampicin-resistant strains (4% in 2010). The rapid increase in resistance makes the discovery of new molecular scaffolds a priority to achieve effective control of the disease [1].
The aim of recent work was to synthesize a series of N-(phenyl)-2-hydroxynaphthalene-1-carboxamides with multiple substitutions on a phenyl ring (Figure 1). Thirty compounds with halogen, trifluoromethyl, methyl, methoxy, and nitro-substituted anilide rings were prepared according to a well-approved microwave-assisted synthetic method [2]. The structure of the novel compounds was confirmed by 1H and 13C NMR, HRMS, and IR spectroscopy. Primary in vitro screening of the synthesized compounds was performed against Mycobacterium marinum, Mycobacterium kansasii, and Mycobacterium smegmatis. Several compounds showed antimycobacterial activity comparable to standard pyrazinamide and rifampicine.
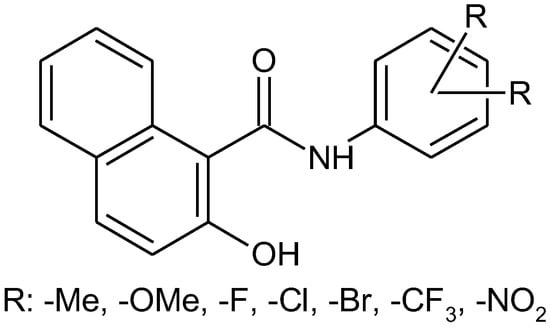
Figure 1.
Substituted N-(phenyl-2-hydroxynaphthalene-1-carboxamides.
References:
- Global Tuberculosis Report 2021, Geneva: WHO; 2021
- Goněc T., Kos J., et al. Molecules 2013, 18, 9397–9419.
31. Insight into the Synthesis of Polyhydroxy 2-Arylbenzofurans
Department of Chemical Drugs, Faculty of Pharmacy, Masaryk University, Palackého tř. 1946/1, 612 00 Brno, Czech Republic
Benzofurans, which possess prenyl or geranyl chains, are naturally occurring compounds that are of pharmaceutical interest thanks to their broad spectrum of biological activity. Mulberrofurans, geranylated polyhydroxy benzofurans, found in the bark root of Morus sp. have been studied mainly for their antiphlogistic, antioxidative, and cytotoxic effects [1,2,3].
Although these compounds have been known for more than 40 years, only a few of these compounds have been synthesized [4,5]. This work aims to prepare mulberrofuran Y, which could then be further analyzed and studied in various biological assays and hopefully used as a template for preparing more potential derivatives.
The first attempts aimed to prepare substituted O-arylhydroxylamine, which could be used as a key intermediary in the process of preparing a 2-arylbenzofuran skeleton, but this pathway led to no success [6]. Another unsuccessful trial was conducted using isovanillin as a starting compound, aiming to prepare polysubstituted 2-iodophenol as another key intermediary [7].
Aiming to prepare the same key intermediary, salicylaldehyde and methyl 4-iodosalicylate were chosen as precursors in other synthetic pathways, which among other things, resulted in the synthesis of several novel compounds as well as finding unpredictable chemical behavior within synthetic pathways.
The study was supported by the project MUNI/A/1181/2021
References:
- Shim, S.; Sung, S. H.; Lee, M.; 2018, 58, 117–124.
- Martins, B. A.; Sande D.; Solares M. D.; Takashi J. A.; Nat. Prod. Res. 2021, 35(24), 5993–5996.
- Zhang, Q.; Tang Y.; Chen R.; Yu D.; Chem & Biodiv. 2007, 4(7), 1533–1540.
- Gunawan, Ch.; Rizzacasa, M. A.; Org. Lett. 2010, 12(7), 1388–1391.
- Kim, Ch. G.; Jun, J.; Bull. Kor. Chem. Soc. 2015, 36(9), 2278–2283.
- Maimone, T. J.; Buchwald S. L.; J. Am. Chem. Soc. 2010, 132(29), 9990–9991.
- Bosiak, M. J.; ACS Catalysis. 2016, 6(4), 2429–2434.
32. Effect of Lactobacilli on 5-Fluorouracil-Induced Intestinal Mucositis In Vitro
Department of Cell and Molecular Biology of Drugs, Faculty of Pharmacy, Comenius University Bratislava, Kalinčiakova 8, 831 41 Bratislava, Slovak Republic
5-Fluorouracil (5-FU) is commonly used for the treatment of various tumors and is the key drug in colorectal cancer treatment (CRC) [1]. Intestinal mucositis is the most frequent side effect of such therapy, which significantly impacts on patient quality of life and delays the treatment cycle, therefore, reducing the overall efficacy of the treatment [2]. Our aim was to study the effect of a potential probiotic strain, Limosilactobacillus reuteri E (LRE; isolated and identified by Bilková et al., 2011 [3]), on 5-FU induced inflammation and its function to strengthen the intestinal barrier in the model human cell line Caco-2 derived from colon adenocarcinoma.
Caco-2 cells were cultivated for 21 days on permeable inserts under standard conditions (DMEM medium, penicillin and streptomycin 1%; 10% FBS; 37 °C; 5% CO2). The differentiation of cells was verified by measuring transepithelial electrical resistance and expression of gene-coding tight junction proteins (CLDN1, CLDN2, OCLN) by qPCR. Caco-2 cells were pretreated with 5-FU (100 µM; 24 h) and treated with LRE (1 × 108 CFU/mL; 4 h). An MTT test was used to research the effect of LRE on 5-FU-induced cytotoxicity in Caco-2 cells. The effect of LRE on inflammation (IL-1β, IL-8, NFκβ, A20, PPARα) and the integrity of the cell barrier were studied by qPCR.
LRE increased the viability (p < 0.05) of Caco-2 cells after 5-FU pretreatment. Moreover, we observed a significant increase in the expression of CLDN1 (p < 0.05), OCLN (p < 0.01), and IL-8 (p < 0.01) in 5-FU + LRE treated cells vs. the control, while a significant decrease was detected in the expression of NFKβ (p < 0.01) and PPARα (p < 0.001). Significant changes in the expression of selected genes were also seen in the Caco-2 cells individually treated with 5-FU or LRE.
Our results indicate that LRE affects inflammation and intestinal mucositis induced by 5-FU in vitro, and therefore clarification of its role in this process is going to be studied further.
The study was supported by grant VEGA 1/0429/21.
References:
- Ochiai, T.; Umeki M.; Miyake H.; et al. Oncology Reports. 2014, 32(3), 887–892.
- Batista, V. L.; Da Silva T. F.; De Jesus L. C. L.; et al. Frontiers in Microbiology. 2020, 11.
- Bilková, A.; Kiňova Sepová H.; Bukovský M. Veterinarni medicina. 2011, 56(7), 319–324.
33. Structural Hybrids of Fluoroquinolones and Their Prospects as New Antimicrobial Medicines
1
Department of Pharmaceutical Chemistry, National University of Pharmacy, Pushkinska str., 53, 61002, Kharkiv, Ukraine
2
Department of Organic Chemistry, V.N. Karazin Kharkiv National University, Svobody sq., 4, 61022, Kharkiv, Ukraine
3
Department of Microbiology, Virology and Immunology, National University of Pharmacy, Pushkinska str., 53, 61002, Kharkiv, Ukraine
4
Department of Medicinal Chemistry, National University of Pharmacy, Pushkinska str., 53, 61002, Kharkiv, Ukraine
Nowadays, the creation of novel antimicrobials still is a challenging task despite the fact that medical scientists all over the world have contributed greatly to this issue. Moreover, new diseases that appear together with the problem of the resistance of microorganisms create a vast area for united chemical and pharmacological research.
Therefore, our scientific team has been fruitfully working with fluoroquinolones and their hybridization based on docking studies with a further investigation of the antibacterial activity of the obtained compounds. Our findings, as well as the results of the other scientists [1,2,3] in this area, prove that representatives of the second generation of fluoroquinolones can be modified in the C-3 or C-7 position with the creation of new biologically active molecules.
Through the course of our research ciprofloxacin and norfloxacin substituted with 1,2,3-triazole moiety at C-7 revealed moderate activity against St. aureus ATCC 25923, E. coli ATCC 25922, B. subtilis ATCC 6633, P. aeruginosa ATCC 27853, and C. albicans NCTC 885-653. Furthermore, C-3-substituted arylsulfonyl derivatives were synthesized, and their activity is under study now.
The study was supported by the Ministry of Health of Ukraine from the state budget according to the topic “Molecular design and microbiological screening of innovative derivatives of fluoroquinolone antibiotics in order to combat resistant strains of microorganisms” (SRN: 0121U109239).
References:
- Mohammed, H.H.; Abuo-Rahma, G.E.-D.A.; Abbas, S.; Abdelhafez, E.-S.M. Current Trends and Future Directions of Fluoroquinolones. Curr. Med. Chem. 2018, 26, 3132–3149.
- Zahoor, A.F.; Yousaf, M.; Siddique, R.; Ahmad, S.; Naqvi, S.A.R.; Rizvi, S.M.A. Synthetic strategies toward the synthesis of enoxacin-, levofloxacin-, and gatifloxacin-based compounds: A review. Synth. Commun. 2017, 47, 1021–1039.
- Zhang, G.-F.; Liu, X.; Zhang, S.; Pan, B.; Liu, M.-L. Ciprofloxacin derivatives and their antibacterial activities. Eur. J. Med. Chem. 2018, 146, 599–612.
34. Efficacy and Safety of the Antidepressant Vortioxetine
Department of Toxicological and Analytical Chemistry, Faculty of Pharmacy, Danylo Halytsky Lviv National Medical University, 79010, Lviv, 69 Pekarska str, Ukraine
Introduction Vortioxetine is an antidepressant that was approved by the FDA for the treatment of major depressive disorder. The aim of the work was to assess the efficacy and safety of vortioxetine in patients with depression.
Materials and methods. The analysis of scientific publications on the effectiveness and tolerability of vortioxetine was conducted in the scientific systems MEDLINE, Google Scholar, and ScienceDirect.
Results and discussion. The most common adverse effects reported from vortioxetine in the studies were nausea, vomiting, and constipation. Nausea frequency was dose-dependent, reaching 32% when taking vortioxetine at a dose of 20 mg/day [1]. Nausea was practically the only reason for the early discontinuation of vortioxetine treatment.
Other important side effects of vortioxetine therapy are hypertensive crisis and increased suicidal risk. A case of acute vortioxetine poisoning with suicidal intent was registered [2].
The proportion of patients with sexual dysfunction was 1.7% in the Vortioxetine 5–10 mg group and 2.3% in the Vortioxetine 15–20 mg group [3].
An analysis of data from short-term studies showed that treatment with vortioxetine did not lead to any clinically significant changes in blood pressure and heart rate. At the same time, serotonergic transmission and modulation play a role in hemostasis. Therefore, it is recommended to use the drug with caution in patients with bleeding disorders.
Conclusions Administration of vortioxetine in clinical trials in the dose range from 40 to 75 mg caused exacerbation of the following side effects: nausea, postural dizziness, diarrhea, abdominal discomfort, generalized pruritus, drowsiness, and facial flushing. Post-marketing experience mainly concerns an overdose of vortioxetine up to 80 mg. The most common side effects are nausea and vomiting. Seizures and serotonin syndrome have been reported after taking doses several times higher than the therapeutic range.
References:
- Gonda, X.; Sharma, S.; Tarazi, F. Expert Opinion on Drug Discovery. 2019, 14(1), 81–89.
- Mazza, M.; Rossetti, A.; Botti E.; et al. Medicine. 2018, 97(25). P. 10788.
- Salagre, E.; Grande, I.; Solé, B.; et al. Revista de Psiquiatría y Salud Mental, 11(1), 2018, P. 48–59.
35. Derivatives of Putrescine and Spermidine from Flowers of Amorpha fruticosa L., Fabaceae
Department of Natural Drugs, Faculty of Pharmacy, Masaryk University, Palackého tř. 1946/1, 612 00 Brno, Czech Republic;
The aim of the assay was the selection of the most potent compounds responsible for the anticholinesterase activity of methanolic extracts of Amorpha fruticosa L., Fabaceae.
Amorpha fruticosa L., Fabaceae is a shrub native to North America, which is known by several common names: false indigo bush and desert false indigo bush. It is used for blue staining [1].
The ethyl acetate extract of flowers was subjected to a silica gel column using chloroform-methanol to afford 26 fractions. Fractions AF_K_E_8 and AF_K_E_9 were selected for separation on semi-preparative HPLC. The six main compounds were obtained. The structure elucidation of compounds 1–6 was measured with UV spectra, HRMS, and NMR spectra.
After evaluating the results by spectral methods and comparing them with the literature, the following substances were identified: Compound (1) as mongolicin A (or N1- (E) -N6- (Z) -di-p-coumaroylputrescine) [2]; compound (2), as N1, N6- (E) -di-p-coumaroylputrescine [3]; compound (3) was identified as safflospermidine B (or N1- (E) -N5, N10- (Z) -tri-p-coumaroylspermidine) [4]; compound (4) was identified as N1, N5-(Z)-N10-(E)-tri-p-coumaroylspermidin [5]. The yield of the compound (5) was only 0.56 mg, making it difficult to identify. However, based on the similarity of the UV spectra of the isolated substances, it is assumed that it is one of the isomers. Compound (6) was obtained in the largest amount and was determined from spectral data. It is an “all trans” isomer—N1, N5, N10-(E)-tri-p-coumaroylspermidin [6].
This type of substance was isolated from flowers of Amorpha fruticosa for the first time. The anticholinesterase activity of the isolated substances will be measured.
References:
- Kozuharova E.; Mastkowski A.; Woźniak D.; Simeonova R. et al. Front. Pharmacol. 2017, 8(333), 1–17.
- Kim SB.; Liu Q.; Ahn JH.; Jo YH. et al. Bioor. Chem., 2018, 81, 127-133
- Choi SW.; Lee SK.; Kim EO.; Oh JH. et al. J. Agric. Food. Chem. 2007, 55(10), 3920-3925.
- Khongkarat P.; Ramadhan R.; Phuwapraisirisan P.; Chanchao C. Heliyon. 2020, 6(3), e03638.
- Zhao G.; Qin GW.; Gyi Y.; Guo LH. Chem. Pharm. Bull. 2010, 58(7), 950–952.
- Jiang JS.; Lü L.; Yang YJ.; Zhang JL.; Zhang PC. J. Asian Nat. Prod. Res. 2008, 10(5), 447–451.
36. Bile Acids—Natural Building Blocks for Porous Coordination Self-Assemblies
1
Department of Natural Drugs, Faculty of Pharmacy, Masaryk University, Palackého tř. 1946/1, 612 00 Brno, Czechia
2
Department of Chemistry, Faculty of Science, Masaryk University, Kamenice 5, 625 00 Brno, Czechia
3
CEITEC, Masaryk University, Kamenice 5, 625 00 Brno, Czechia
4
RECETOX, Masaryk University, Kamenice 5, 625 00 Brno, Czechia
Bile acids are relatively rigid amphiphilic molecules naturally contained in bile, which are responsible for the digestion and transportation of lipids and lipid-soluble nutrients [1]. Inspired by their properties, behaviors, and biological activities, we have decided to employ bile acids in the design and preparation of ligands and their supramolecular coordination self-assemblies [2,3]. Unsymmetric and inherently chiral pyridyl ligands (L) were used for coordination with model palladium (II) nodes forming various cationic complexes in a controlled fashion, e.g., Pd3L6 (Figure 1), Pd6L12, or even Pd12L16. These large concave complexes containing metals and numerous chiral centers can resemble enzymatic binding pockets or transmembrane channels. Some of the complexes can further self-organize upon external stimulus into nano- or microscopic particles (Figure 1).
Figure 1.
Self-assembly of bile acid-based coordination complexes Pd3L6 and their self-organization into microparticles [3].
Our work introduces sustainable natural products, bile acids, as main components for the coordination of ligands for the preparation of large chiral coordination self-assemblies. This further leads to the development of a new family of porous metal-organic materials with intriguing properties and applications.
References:
- Durník, R.; Šindlerová, L.; Babica, P.; Jurček, O. Molecules 2022, 27(9), 2961.
- Jurček, O.; Bonakdarzadeh, P.; Kalenius, E.; et al. Angew. Chem. Int. Ed. 2015, 54, 15462–15467.
- Jurček, O.; Nonappa; Kalenius, E.; et al. Cell Rep. Phys. Sci. 2021, 2, 100303.
37. Molecular Docking of 3 Series of Potential Cancerostatics to Cas and Hdacs
1
Department of Pharmaceutical chemistry, Faculty of Pharmacy, Comenius University, Odbojarov 10, 832 32 Bratislava, Slovakia
2
Department of Chemical Drugs, Faculty of Pharmacy, Masaryk University, Czech Republic
Histone deacetylase and carbonic anhydrase are both zinc-dependent metalloenzymes involved in cancer progression. New inhibitors of these enzymes, especially isoenzymes CA IX, CA XII and HDAC 1, 2, 3, and 8, could act as small molecular cancerostatistics [1,2]. A series of three groups of potential inhibitors were docked to the carbonic anhydrase isoenzymes CA I, CA II, CA III, CA IV, CA VII, CA IX, CAXII, CA XII, CAXIV, and the isoenzymes of histone deacetylase HDAC 1, HDAC 2, HDAC 3, HDAC 4, HDAC 6, HDAC 7, and HDAC 8. The series of ligands consists of (2E)-3-phenylprop-2-enhydroxamates, (2E)-3-phenylprop-2-enthiols, and (2E)-N-(2-aminophenyl)-3-phenylprop-2-enamides. Various substituted 2-hydroxy-N-phenylacetamides were attached to the phenyl in position four. The docking was performed in the program Schrodinger Glide [3] with XP precision for all ligands. Ligands belonging to the hydroxamate group were docked to HDACs in monodentate and also bidentate binding modes. Whereas the best docking scores for the poses docked in isoenzymes HDAC 1, 4, and 7 were achieved by the bidentate binding mode, the poses in complexes with isoenzymes HDAC 2, 3, and 6 had a higher docking score in monodentate binding mode. In general, it is very convenient if the imine nitrogens of the two closest histidines, HIS140 and HIS141 (numbering according to HDAC 1 [PDB: 5ICN [4]]), are oriented to the hydrogens of the hydroxamate functional group. Poses of the ligands MO22, MO23, and MO19 show the highest docking scores in complexes with isozymes HDAC 1, 2, 3, and 8.
The thiol derivatives were docked with constraints on the coordination bond between zinc di-cation and sulfur of the thiol group. The average docking score of poses of the ligand SH03 for HDAC 1, 2, 3, and 8 isoenzymes was -8,71. Although the docking scores of this ligand are slightly lower for CA isoenzymes (-7,43 for CA IX and -6,89 for CA XII), the molecule could be a significant target of interest for dual inhibitor design.
For the docking of the group of benzamide ligands, the position of the functional group of the ligand from crystal structure HDAC2 with a benzamide [PDB: 5IWG [5]] was used as a core pattern for the pose comparison. The highest average docking score was obtained by the poses of molecule BA14 with a value of -7,92 for HDAC 1, 2, 3, and 8. The lower average docking score of benzamide derivatives could be explained by a larger volume of the functional group.
References:
- C.T. Supuran, J.-Y. Winum, Carbonic Anhydrase Ix Inhibitors in Cancer Therapy: An Update, Future Med. Chem. 7 (2015) 1407–1414.
- P. Bertrand, Inside Hdac with Hdac Inhibitors, Eur. J. Med. Chem. 45 (2010) 2095–2116.
- R.A. Friesner, Et Al. Extra Precision Glide: Docking and Scoring Incorporating a Model of Hydrophobic Enclosure for Protein−Ligand Complexes, J. Med. Chem. 49 (2006) 6177–6196.
- P.J. Watson, Et Al. Insights into The Activation Mechanism of Class I Hdac Complexes by Inositol Phosphates, Nat. Commun. 7 (2016) 11262.
- F.F. Wagner, Et Al. Kinetic and Structural Insights into The Binding of Histone Deacetylase 1 And 2 (Hdac1, 2) Inhibitors, Bioorg. Med. Chem. 24 (2016) 4008–4015.
38. Synthesis and Bioevaluation of Fluorescent Molecular Probes for the Early Detection and Progression of Alzheimer’s Disease
1
University of Ljubljana, Faculty of Chemistry and Chemical Technology, Večna pot 113, Ljubljana, Slovenia
2
University of Ljubljana, Faculty of Pharmacy, Aškerčeva 7, Ljubljana, Slovenia
3
University of Ljubljana, Faculty of Medicine, Vrazov trg 2, Ljubljana, Slovenia
Alzheimer’s disease (AD) is a chronic, irreversible, progressive neurodegenerative disease with the most common symptom of dementia [1]. Currently, there are neither effective therapeutics nor easily accessible, non-invasive, reliable, and inexpensive diagnostic tools. Therefore, the development of a diagnostic method that allows widespread early AD detection and disease progression will be a defining moment that will lead to the identification of vulnerable but asymptomatic populations that may be more susceptible to existing therapeutics. Moreover, such a diagnostic method will certainly accelerate the development of improved AD therapeutic options [2].
The Košmrlj group is focused on the design and synthesis of fluorescent molecular probes for the ex vivo detection of AD-related proteins, such as amyloid β, tau protein, etc., in the blood of AD patients. The molecular probes consist of three crucial moieties: (a) an electron donor group, (b) an aromatic π-linker, and (c) an electron-withdrawing group, which are essential for the fluorescent properties due to the push-pull electron effect. This diagnostic method is based on the differences in molecular probes’ optical properties, as the absorption, emission, and excitation maxima are red-shifted in the presence of AD proteins compared to unbound probes. In vitro determination of the dissociation constants (Kd) of molecular probes to amyloid β and tau protein in buffer, as well as ex vivo staining of these proteins on post-mortem brain slices from patients with AD pathology, are currently under investigation. We expect that such a stepwise approach will lead to the design of a “smart molecular probe” that will allow the detection of AD-related proteins in the blood of patients [3].
The study was supported by the Slovenian Research Agency (ARRS; Research Core Funding grant P1-0230 and project J1-3018).
References:
- Zvěřová, M. Clin. Biochem. 2019, 72, 3–6.
- Winder, Z.; Sudduth, T.L.; Anderson, S.; et al. Alzheimer’s Dement. 2022, 1–12.
- Rejc, L.; Šmid, L.; Kepe, V.; et al. J. Med. Chem. 2017, 60(21), 8741–8757.
39. Determination of Drug-Protein Binding Parameters by Ce-Fa Method with Conductivity and UV-VIS Detection
Department of Biochemistry, Faculty of Science, Masaryk University, Kamenice 5, 625 00 Brno, Czech Republic
Plasma proteins increase drug solubility and act as a transport system; however, the bound drug is not able to permeate through the membrane to tissues and provide its pharmacological activity. The strength of interaction affects the pharmacological profile of the drug. In this regard, it is important to know the binding parameters, such as the binding constant (Kb) and stoichiometry of the interaction (n). Nowadays, there is an increasing emphasis on the development of sensitive and highly efficient laboratory methods that will help to understand and describe these interactions in detail. In newly created methods, the emphasis is on the low consumption of samples and chemicals, physiological conditions of studied interactions or in conditions close to physiological ones, and finally, that neither of the interaction partners is immobilized or tagged, as this may lead to the binding parameters. One of the methods for fulfilling these criteria is the capillary electrophoresis-frontal analysis (CE-FA) [1,2,3]. The aim of this study was to optimize a new approach that combines the CE-FA method with contactless conductivity detection (C4D) for studying plasma protein-drug.
The model of salicylic acid (SA) and human serum albumin (has) was chosen as the interaction pair of drug and plasma protein. The binding parameters were calculated from experimental data by nonlinear regression analysis. The binding parameters of SA-HSA measured by the CE-FA C4D method were log Kb = 4.24 ± 0.05 and n = 3.23 ± 0.10. The obtained binding values corresponded to the results of the conventional CE-FA with ultraviolet-visible (UV-VIS) detection and the range reported in the literature. Optimized CE-FA C4D analysis lasted less than 5 min, and the binding data were characterized by good repeatability, RSD = 1.09% for log Kb and RSD = 3.04% for n.
The study was supported by grant GA19-08358S from the Czech Science Foundation.
References:
- Mlčochová, H.; Ratih, R.; Michalcová, L. et al. Electrophoresis. 2022; doi.org/10.1002/elps.202100320
- Mlčochová, H.; Michalcová, L.; Glatz, Z. Electrophoresis. 2022, 43(9–10), 955–963.
- Michalcová, L.; Mlčochová, H.; Glatz, Z. J. Chromatog. A. 2021, 1635, 461734–461742.
40. Comparative Study of Ibuprofen Liberation from Cationic and Anionic Polysaccharide Hydrogels
1
Department of Pharmaceutical Analysis and Nuclear Pharmacy, Faculty of Pharmacy, Comenius University Bratislava, Odbojárov 10, 83232 Bratislava, Slovakia
2
Toxicological and Antidoping Centre, Faculty of Pharmacy, Comenius University Bratislava, Odbojárov 10, SK-832 32 Bratislava, Slovakia
3
Department of Galenic Pharmacy, Faculty of Pharmacy, Comenius University Bratislava, Odbojárov 10, 83232 Bratislava, Slovakia
Polysaccharide-based gelling agents are attractive in pharmacies due to their safety, biocompatibility, biodegradability, relatively easy method of preparation, and low price. Due to their variable physical-chemical properties, polysaccharides have the potential to be used for designing new drug delivery systems for controlled drug release. In this work, rheologic and texture properties, and the drug release of model drug ibuprofen, were investigated in cationic (high molecular weight chitosan CS HMW, chitosan derived from crab shells CSc, and chitosan derived from shrimp shells CSs) and anionic (carboxymethylcellulose CMC, sodium salt of carboxymethylcellulose NaCMC, tragacanth TRG, carrageenan CRG, xanthan gum XTG) polysaccharide-based hydrogels.
Due to their favorable properties for topical applications (structural viscosity > 10.0 N.S m−2) 6% NaCMC, 8% and 10% CMC, 2%, 4% and 6% TRG, and 8% and 10% CRG, 4% and 6% TXG, 2% and 3% CSc, 2% and 3% CSs, 2% and 3% CS HMW were studied for texture properties and IBU release. The most promising tested IBU hydrogels for topical administration, considering the hardness and adhesiveness, were 6% NaCMC, 6% TRG, 4% and 6% XTG, 2% and 3% CSc, 3% CSs, and 2% and 3% CS HMW with hardness > 5 g, adhesiveness > 5 g.s., and minimal retracting force > 5 g. The amount of released IBU in 180 min increased in order: 3% CS HMW (15.5%), 3% CSs (16.9%), 3% CSc (17.0%), 2% CS HMW (20.9%), 2% CSc (20.1%), 2% CSs (21.0%), 10% CMC (51.2%), 10% CRG (55.5%), 8% CRG (56.6%), 8% CMC (56.8%), 6% TRG (58.2%), 6% XTG (58.3%), 6% NaCMC (61.8%), 4% XTG (62.4%), 4% TRG (63.4), and 2% TRG (64.5%). CS hydrogels showed sustained release following zero-order kinetics. In general, a higher IBU diffusion was observed from anionic hydrogels in comparison with the neutral ones (probably due to the repulsive Coulomb forces between negatively charged groups in the ionic polymers and negatively charged (carboxylate) group of IBU). Although 2% TRG showed the fastest release of IBU, 4% hydrogel of XTG was chosen as optimal for topical dermal application because of its better adhesiveness, hardness, and viscosity, along with a still relatively fast release of IBU.
This work was supported by the projects VEGA 1/0514/22, KEGA 027UK-4/2020, APVV-15-0585, FaF/21/2022, and UK/73/2022.
This poster was created thanks to support from the Operational Program Integrated Infrastructure for the project: National infrastructure for supporting technology transfer in Slovakia II—NITT SK II, co-financed by the European Regional Development Fund.
41. Comparative Study of Ibuprofen Liberation from Cationic and Neutral Polysaccharide Hydrogels
1
Department of Galenic Pharmacy, Faculty of Pharmacy, Comenius University Bratislava, Odbojárov 10, SK-83232 Bratislava, Slovakia
2
Department of Pharmaceutical Analysis and Nuclear Pharmacy, Faculty of Pharmacy, Comenius University Bratislava, Odbojárov 10, SK-83232 Bratislava, Slovakia;
3
Toxicological and Antidoping Centre, Faculty of Pharmacy, Comenius University Bratislava, Odbojárov 10, SK-832 32 Bratislava, Slovakia
Polysaccharide-based gelling agents show a potential to be used for designing new drug delivery systems with controlled, prolonged, or sustained drug release. In this study, rheological and texture properties, as well as the in vitro release of model drug ibuprofen of cationic (high molecular weight chitosan CS HMW, chitosan derived from crab shells CSc and chitosan derived from shrimp shells CSs and neutral hydrogels (methylcellulose MC, hydroxypropylmethylcellulose HPMC, hydroxyethylcellulose HEC) were investigated.
Based on values of structural viscosity, medium-viscous (10.0–50.0 N.S.m−2), and high-viscous (>50.0 N.S.m−2) hydrogels (including 6% MC, 2% HPMC, 4% and 6% HEC, 3% CSc, 2% and 3% CSs, 2% and 3% CS HMW) were further studied for texture properties. The texture profile analysis was used to investigate the effect of the concentration of the gelling substance and the effect of IBU content on the mechanical properties of the resulting hydrogel. Hardness, adhesiveness, and minimal retracting force were evaluated, and the most promising tested IBU-hydrogels for dermal application were: 2% HPMC, 6% HEC, 2% and 3% CSc, 3% CSs, and 2% and 3% CS HMW with hardness > 5 g, adhesiveness > 5 g.s and minimal retracting force > 5 g. The amount of released IBU after 180 min increased in order 3% CS HMW (15.5%), 3% CSs (16.9%), 3% CSc (17.0%), 2% CS HMW (20.9%), 2% CSc (20.1%), 2% CSs (21.0%), 4% MC (52.5%), 6% HEC (53.3%), 6% MC (54.3%), 2% HPMC (56.9%), and 4% HEC (57.0%). The drug release from CS hydrogels follows zero-order kinetics and is sustained without initial burst release. The amount of released IBU after 330 min increased in order: 3% CSs (34.4%), 3% CS HMW (34.8%), 3% CSc (43.4%), 2% CSs (45.1%), 2% CSc (47.9%), and 2% CS HMW (51.3%).
Significant differences (p < 0.01) in IBU diffusion were statistically confirmed between the groups of the hydrogels composed of the positively charged polysaccharides and the neutral ones. This behavior can be explained via the stabilization of CS-IBU associates by attractive Coulomb interactions and, thus, a slower release of IBU from the CS hydrogels.
This work was supported by the projects FaF/21/2022, UK/73/2022, VEGA 1/0514/22, KEGA 027UK-4/2020, and APVV-15-0585.
This poster was created thanks to support from the Operational Program Integrated Infrastructure for the project: National infrastructure for supporting technology transfer in Slovakia II—NITT SK II, co-financed by the European Regional Development Fund.
42. Synthesis and Biological Screening of Phenylpiperazine Derivatives of 3-Substituted 1H-Indole-2-Carboxylic Acids and Their Quaternary Salts
1
Department of Chemical Drugs, Faculty of Pharmacy, Masaryk University, Palackého tř. 1946/1, 612 00 Brno, Czech Republic
2
Department of Natural Drugs, Faculty of Pharmacy, Masaryk University, Palackého tř. 1946/1, 612 00 Brno, Czech Republic
3
Department of Molecular Pharmacy, Faculty of Pharmacy, Masaryk University, Palackého tř. 1946/1, 612 00 Brno, Czech Republic
Aryloxyphenylpiperazinylpropanols are a wide group of synthetic compounds that have been prepared as potential drugs affecting the central nervous and cardiovascular systems, and some also exhibit strong activity against various microorganisms [1].
A series of new 3-(4-arylpiperazin-1-yl)-2-hydroxypropylesters of 3-methyl- or 3-ethyl-1H-indole-2-carboxylic acids and their quaternary salts (Figure 1) were synthesized by multistep reactions. In the first phase, 3-substituted 1H-indole-2-carboxylic acids were synthesized, 3-methyl derivatives by the methods we described previously [2] and 2-ethyl derivatives by a new, different procedure. In the second phase, the corresponding indole-2-carboxylic acids were converted into their potassium salts, which reacted with glycidyl tosylate to give oxirane intermediates. The oxirane ring was then opened with appropriate phenylpiperazine. In the third phase, hydrochloride salts and quaternary ammonium salts (QUATs) were prepared from the final bases. The structure and purity of the prepared compounds were verified by the available methods of instrumental analysis (1H-NMR, 13C-NMR, FT-IR, TLC, HPLC).
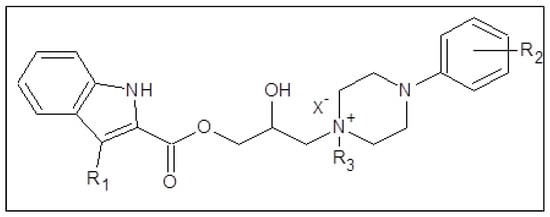
Figure 1.
The common structure of synthesized compounds.
The hydrochloride salts and QUATs were tested for their acetylcholinesterase and butyrylcholinesterase inhibition activity by a modified Ellman method. The QUATs were tested for antimicrobial activity against both Gram-negative and Gram-positive bacteria, as well as anti-biofilm activity and anti-fungal activity. Finally, compounds displaying antimicrobial activity were tested for cytotoxicity against mammalian cell cultures.
References:
- Marvanová, P.; Padrtová, T.; Mokrý, P. Mini Rev. Med. Chem. 2020, 20(17), 1719–1731.
- Padrtová, T. et al. Curr. Org. Synth. 2020, 17(7), 576–587.
43. Isolation and Identification of Phlorotannins from Brown Algae Ecklonia radiata (C. Agardh) J. AGARDH
1
Department of Natural Drugs, Faculty of Pharmacy, Masaryk University, Palackého tř. 1946/1, 612 00 Brno, Czech Republic
2
Department of Food Quality and Safety, Czech University of Life Sciences Prague, Kamýcká 129, 165 00 Prague 6–Suchdol, Czech Republic
Ecklonia radiata (C.Agardh) J.Agardh (Lessoniaceae) is a small kelp found abundantly in the warm-temperate parts of South Africa, Australia, and New Zealand. Brown seaweeds, especially those of the Ecklonia genus, are known to be a rich source of phlorotannins [1]. Phlorotannins are polyphenolic compounds that are formed through the polymerization of phloroglucinol (1,3,5-trihydroxybenze-ne) [2]. Studies over the past decades have shown that phlorotannins possess several bioactivities, including anti-herbivory, antioxidants, anti-inflammatory, antimicrobial, antiproliferative, antidiabetic, radioprotective, adipogenic, antiallergic, and antiviral, demonstrating promising potential in commercial applications in areas, such as food, nutraceutical, and pharmaceutical applications [2,3].
The dried water-ethanolic extract of different parts of E. radiata was separated by liquid-liquid extraction into several portions. Ethyl acetate portion was subjected to further separation using column chromatography. Selected subfractions were subsequently separated using semi-preparative reversed-phase high-performance liquid chromatography. Identification of the isolated compounds was conducted using available spectral methods. Basic information about the structure was provided by ultraviolet and infrared spectroscopy. Exact mass and molecular formulas were determined using high-resolution mass spectrometry. Structures of the isolated compounds were elucidated using one- and two-dimensional nuclear magnetic resonance spectroscopy.
The study was supported by the Czech Sciences Foundation (CSF Bilateral AT-CZ 21-38204L—Complexes of selected transition metals with plant-derived compounds with anti-NF-kappa B and pro-PPAR dual activities; K.Š.) and Masaryk University, Specific research—support for student projects (MUNI/ A/1688/2020). We thank Dr. Kannan R.R. Rengasamy (Faculty of Natural and Agricultural Sciences, North-West University, South Africa) for providing the plant material.
References:
- Shrestha, S.; Zhang, W.; Begbie, A.J.; et al. Food Funct. 2020, 11, 6509–6516.
- Okeke, E.S.; Nweze, E.J.; Chibuogwu, C.C.; et al. Nat. Prod. Commun. 2021, 16, 1–23.
- Shrestha, S.; Zhang, W.; Smid, S.D. Food Biosci. 2021, 39, 100832.
44. Changes in Bioactive Compounds Content of Crocus Sativus Stigma during Pre-Sowing Corms Treatment
1
Faculty of Natural Sciences, Vytautas Magnus University, Vileikos str. 8, Kaunas LT-44404, Lithuania
2
Department of Pharmaceutical Chemistry, National University of Pharmacy, 4-Valentinivska st., 61168, Kharkiv, Ukraine, mykhailenл
3
Lithuanian University of Health Sciences, Department of Analytical and Toxicological Chemistry, A. Mickevičiaus g. 9, LT 44307 Kaunas, Lithuania,
The current report, for the first time, demonstrated the impact of treatment of Crocus sativus corms with plasma (CP, 3 and 5 min), vacuum (3 min), and electromagnetic field (EMF, 5 min) on a number of bioactive components in Crocus sativus stigma. Saffron spice is obtained from the dried stigmas of Crocus, which is a natural food coloring and medicinal raw material [1]. The influence of corms processing on emergence kinetics in field conditions and on growth and morphometric parameters of plants was determined; changes in antioxidant activity and the total amount of phenolic compounds in leaves and stigma, changes in the number of leaf trichomes, and changes in the number of secondary metabolites in stigma. It was found that the maximum emergence (viability) in the field of control and treated corms was 100%. However, the longer (5 min) treatment with CP retarded emergence, while vacuum treatment increased emergence uniformity by 28%. The growth of plants in the CP5 and EMF groups was retarded as compared with the control. Treatment with all stressors significantly (42–74%) increased the number of lower leaf trichomes. CP3 significantly reduced the length and dry weight (43 and 60%, respectively) of flowers, and EMF treatment resulted in 27% longer flowers compared with controls. CP5 increased the antioxidant activity of leaves, and the total content of phenolic compounds in leaves of the CP5 group was 44%, in EMF- 27% higher compared with controls. The treatment of corms with stressors caused significant changes in the number of secondary metabolites in the pistils. HPLC analysis was used to identify and quantify 26 compounds in the raw material, of which 23 compounds are crocetin (crocin) esters, as well as rutin, picrocrocin, and safranal. The main esters of crocetin were trans-4GG (about 40% of the total content), trans-3Gg, trans-2G, cis-4GG, trans-2-gg, and cis-3Gg. The changes induced by vacuum treatment were smaller compared with those induced by CP and EMF. Treatment with CP reduced the number of metabolites. Meanwhile, EMF treatment resulted in a significant positive effect on the synthesis of secondary metabolites. We conclude that the treatment of corms with EMF has the potential to be applied to increase the production of valuable saffron secondary metabolites.
References:
- Butnariu, M.; Quispe, C.; et al. Oxid. Med. Cell. Longev. 2022, Article ID 8214821.
45. Anti-Inflammatory Potential of Selected Phenolic Compounds Isolated from Morus alba
1
Department of Molecular Pharmacy, Faculty of Pharmacy, Masaryk University, Palackého tř. 1946/1, 612 00 Brno, Czech Republic
2
Department of Natural Drugs, Faculty of Pharmacy, Masaryk University, Palackého tř 1946/1, 612 00 Brno, Czech Republic
Morus alba L. (Moraceae), also known as white mulberry, is a medium-sized tree originally from China. It is mainly cultivated for silk farming and fruit production, and its parts are ingredients in traditional Chinese medicine for the therapy of inflammation. M. alba has been a rich source of plant phenolics with various biological effects. Recently, a series of compounds were isolated: three geranylated 2-arylbenzofurans mulberrofuran A and B, and mulberroside C, two flavonoids kuwanon C and U, and a Diels-Alder adduct kuwanon H [1].
Regarding traditional usage, we decided to experimentally verify the anti-inflammatory activity of the isolated compounds. Firstly, the effect of the compounds on the viability of the THP-1-XBlue™-MD2-CD14 cell line (Invivogen, San Diego, CA, USA) was measured using WST-1 assay kit (Roche, Switzerland). Further, the inhibitory effect of the test compounds in a non-toxic concentration (1 μM) after 24 h incubation on the NF-κB cellular pathway was assessed using a QUANTI-Blue™ assay [2].
Among the test compounds, the highest inhibitory effect was observed after incubation of mulberrofuran B (Figure 1), and the activity of NF-κB was decreased to 69 ± 17% (p ≤ 0.0001), which is comparable to the effect of positive control, prednisolone. The anti-inflammatory effect of mulberrofuran B was also described by Čulenová et al., although in a different method—in vivo assay using carrageenan-induced paw edema [1].
Figure 1.
The structure of mulberrofuran B.
The study was supported by Masaryk University, Specific research—support for student projects [grant number MUNI/A/1654/2020—Testing of biological activity of selected natural substances in vitro].
References:
- Čulenová, M.; Sychrová, A.; Hassan, S. T. S. et al. J. Ethnopharmacol. 2020, 248(10), 112296.
- Malaník, M.; Treml, J.; Leláková, V. et al. Bioorg. Chem. 2020; 104, 104298.
46. Targeting Metalloenzymes with Newly Designed Inhibitors for Potential Therapeutic Intervention
1
Department of Chemical Drugs, Faculty of Pharmacy, Masaryk University, Palackého tř. 1946/1, 612 00 Brno, Czech Republic
2
Department of Pharmaceutical Chemistry, Faculty of Pharmacy, Comenius University, Ulica odbojárov 10, 83232 Bratislava, Slovakia
Almost half of all enzymes must cooperate with metal ions to function. Most metalloproteins play a crucial role in metabolically essential processes [1]. They also significantly impact the progression of various human diseases, including cancer, heart disease, and HIV/AIDS [2]. Due to this reason, they are an attractive medical target for the treatment of a wide range of human disorders.
In this work, we focused on zinc metalloproteins such as carbonic anhydrase (CA) and histone deacetylase (HDAC), which are cancer-related enzymes [3,4].
We designed structures for promising metalloenzyme inhibitors. The zinc-binding group was thiol, benzamide, hydroxamate, or other functional groups with good chelating properties. As a linker part of the designed structure, we used the p-Coumaric acid motif. Molecular docking was performed in the Schrodinger Glide program with XP precision for all molecules [5]. Only physiologically relevant human CA and HDAC isoforms were chosen for the docking. According to in silico results, we have synthesized compounds with considerably high docking scores, indicating their satisfying binding at the active site of selected metalloenzymes.
The study was supported by the project MUNI/IGA/1339/2021
References:
- Laity, J.H., Lee, B.M., Wright, P.E., 2001. Zinc finger proteins: new insights into structural and functional diversity. Curr. Opin. Struct. Biol., 2001, 11, 39–46.
- Jiang, Z., You, Q., Zhang, X., 2019. Medicinal chemistry of metal chelating fragments in metalloenzyme active sites: A perspective. Eur. J. Med. Chem., 2019, 165, 172–197.
- Supuran, C.T., 2008. Carbonic anhydrases--an overview. Curr. Pharm. Des., 2008, 14, 603–614.
- Ma, X., Ezzeldin, H.H., Diasio, R.B., 2009. Histone deacetylase inhibitors: current status and overview of recent clinical trials. Drugs, 2009, 69, 1911–1934.
- Friesner, R.A., Murphy, R.B., Repasky, M.P., Frye, L.L., et al., 2006. Extra Precision Glide: Docking and Scoring Incorporating a Model of Hydrophobic Enclosure for Protein−Ligand Complexes. J. Med. Chem., 2006, 49, 6177–6196.
47. Silver(I) Compounds with Bioactive Molecules as Carriers for Antimicrobial and Cytotoxic Silver(I) Ion
1
Department of Inorganic Chemistry, Faculty of Science, P. J. Šafárik University, Moyzesova 11, 041 54 Košice, Slovak Republic
2
Department of Microbiology and Immunology, University of Veterinary Medicine and Pharmacy, Komenského 73, 041 81 Košice, Slovak Republic
3
Department of Inorganic Chemistry, Faculty of Science, Charles University, Hlavova 2030, 128 00 Prague, Czech Republic
4
NMR Laboratory, Faculty of Science, P. J. Šafárik University, Moyzesova 11, 041 54 Košice, Slovak Republic
5
Department of Pharmacology, Faculty of Medicine, P. J. Šafárik University, Trieda SNP 1, 040 11 Košice, Slovak Republic
We have been fighting the global pandemic caused by COVID-19 for the third year in a row, but the worrying fact is that other serious diseases and their treatment remain in the shadow of the pandemic. Antibiotic resistance is still a threat to public health during this pandemic, and the forecasts of the deaths caused by antimicrobial resistance infections are relatively pessimistic for the coming years. Thus, it is very important to continue to develop and improve the treatment of emerging infectious diseases and to face the growing number of resistant microbial pathogens. In addition, research on new anticancer drugs and other therapeutics should not be left behind.
The aim of our experimental study was the synthesis of solid-state characterization and in vitro biological evaluation of the new silver (I) coordination compounds with bioactive molecules such as amino acids and N-heterocyclic compounds. Some of the prepared compounds exhibit higher levels of biological activity than commercially used drugs, silver (I) sulfadiazine and cisplatin, and even selective inhibition of the growth of pathogenic and probiotic bacteria, as well as cancer and healthy cells, has been observed [1,2,3]. Based on the obtained results, it is appropriate to consider carrying on other pharmacology studies in the future.
The study was supported by P. J. Šafárik University in Košice vvgs-2022-2123 and Slovak grant agency KEGA 006UPJŠ-4/2021.
References:
- Rendošová, M.; Gyepes, R.; Maruščáková, I. C.; et al. Dalton Trans. 2021, 50, 936–953.
- Rendošová, M.; Vargová, Z.; Sabolová, D.; et al. J. Inorg. Biochem. 2018, 186, 206–216.
- Kuzderová, G.; Rendošová, M.; Gyepes, R.; et al. Molecules 2021, 26, 6335.
48. Critical Micelle Concentration and Cytotoxicity of Phenylcarbamidacid Derivates
1
Department of Pharmaceutical Chemistry, Faculty of Pharmacy, Comenius University, Odbojárov 10, 832 32, Bratislava, Slovak Republic
2
Department of Microbiology and Virology, Faculty of Natural Sciences, Comenius University, Mlynska dolina, Ilkovičova 6, 842 15, Bratislava 4, Slovak Republic
The critical micellar concentration (CMC) of base esters of various substituted phenylcarbamic acid (occupationally labeled 6a, 6b, 6c, 6d, 6e, 6i, 6j, 6k, and 6l) in the aqueous solution was determined using absorption spectroscopy in the UV/VIS spectrum area using pyrene as a probe in the environment. This dependence had a sigmoidal character, and the CMC was the center of the sigmoid [1,2].
The cytotoxicity and inhibitory effects of the individual substances were determined using the MTT test on the Hela cell line. Test substances 6a, 6b, 6c, 6d, and 6e showed significant cytotoxicity at 100 and 2500 μg/mL concentrations. Substances 6i, 6j, 6k, and 6l were more toxic than the previous substances; a 5 μg/mL concentration showed medium cytotoxicity. No inhibitory activity was observed at the lower concentrations of tested substances. IC50 values were calculated using nonlinear regression analysis using GraphPad Prism 7. Substances 6a, 6b, 6c, 6d, and 6e had a lesser effect on inhibiting HeLa cell proliferation than substances 6i, 6j, 6k, and 6l. The CMC values for all study substances were more significant than their corresponding IC50 values. Depending on the -logc (-logCMC and -logIC50) of the number of carbon atoms (n) in the hydrophobic chain, the analytes’ CMC was not responsible for inhibition because all the studied substances act in the monomer state.
KEYWORDS: Critical micelle concentration. Phenylcarbamates. Absorption spectroscopy. Cytotoxicity
The study was supported by the Grant FaF/19/2022.
References:
- Salanci, E., Andriamainty, F., Malík, I., Šándrik, R. et al. Chem. Pap. 2021, 75, 3081–3090.
- Stopková, L., Gališinová, J., Šuchtová, Z., Andriamainty, F. et al. Molecules. 2018, 23(5), 1064.
49. The Effect of Poorly Compressible Ascorbic Acid on Dilution Potential of Co-Processed Excipients
Department of Pharmaceutical Technology, Faculty of Pharmacy, Masaryk University, Palackého tř. 1946/1, 612 00 Brno, Czech Republic
Direct compression is a common method consisting of the compression of a dry powder blend of active pharmaceutical ingredients (API) and excipients into a tablet. An appropriate choice of excipients and their optimal ratio can compensate for the poor compressible and tablet properties of API. An example of such a substance is ascorbic acid (AA) [1,2]. The ability of excipients to retain their tabletability, even after dilution with another ingredient, is called dilution potential [1,3].
This study aimed to find the dilution potential of six co-processed excipients (CPE)—Advantose®® FS95, Cellactose®® 80, CombiLac®®, MicroceLac®® 100, Pearlitol®® Flash, and StarLac®®. From each CPE, four batches were prepared containing 1% of sodium stearyl fumarate and 0, 25, 50 or 75% of AA. Tablets from each batch were compressed at five compaction forces (3, 4, 5, 6, 7 kN) and were evaluated on the pharmacopoeial requirement. The results of work potential (determined by Minchom and Armstrong), MA index and dilution capacity index (proposed by Habib et al.) were calculated by determination of area under the curve of tensile strength versus compression pressure profile [3,4].
The obtained values of dilution potential of all CPE were over 70 weights % of AA. However, none of the batches of tablets containing 75% AA met the required properties (friability and disintegration). The tablets which met these requirements were: Cellactose®® 80 with 25% AA compressed by forces 5–7 kN; CombiLac®® with 25% AA compressed by forces 4–7 kN, MicroceLac®® 100 with 25% AA compressed by forces 3–6 kN and MicroceLac®® 100 with 50% AA compressed by force 7 kN.
References:
- Gupta, P.; Nachaegari, S.K.; Bansal, A.K. Excipient Development for Pharmaceutical, Biotechnology and Drug Delivery Systems, 2006, 109–126.
- Jagtap, S.; Amrita, B.; Rina, M.; Jain, D. J. Adv. Sci. Res. 2012, 3(3), 15–24.
- Habib, Y.; Augsburger, L.; Reier, G.; Wheatley, T.; Shangraw, R. Pharm. Dev. Technol. 1996, 1(2), 205–212.
- Malkowska, S.; Khan, K.A. Drug Dev. Ind. Pharm. 1983, 9(3), 331–347.
50. Isatin Hydrazones-Biologically Active Compounds
Department of Pharmaceutical Chemistry, Faculty of Pharmacy, Comenius University, Odbojárov 10, 832 32, Bratislava, Slovak Republic
This research is focused on the synthesis, characterization, and study of the biological properties of isatin hydrazones as suitable candidates for new drugs. Isatin and its derivatives belong to the organic compounds used in chemical practice. They are used primarily in areas such as medicinal chemistry (e.g., as antibiotics, antimalarials, etc.), in the field of nanotechnology, development of analytical reagents and dyes, and in the field of synthesis of heterocyclic compounds and stereoselective procedures. The high application potential of isatin and its derivatives, their occurrence, and the occurrence of their metabolites in plants and the human body has aroused the interest of chemists, doctors, and pharmacists in the study of their chemical reactivity. [1] The aim of the work was the synthesis of new, not yet described in the literature, derivatives of 3-(phenylhydrazono)isatin with a methylmorpholine substituent in the N-1 position (Figure 1) [2].
Figure 1.
Reaction scheme for obtaining 3-(phenylhydrazono)isatin with a methylmorpholine substituent in the N-1 position.
The study was supported by FaF/9/2022
References:
- Nath, R.; Pathania, S.; Grover, G.; Akhtar, M. J. J. Mol. Struct. 2020, 1222, 128900.
- Polychronopoulos, P.; Magiatis, P.; Skaltsounis, A. L.; et al. J. Med. Chem. 2004, 47, 935–946.
51. Texturometric Analysis of Mucoadhesive Lyophilisates Containing Gentamicin
1
Comenius University, Faculty of Pharmacy, Department of Galenic Pharmacy, Odbojárov 10, SK-832 32 Bratislava, Slovakia,
2
hameln rds s.r.o., Horná 36, SK-900 01 Modra, Slovakia,
Texture profile analysis (TPA), texturometry, is used to determine the textural properties of food, packaging material, and pharmaceutical and cosmetic products. Structural and physical properties are quantitatively tested: cohesion, resistance, hardness, elasticity, brittleness, breakability, elasticity, adhesion, compressibility, expandability, elongation, spreadability, and chewability. The instrument intended for TPA, the texturometer, simulates the stress situations during the use or consumption of the tested sample. Testing takes place in one or more cycles through pressure and tension [1,2].
The pilot study was focused on the formulation of lyophilisates that could serve as mucoadhesive dosage forms for the treatment of aphthae, containing gentamicin as an antibacterial agent. Gentamicin is an aminoglycoside broad-spectrum antibiotic used in 0.5% w/w concentration [3]. The initial aim of the study was to determine the mechanical and texturometric properties of individual lyophilizates and to evaluate their potential applicability when applied to oral mucosa. Lyophilisates, prepared by freeze-drying of hydrogels based on chitosan (high molecular weight) and semi-synthetic cellulose derivatives (hydroxypropylmethylcellulose, hydroxyethylcellulose), were evaluated texturometrically. For the evaluation, a texturometer interfaced with Stable Micro Systems’ Exponent computer software was used.
Each type of lyophilizate was texturometrically measured and evaluated as having good mechanical strength. Not a single sample was disrupted due to mechanical stress during the measurement itself, and it kept its integrity and completeness. The best results in strength were observed with 3% chitosan lyophilizate.
References:
- Texture Analysis and Texture Profile Analysis. Rheology Lab [online]. [cit. 2022-04- 04]. Available online: https://www.rheologylab.com/services/texture-analysis/
- Texture Profile Analysis. Texture Technologies [online]. [cit. 2022-04-04]. Available online: https://texturetechnologies.com/resources/texture-profile-analysis#tpa-measurements
- Kuželová, M.; Kovácsová, B.; Švec, P.: Farmakológia antiinfekčných liečiv. Martin: Vydavateľstvo Osveta,spol. s. r. o., 2009. ISBN 978-80-8063-327-1.
52. Medicinal Mushrooms Ophiocordyceps Sinensis and Cordyceps Militaris
Department of Chemistry, Biochemistry and Biophysics, University of Veterinary Medicine and Pharmacy in Košice, Komenského 73 Košice, Slovak Republic,,
The main component of Ophiocordyceps sinensis and Cordyceps militaris extracts are polysaccharides. These natural biopolymers represent a large class of biologically active components and contribute to the total pharmacological activity and effect on health. They contain monosaccharides, such as rhamnose, ribose, arabinose, xylose, mannose, glucose, galactose, mannitol, fructose, or sorbose. The exopolysaccharide fraction has a large number of pharmacological effects; two of the most important of them are immunomodulatory and antitumor effects. Among the polysaccharides also belongs mannoglucan, which shows weak cytotoxic activity against the SPC-I cancer line [1]. More than ten nucleosides and their related compounds, including adenine, adenosine, inosine, cytidine, cytosine, guanine, uridine, thymidine, uracil, hypoxanthine and guanosine, have been successively isolated from Ophiocordyceps sinensis. It contains many amino acids and polypeptides, with the expected effect on the cardiovascular system. They also have a sedative and hypnotic effect, with tryptophan as the most effective component among them [2].
Polysaccharides were extracted from four samples: sample no. 1 (grown on Oryza sativa indica substrate, Ophiocordyceps sinensis strain), sample no. 2 (grown on Oryza sativa japonica substrate, Ophiocordyceps sinensis strain), sample no. 3 (grown on Oryza sativa indica substrate, Cordyceps militaris strain), sample no. 4 (grown on Oryza sativa japonica substrate, Cordyceps militaris strain). The stable 2,2-diphenyl-1-picrylhydrazyl radical has been used to determine the antioxidant activity. From the results for samples 1–4, we can see the highest antioxidant activity of sample no. 3 (IC50 = 2.85 ± 0.04 mg.mL−1). Through NMR spectroscopy and according to the literature, we found out that the majority of the chemical compounds in the deproteinized extracts was a hydrophilic polyglucan signed as CBHP.
References:
- Zhang, J.; Wen Ch.; Duan Y.; Zhang H.; Ma H. Int. J. Biol. Macromol. 2019, 132, 906–914.
- Liu, Y.; Wang J.; Zhang H.; Zhang X.; Han CH. eCAM. 2015.
53. Determination of Fireweed Organic Acid Composition
1
Zhytomyr Basic Pharmaceutical Professional College, Chudnivska str., 99, Zhytomyr, Ukraine, 10005
2
Lithuanian University of Health Sciences, Department of Analytical and Toxicological Chemistry, A. Mickevičiaus g. 9, LT 44307 Kaunas, Lithuania,
3
Department of Pharmaceutical Chemistry, National University of Pharmacy, 4-Valentinivska st., 61168, Kharkiv, Ukraine,
The study of organic acid content is necessary to determine the dynamics of metabolism of plants that are promising for the treatment of diseases associated with metabolic disorders, inflammatory diseases, and others. Chamaenerion angustifolium (L.) Scop., commonly known in North America and Europe as “fireweed” or “rosebay willow-herb”, is a very widespread and variable species of the willowherb family (Onagraceae) [1]. Fireweed is a traditional food and medicinal plant in Europe. The current work included the study of organic acids of aerial part and rhizomes of C. angustifolium occurred in Ukraine and Poland by gas chromatography-mass spectrometry (GC–MS) method using a Shimadzu GC-MS-QP2010 equipped with a Rxi-5ms (Restek Corporation capillary column (30 m × 0.25 mm, 0.25 µm) with a liquid stationary phase (5% diphenyl and 95% polysiloxane) after derivatization with MTBSTFA reagent. The method was discarded early [2]. The study results confirm the present 26 free carboxylic acids in raw plant material of C. angustifolium, among them 10 amino acids, 10 low-chain organic acids, three phenolic acids, one heterocyclic acid, and two fatty acids. The low-molecular-weight organic acids were represented as mono-, di- and tricarboxylic acids. The number of monocarboxylic acids prevailed among other groups in fireweed raw materials and were represented by lactic acid, glycolic acid, hydracrylic acid, propanoic acid, glyceric acid. Among dicarboxylic acids, oxalic acid, succinic acid, fumaric acid, and malonic acid amid contained in the aerial part and rhizomes of C. angustifolium. 3-Hydroxybenzoic acid, shikimic acid, and caffeic acid were identified in fireweed raw materials among phenolic compounds. Major organic acids in the highest concentrations in the aerial parts of samples were glyceric acid (0.74 ± 0.02–2.01 ± 0.05% of total organic acids amount) and citric acid (1.49 ± 0.87–3.87 ± 1.32% from total organic acids amount). The citric acid (3.19 ± 1.07–5.09 ± 1.33% of total organic acids amount) also dominated in the underground parts. An aerial part of C. angustifolium is the more promising raw material for the development of medicines and dietary supplements according to organic acids content.
References:
- Granica, S.; Piwowarski, J.P.; Czerwińska, M.E.; Kiss, A.K. J. Ethnopharmacol. 2014, 156, 316.
- Mykhailenko, O; Ivanauskas, L.; Bezruk, I.; Lesyk, R.; Georgiyants, V. Sci. Pharm. 2020, 88, 8.
54. Synthesis of New Derivatives with Aryloxyaminopropanol and Arylaminoethanol Fragment and Study of Their Potential Biological Activities
1
Department of Chemical Drugs, Faculty of Pharmacy, Masaryk University, Palackého tř. 1946/1, 612 00 Brno, Czech Republic,
2
Department of Chemical Drugs, University of Veterinary Medicine and Pharmacy in Košice, Komenského 73, 041 81 Košice, Slovak Republic
3
Department of Natural Drugs, Faculty of Pharmacy, Masaryk University, Palackého tř 1946/1, 612 00 Brno, Czech Republic
Aryloxyaminopropanols drugs are used as antiarrhythmics, antihypertensives, and beta-blockers and are also characterized by a lipolytic effect [1]. Drugs with antagonistic effects on alpha- and beta-receptors also include derivatives with an arylaminoethanol fragment [2,3]. The work deals with the synthesis of new aryloxyaminopropanol and arylaminoethanol derivatives with a carbamate functional group in the aromatic part of the molecule and a modified amino group, which is a part of piperidine. The wide possibility of substitution of the aromatic ring and substituents on the nitrogen can leads to the preparation of compounds with combined pharmacological properties. Aryloxyaminopropanol derivatives were prepared by a three-step synthesis from para- and meta-substituted aminophenol. Arylaminoethanol carbamates were prepared by a four-step synthesis from 1-(4-aminophenyl)ethan-1-one. Selected synthesized compounds were tested for anticholinesterase activity (inhibition of acetylcholinesterase and butyrylcholinesterase) and antimicrobial activity (Escherichia coli, Staphylococcus aureus). The tests showed that an aryloxyaminopropanol meta-derivative with an ethyl substituent on the carbamate functional group reduced acetylcholinesterase activity (28.1 ± 0.8%) and butyrylcholinesterase activity (24.5 ± 2.8%). Antimicrobial activity of Escherichia coli and Staphylococcus aureus strains showed that all meta-substituted carbamate derivatives suppressed the growth of Staphylococcus aureus bacteria over para-substituted aryloxyaminopropanol derivatives. The highest ability to inhibit the growth of bacteria of the Staphylococcus aureus strain was observed in the meta-derivative with a methyl substituent on the carbamate functional group (128.5% RIZD).
References:
- Mlynárová, R., Celková, H., Račanská, E. Ceska Slov. Farm. 1996, 45(5), 260–4.
- Gruetter, C. A. xPharm: The Comprehensive Pharmacology Reference 2007, 1–7.
- Franzosi, M. G., Latini, R. Side Eff. Drugs Annu. 2012, 34, 303–310.
55. Bioavailable Silver(I) and Zinc(II) Complexes and Their Biological Response
1
Faculty of Science, P. J. Šafárik University, Moyzesova 11, 041 54 Košice, Slovak Republic
2
Department of Pharmacology, Faculty of Medicine, P. J. Šafárik University, Trieda SNP 1, 040 11 Košice, Slovak Republic
3
Department of Inorganic Chemistry, Faculty of Science, Charles University, Hlavova 2030, 128 00 Prague, Czech Republic
4
Department of Biochemistry and Microbiology, Slovak University of Technology, Radlinského 9, 812 37 Bratislava, Slovak Republic
5
Department of Microbiology and Immunology, University of Veterinary Medicine and Pharmacy, Komenského 73, 041 81 Košice, Slovak Republic
Pathogenic bacteria and viruses have endangered people every day. In conjunction with growing knowledge in the field of biology, medicine and pharmacy, numerous drugs have been developed that are capable of inhibiting the growth of these microorganisms. However, since drug resistance is a ubiquitous phenomenon, new antimicrobials are still developed continuously. In addition to current antimicrobials that are isolated either from various strains of microorganisms or synthetically (semi-synthetically) prepared, metal ion-based antiseptics have also recently emerged as promising alternatives.
It is known that compounds based on Ag(I), Zn(II) and Cu(II) are the ones antimicrobially most active, but there are currently also tests with Ga(III), In(III), Ru(II) and others samples [1]. However, finding a suitable form of their application to organisms is a difficult process. One of the ideas is to combine biologically active metal ions with effective organic ligands, such as N-heterocyclic carbenes, phosphines, peptides and carboxylic acids have so far been most frequently used [2].
The contribution will present the antimicrobial and anticancer effects of selected silver(I) and zinc(II) complexes prepared in our laboratory with a focus on the relationship between their structure and biological activity [3,4].
The study was supported by the Slovak grant agency KEGA 006UPJŠ-4/2021.
References:
- Claudel, M.; Schwarte, J. V.; Fromm, K. M; Chemistry 2020, 2, 849–899.
- Medici, S; Peana, M.; Crisponi, G.; et al. Coord. Chem. Rev. 2016, 327–328, 349–359.
- Kuzderová, G.; Rendošová, M.; Gyepes, R.; et al. J. Inorg. Biochem. 2020, 210, 111170.
- Kuzderová, G.; Rendošová, M.; Gyepes, R.; et al. Molecules 2021, 26, 6335.
56. Benzyl Amides of 4-Methylthienopyrimidine as Novel Antibacterials
1
Department of Pharmaceutical Chemistry, National University of Pharmacy, Pushkinska St. 53, 61002, Kharkiv, Ukraine,,,,
2
Enamine Ltd., Chervonotkatska St. 78, 02094 Kyiv, Ukraine
Recent studies show that thieno[2,3-d]pyrimidine core modified with amide functional group at 4, 5 or 6 of the core heterocyclic structure is a promising structure for antibacterial. Some recent studies confirmed their ability to be inhibitors of bacterial TrmD and, as a result, make them interesting compounds for antibacterial activity screening [1].
In the present research, we focused our efforts on the introduction of a methyl group at position four of the thieno[2,3-d]pyrimidine and carboxamide group at position 6. We used Suzuki coupling with the corresponding chloride 1 to replace chlorine with a 4-methyl substituent (2). Further, core acid 3 was obtained by hydrolysis of an ester 2. Acid 3 showed good reactivity in peptide coupling regents’ promoted reactions with amines (4) (Figure 1).
Figure 1.
Synthesis of thieno[2,3-d]pyrimidine core modified with amide functional group.
The study of antibacterial activity for the synthesized amides 4 revealed high antimicrobial activity for the benzyl amides, which shoved growth inhibition of Pseudomonas aeruginosa АТСС 27853 strain.
The study was supported by the Ministry of Health Care of Ukraine at the expense of the State Budget in framework #2301020, “Scientific and scientific-technical activity in the field of health protection” on the topic “Synthesis and study of new thienopyrimidines for the detection of antimicrobial and related types of pharmacological activity” (State registration number: 0121U109472. Order of the Ministry of Health of Ukraine of 17 November 2020 № 2651). The authors acknowledge Enamine Ltd. for the measurement of 1H, 13C NMR and LC-MS spectra of the obtained substances. We are grateful to T.P. Osolodchenko (Mechnikov Institute of Microbiology and Immunology of the NAMS of Ukraine, Kharkiv, Ukraine) for her assistance in conducting the antimicrobial activity study.
References:
- Zhong, W.; Pasunooti, K. K.; Balamkundu, S.; et al. J. Med. Chem., 2019, 62(17), 7788.
57. Study of Surfactant Partitioning into Mammalian and Bacterial Model Membranes
Department of Physical Chemistry of Drugs, Faculty of Pharmacy, Comenius University Bratislava, Odbojárov 10, 832 32 Bratislava, Slovak Republic
Due to the alarming increase in the number of cases of antibiotic-resistant bacterial infections, it is necessary to study the mechanism of antimicrobial action. Solubilization of bacterial membranes is the mechanism of the antimicrobial effect of surfactants. The interaction of surfactant N,N-dimethyl-1-dodecanamine-N-oxide (DDAO) with mammalian and bacterial model membranes was studied using static light scattering and fluorescence spectroscopy. Unilamellar liposomes (ULLs), consisting of palmitoyloleoylphosphatidylcholine (POPC) with cholesterol (CHOL), were used to mimic the lipid part of the mammalian cytoplasmic membrane. A mixture of palmitoyloleoylphosphatidyl-ethanolamine (POPE) and palmitoyloleoylphosphatidylglycerol (POPG) was used to mimic the inner membrane of Escherichia coli. The solubilization is a complex process of liposome-mixed micelle transformation. The process is accompanied by decreasing particle sizes, which were nephelometrically observed on ULLs. Critical DDAO concentrations causing saturation of the bilayers (DtSAT), complete solubilization (DtSOL), as well as DDAO concentration causing a 50% decrease in the scattering intensity (DtMID), were calculated. The first phase of the solubilization process (membrane perturbation) was observed using fluorescence spectroscopy. Leakage of the fluorescent probe calcein, which was encapsulated inside the ULLs, through the pores in the bilayer created by DDAO, was detected by the increasing values of fluorescence intensity. Critical DDAO concentrations causing heavy (DtPERT) and full (DtRLS) release of the probe were calculated. All experiments were performed at various lipid concentrations. Using the dependences of all critical DDAO concentrations on the lipid concentration, we have calculated the partition coefficients of DDAO for both types of model membranes, as well as the effective ratios of the amount of DDAO integrated into the bilayer to the amount of lipid. Our data show that the partition coefficient of DDAO is higher when interacting with bacterial model membranes and the bacterial model membranes need a more incorporated surfactant to undergo the solubilization.
The study was supported by the VEGA grants 1/0223/20 (prof. RNDr. Daniela Uhríková, CSc.), 1/0228/17, APVV project 17-0239 (prof. Ing. Vladimír Frecer, DrSc.) and JINR topical themes 04-4-1142-2021/2025.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).