
Primary Cutaneous Gamma-Delta T-Cell Lymphoma Initially Diagnosed as Subcutaneous Panniculitis-like T-Cell Lymphoma with Dermatomyositis

 ,
, 
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
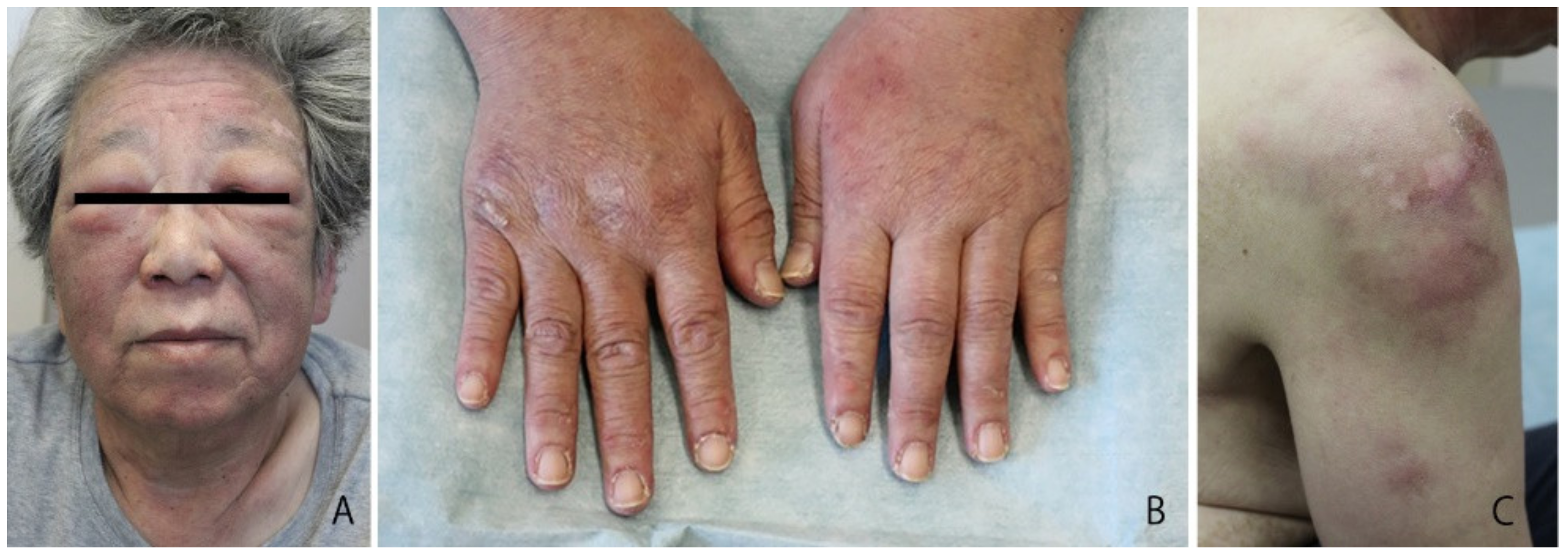
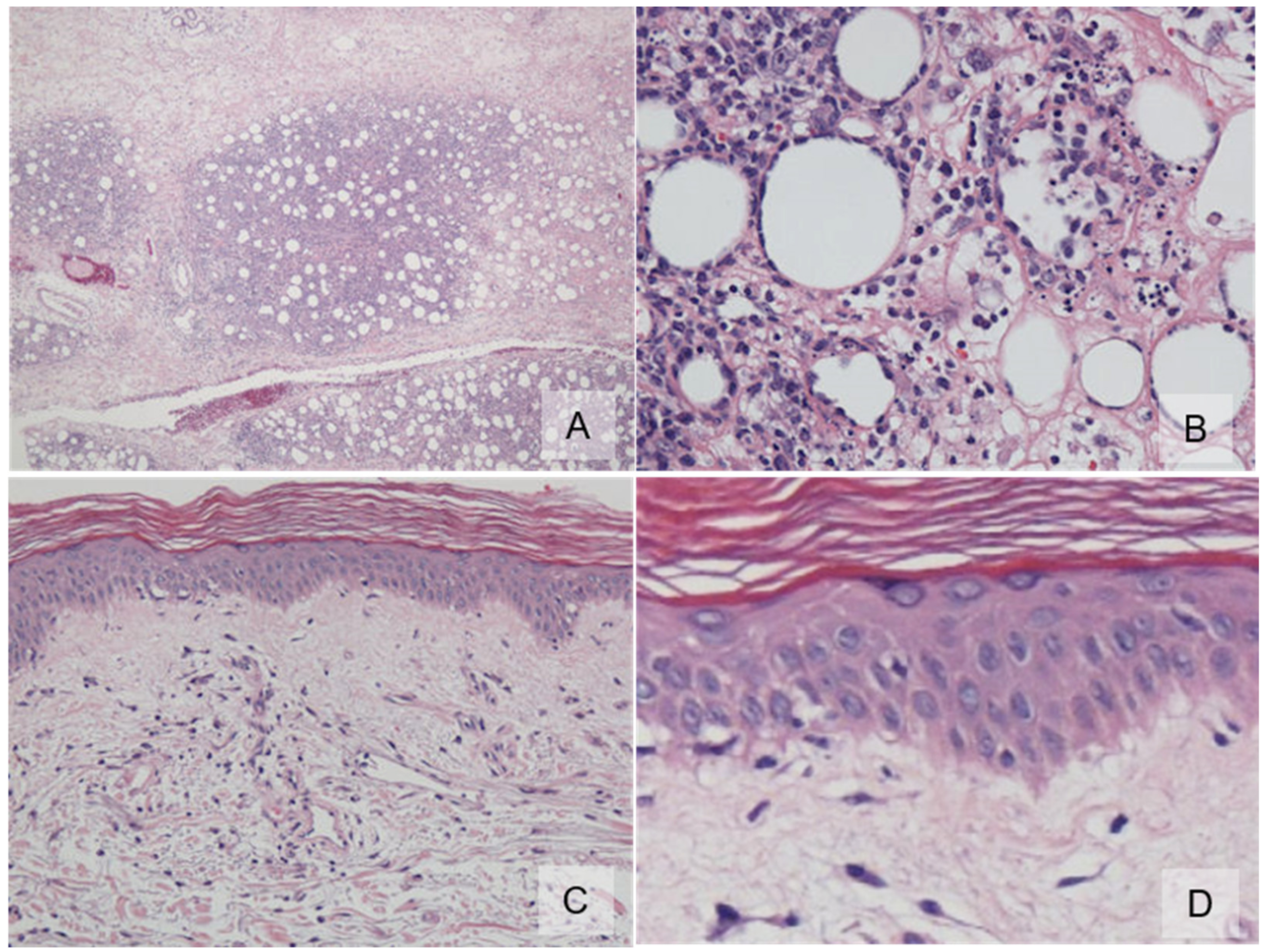
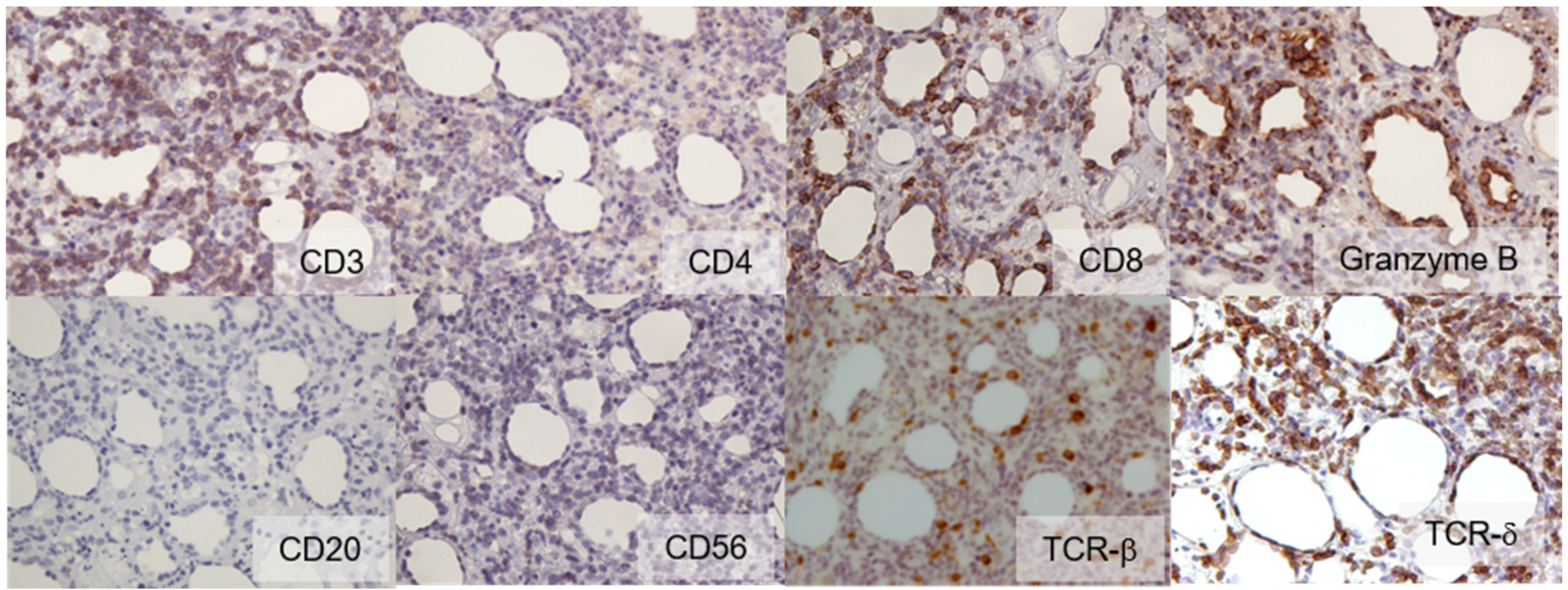
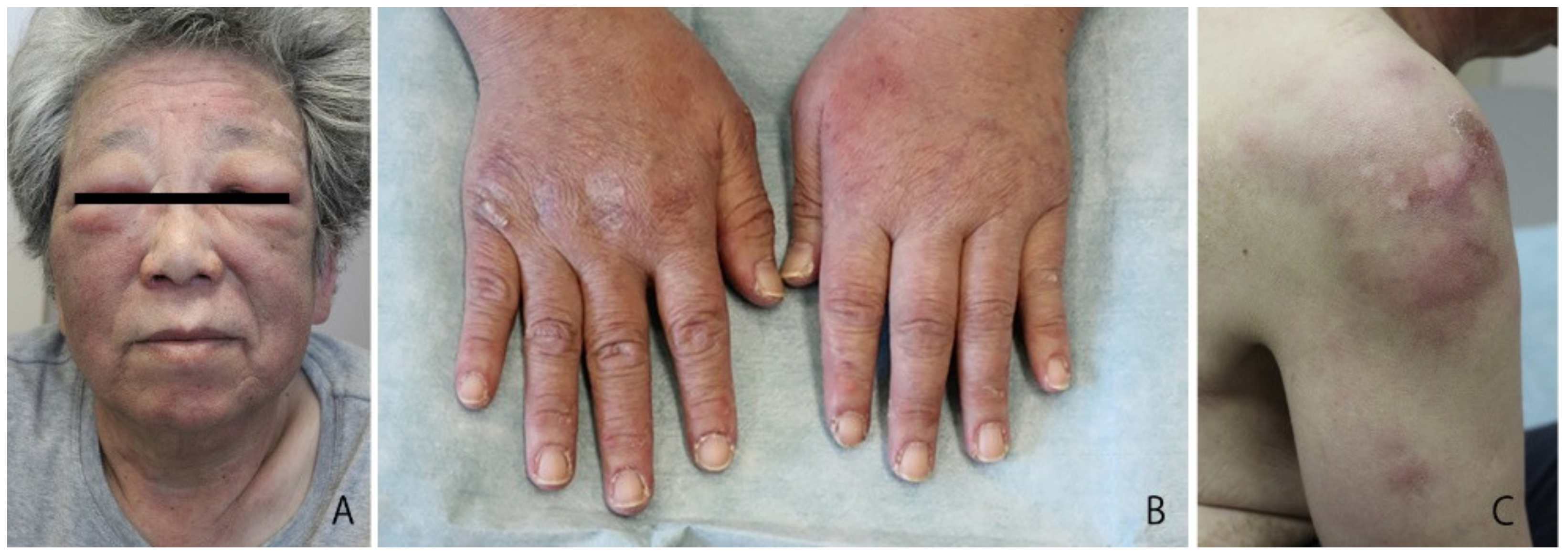
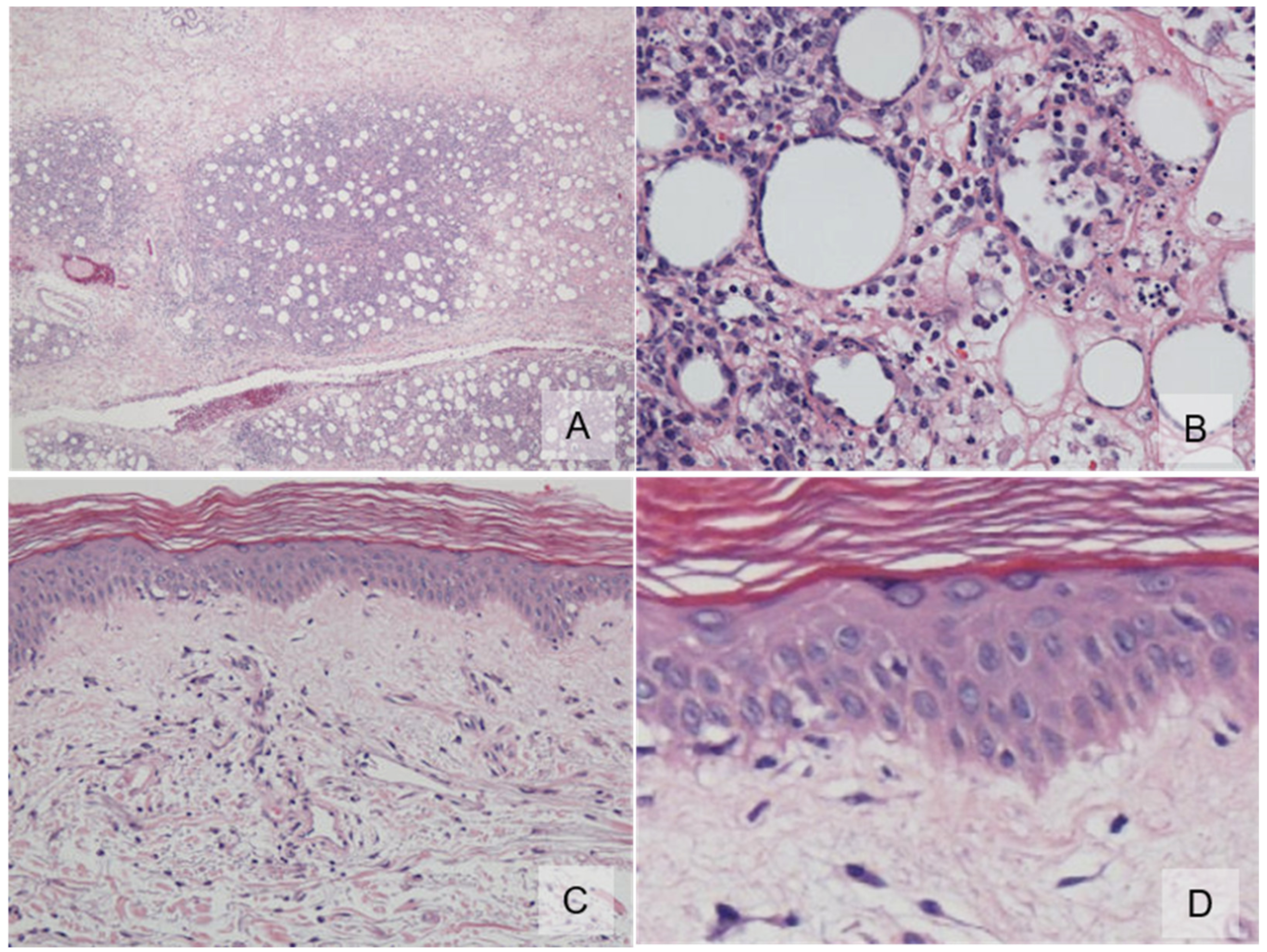
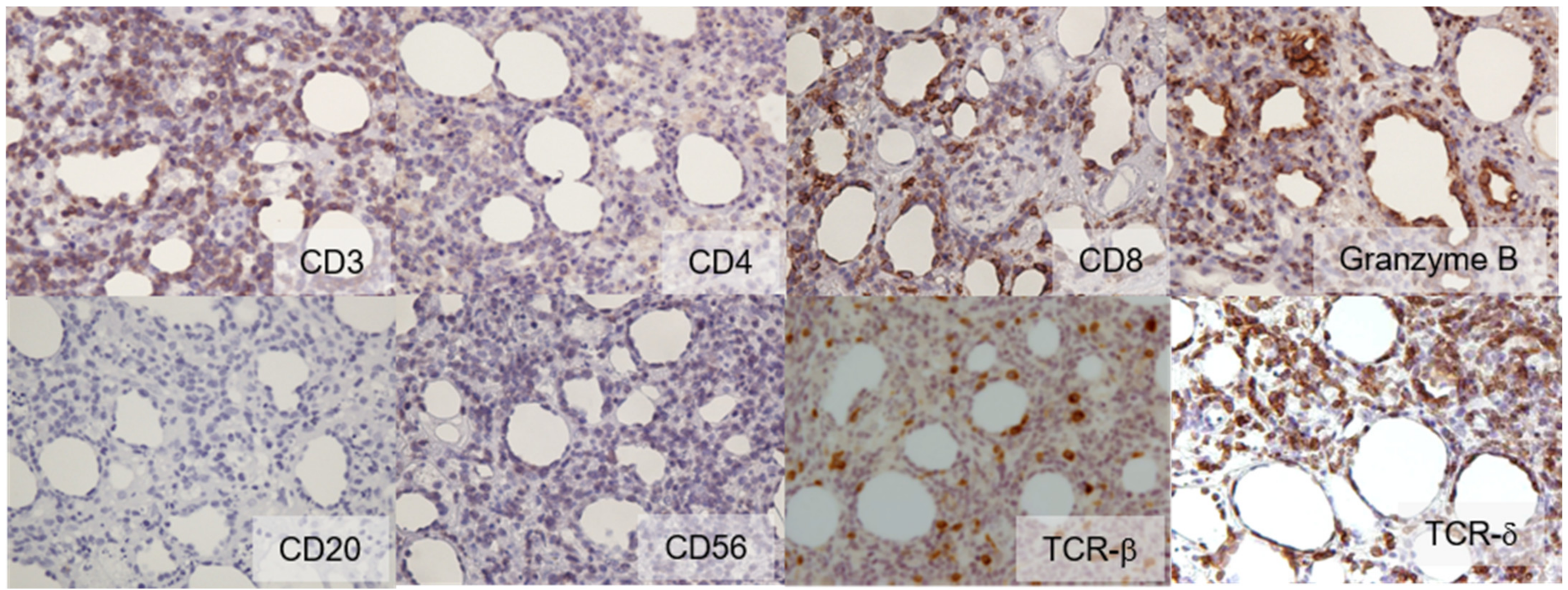
2. Case Report
3. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Berti, E.; Willemze, R.; Guitart, J.; Jaffe, E.S.; Kempf, W.; Petrella, T.; Robson, A.; Cerroni, L.; Torres-Cabala, C.A. Primary cutaneous periferal T-cell lymphomas, rare subtypes. In WHO Classification of Skin Tumours, 4th ed.; Elder, D.E., Massi, D., Scolyer, R.A., Eds.; IARC Press: Lyon, France, 2018; pp. 248–253. [Google Scholar]
- Willemze, R.; Jaffe, E.S.; Burg, G.; Cerroni, L.; Berti, E.; Swerdlow, S.H.; Ralfkiaer, E.; Chimenti, S.; Diaz-Perez, J.L.; Duncan, L.M.; et al. WHO-EORTC classification for cutaneous lymphomas. Blood 2005, 105, 3768–3785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Willemze, R.; Jansen, P.M.; Cerroni, L.; Berti, E.; Santucci, M.; Assaf, C.; Dijk, M.R.C.-V.; Carlotti, A.; Geerts, M.-L.; Hahtola, S.; et al. Subcutaneous panniculitis-like T-cell lymphoma; definition, classification, and prognostic factors: An EORTC Cutaneous Lymphoma Group Study of 83 cases. Blood 2008, 111, 838–845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taghon, T.; Rothenberg, E.V. Molecular mechanisms that control mouse and human TCR-alphabeta and TCR-gammadelta T cell development. Semin. Immunopathol. 2008, 30, 383–398. [Google Scholar] [CrossRef] [PubMed]
- Jaffe, E.S.; Willemze, R.; Cerroni, L.; Guiart, J. Subcutaneous panniculitis-like T-cell lymphoma. In WHO Classification of Skin Tumours, 4th ed.; Elder, D.E., Massi, D., Scolyer, R.A., Eds.; IARC Press: Lyon, France, 2018; pp. 242–243. [Google Scholar]
- Magro, C.M.; Wang, X. Indolent primary cutaneous gamma/delta T-cell lymphoma localized to the subcutaneous panniculus and its association with atypical lymphocytic lobular panniculitis. Am. J. Clin. Pathol. 2012, 138, 50–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Endly, D.C.; Weenig, R.H.; Peters, M.S.; Viswanatha, D.S.; Comfere, N.I. Indolent course of cutaneous gamma-delta T-cell lymphoma. J. Cutan. Pathol. 2013, 40, 896–902. [Google Scholar] [CrossRef] [PubMed]
- Alexander, R.E.; Webb, A.R.; Abuel-Haija, M.; Czader, M. Rapid progression of primary cutaneous gamma-delta T-cell lymphoma with an initial indolent clinical presentation. Am. J. Dermatopathol. 2014, 36, 839–842. [Google Scholar] [CrossRef] [PubMed]
- Von Dücker, L.; Fleischer, M.; Stutz, N.; Thieme, M.; Witte, M.; Zillikens, D.; Sadik, C.D.; Terheyden, P. Primary Cutaneous Gamma-Delta T-Cell Lymphoma With Long-Term Indolent Clinical Course Initially Mimicking Lupus Erythematosus Profundus. Front. Oncol. 2020, 10, 133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yi, L.; Qun, S.; Wenjie, Z.; Wen, Z.; Jian, L.; Yan, Z.; Fengchun, Z. The presenting manifestations of subcutaneous panniculitis-like T-cell lymphoma and T-cell lymphoma and cutaneous γδ T-cell lymphoma may mimic those of rheumatic diseases: A report of 11 cases. Clin. Rheumatol. 2013, 32, 1169–1175. [Google Scholar] [CrossRef] [PubMed]
- Tudorancea, A.D.; Ciurea, P.L.; Vreju, A.F.; Turcu-Stiolica, A.; Gofita, C.E.; Criveanu, C.; Musetescu, A.E.; Dinescu, S.C. A Study on Dermatomyositis and the Relation to Malignancy. Curr. Health Sci. J. 2021, 47, 377–382. [Google Scholar] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hirata, C.; Nakai, K.; Kurasawa, Y.; Maekawa, N.; Kuniyuki, S.; Yamagami, K.; Ohsawa, M.; Tsuruta, D. Primary Cutaneous Gamma-Delta T-Cell Lymphoma Initially Diagnosed as Subcutaneous Panniculitis-like T-Cell Lymphoma with Dermatomyositis. Dermatopathology 2022, 9, 143-147. https://doi.org/10.3390/dermatopathology9020018
Hirata C, Nakai K, Kurasawa Y, Maekawa N, Kuniyuki S, Yamagami K, Ohsawa M, Tsuruta D. Primary Cutaneous Gamma-Delta T-Cell Lymphoma Initially Diagnosed as Subcutaneous Panniculitis-like T-Cell Lymphoma with Dermatomyositis. Dermatopathology. 2022; 9(2):143-147. https://doi.org/10.3390/dermatopathology9020018
Chicago/Turabian StyleHirata, Chika, Kozo Nakai, Yusuke Kurasawa, Naoki Maekawa, Shuichi Kuniyuki, Keiko Yamagami, Masahiko Ohsawa, and Daisuke Tsuruta. 2022. "Primary Cutaneous Gamma-Delta T-Cell Lymphoma Initially Diagnosed as Subcutaneous Panniculitis-like T-Cell Lymphoma with Dermatomyositis" Dermatopathology 9, no. 2: 143-147. https://doi.org/10.3390/dermatopathology9020018
APA StyleHirata, C., Nakai, K., Kurasawa, Y., Maekawa, N., Kuniyuki, S., Yamagami, K., Ohsawa, M., & Tsuruta, D. (2022). Primary Cutaneous Gamma-Delta T-Cell Lymphoma Initially Diagnosed as Subcutaneous Panniculitis-like T-Cell Lymphoma with Dermatomyositis. Dermatopathology, 9(2), 143-147. https://doi.org/10.3390/dermatopathology9020018