1. Introduction
Despite policymakers’ efforts to regulate emissions of greenhouse gases, the concentration of carbon dioxide in in the atmosphere continues to rise. In 2014, the International Panel on Climate Change (IPCC) recommended that efforts to reduce carbon emissions be coupled with carbon removal from the atmosphere [
1]. Similarly, the 2015 Climate Summit in Paris concluded that nations should strive for a balance between anthropogenic emissions by sources and removals by sinks of greenhouse gases [
2].
Carbon emissions from the transportation, industrial and residential sectors can be substantially addressed through energy conservation and efficiency measures and switching to renewable energy. Carbon emissions from agriculture can be addressed through adoption of practices that reduce emissions of methane (CH
4), nitrous oxide (N
2O) and carbon dioxide (CO
2) [
3]. Technologies for carbon reduction are well established and commercially available but their implementation has been slowed by unfavorable economics compared to the status quo.
In contrast, carbon removal technologies are in their infancy and face even more daunting economic challenges than carbon reduction [
4]. If carbon dioxide at a concentration of 410 ppm is to be removed from the atmosphere (referred to as direct carbon removal or DCR), the energy requirement is substantial. To minimize the energy required to remove CO
2 from air, Lackner proposed to “only skim the [air] stream for the highest available concentration of CO
2” [
5].
Carbon removal can be generally categorized as biological or engineered approaches [
4,
6]. An appealing example of the former is shellfish aquaculture, which sequesters 0.22–0.44 tons of CO
2 as calcium carbonate in shells per ton of harvested shell fish [
7]. Global mollusk harvest in 2014 was 16.1 million tons, thus sequestering 1–2 million tons of carbon annually. A prominent example of an engineering approach are the use of giant fans to move air past streams of aqueous sorbents to capture and concentrate CO
2 for geological storage [
8].
The shellfish example has two attractive features. First, it sequesters carbon as calcium carbonate, a solid that can be easily and safely stored for millions of years (as the White Cliffs of Dover in England testify). Second, seafood is a coproduct of the process, making it a potentially profitable enterprise even in the face of relatively modest prices for the sequestered carbon. In contrast, the only product from direct carbon capture powered by wind or solar energy is a concentrated stream of gaseous CO
2, which is more difficult to securely and safely store than a solid carbonaceous product. Furthermore, the full burden of the enterprise’s profitability is the price society is willing to pay for sequestered carbon. The cost of this DCR process is highly uncertain with estimates ranging from
$200–
$600 per metric ton carbon dioxide (
$/MT CO
2) [
9,
10,
11] while markets for carbon abatement are well less than
$100/MT CO
2.
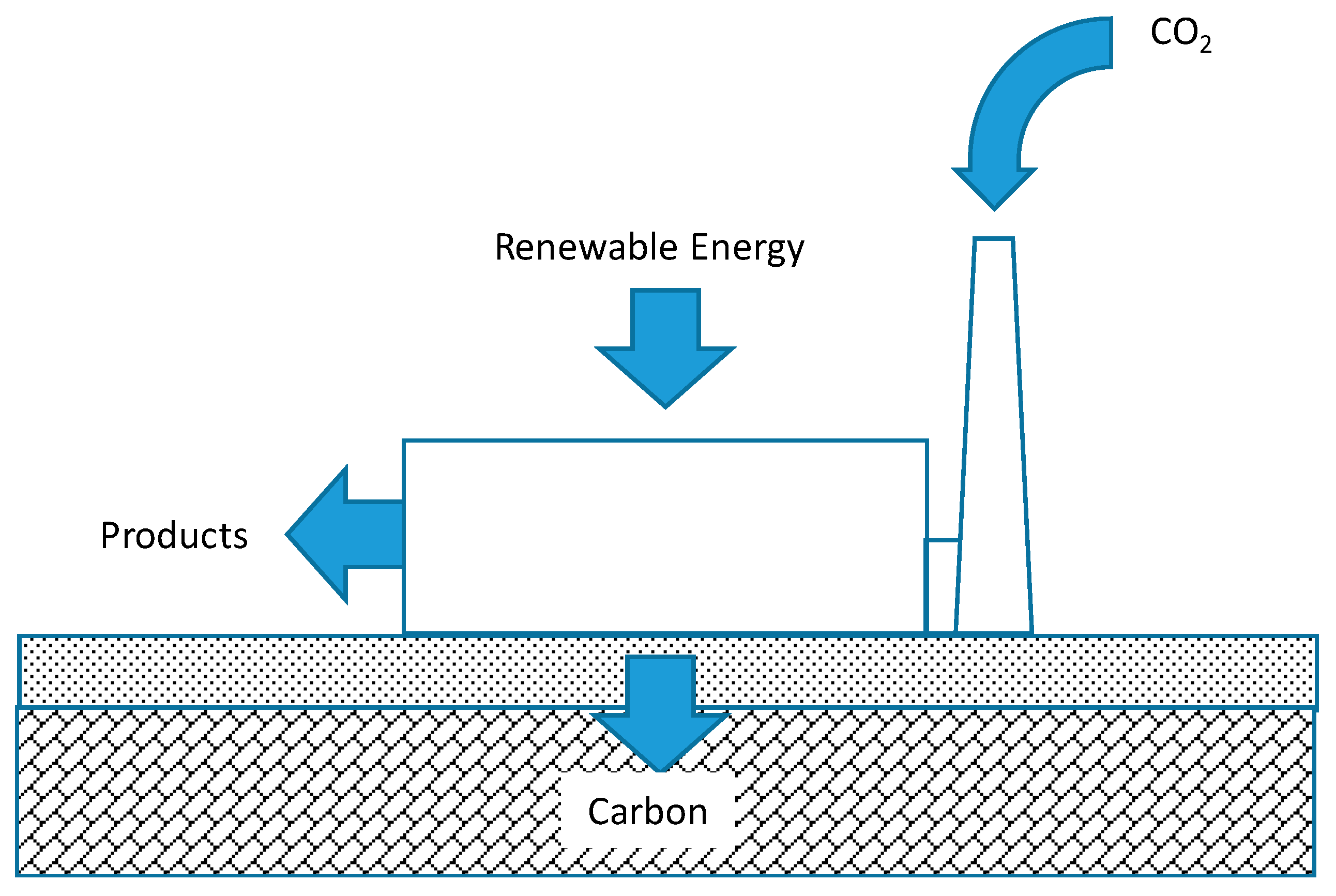
The contrast between these two examples illustrates the attractiveness of carbon negative energy, a system that combines net carbon removal with the production of energy products or other revenue-generating products beyond sequestered carbon, as illustrated in
Figure 1. The prospect of not only reducing but reversing the flow of carbon between the lithosphere and the atmosphere might appear to violate the Second Law of Thermodynamics. However, by powering the process with solar or other forms of renewable energy the process is perfectly feasible. If widely adopted by society, carbon negative energy would result in a carbon negative economy in which economic activity removed rather than added CO
2 to the atmosphere. It should be noted that natural ecosystems are frequently carbon negative energy systems such as marshlands building soil carbon and coral reefs building limestone formations.
Notably, natural ecosystems store carbon as relatively stable solids with the prominent exceptions of methane clathrates and carbon dioxide hydrates, which are stable only at the low temperatures and high pressures found in deep ocean trenches or artic environments [
12]. Ideally, human-constructed carbon negative energy systems would sequester carbon as recalcitrant solids. However, many proposed systems would sequester carbon as gaseous CO
2 in depleted gas and oil wells, saline aquifers or deep ocean water, as subsequently discussed.
Both biochemical and thermochemical processes could be combined with carbon removal to achieve carbon negative energy. A prominent example of the former is the geological sequestration of the almost pure co-product stream of CO
2 from grain ethanol plants [
13]. However, the focus of this paper is thermochemical carbon negative energy systems based on either pyrolysis or gasification of biomass. The fundamentals of these processes are briefly outlined, their strengths and weaknesses in converting biomass into energy and chemical products described and the prospects for integrating them with carbon removal reviewed.
2. Thermochemical Processes for Carbon Negative Energy
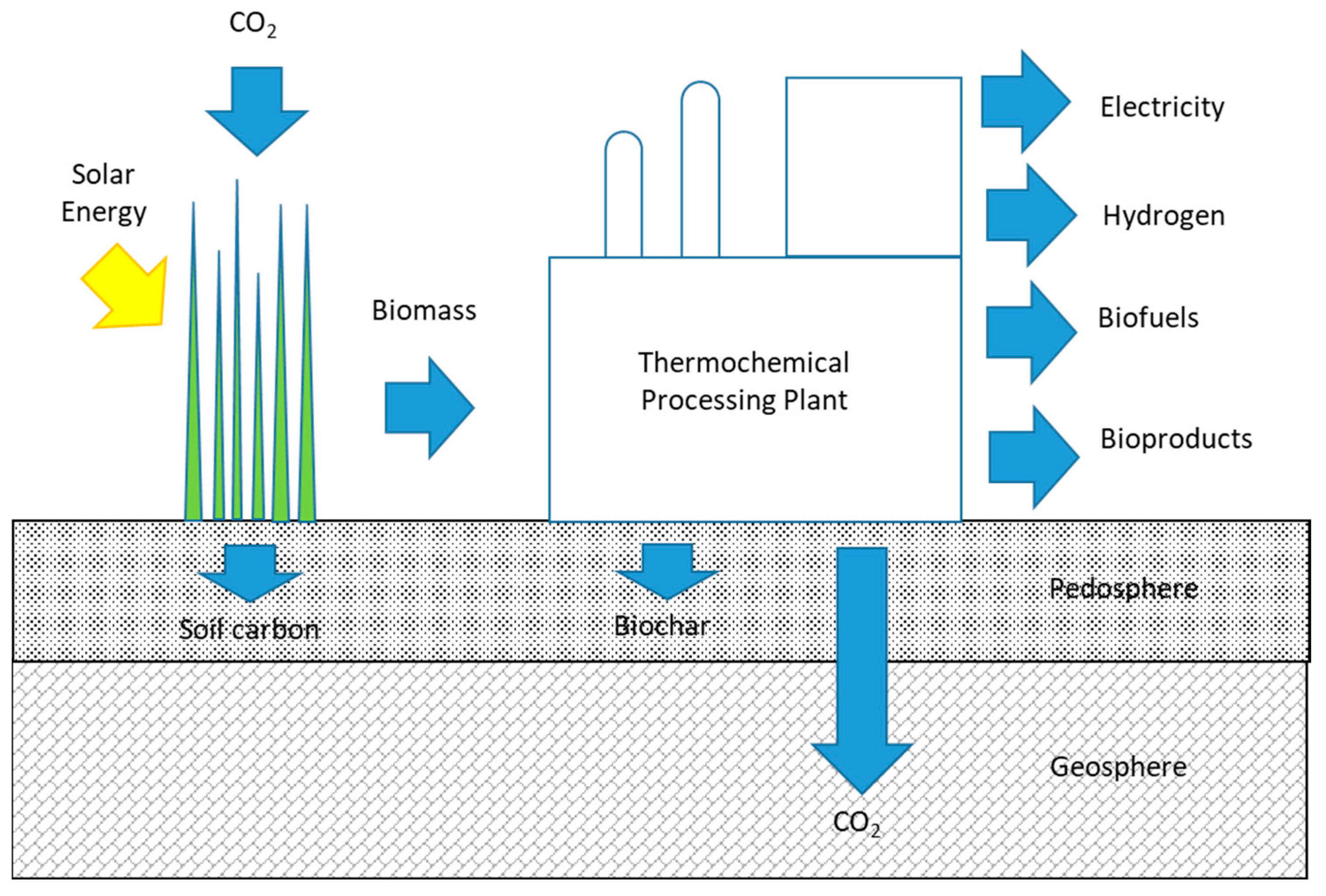
In principle, purely thermochemical processes can be envisioned that employ solar or wind power to remove carbon dioxide from the atmosphere and transform it into both carbon sequestration agent and marketable products. In practice, most thermochemical processes are energetically driven by the chemical energy of their feedstocks. Thus, thermochemical processes for carbon negative energy typically exploit natural photosynthesis in plants to fix carbon from the atmosphere as energy-rich chemical bonds in biomass. This biomass can then be thermochemically processed into power, fuels, commodity chemicals or materials. Part or even all of the carbon in the feedstock is extracted and stored in the biosphere as char (commonly referred to as biochar to emphasize its value in providing ecosystem services) or in deep oceans or geological formations as gaseous or dissolved CO
2 (see
Figure 2). In this way, thermochemical processing is able to couple bioenergy production with carbon removal to achieve carbon negative energy.
Of course, the availability of biomass is just as much a question for carbon negative energy as it is for biofuels. The U.S. Department of Energy (DOE) released a comprehensive report on this subject in 2005 that estimated 1.4 billion tons of biomass would be annually available in the U.S. by 2030 from a combination of forest and agricultural biomass and wastes and dedicated energy crops [
14]. A 2011 update on this report concluded that up to 1.6 billion tons of biomass were potentially available despite reducing the contribution of agricultural residues to mitigate soil erosion from excessive removal of residues [
15]. This was followed by a similarly comprehensive report by the U.S. DOE in 2016 that estimated 1.2–1.5 billion tons would be available at less than
$60/ton by 2040 [
16]. Assuming 47% of this mass is carbon, then thermochemical processing of biomass could remove 200–2750 million tons of CO
2 from the atmosphere annually, depending on the extent that feedstock supply develops and the thermochemical technology employed.
The three major thermochemical technologies considered in this review are slow pyrolysis, fast pyrolysis and gasification. Solvent liquefaction in both organic liquids and water (the latter referred to as hydrothermal processing) is a unique category of thermochemical processing [
17] that is beyond the scope of this review although it also has potential for carbon negative energy.
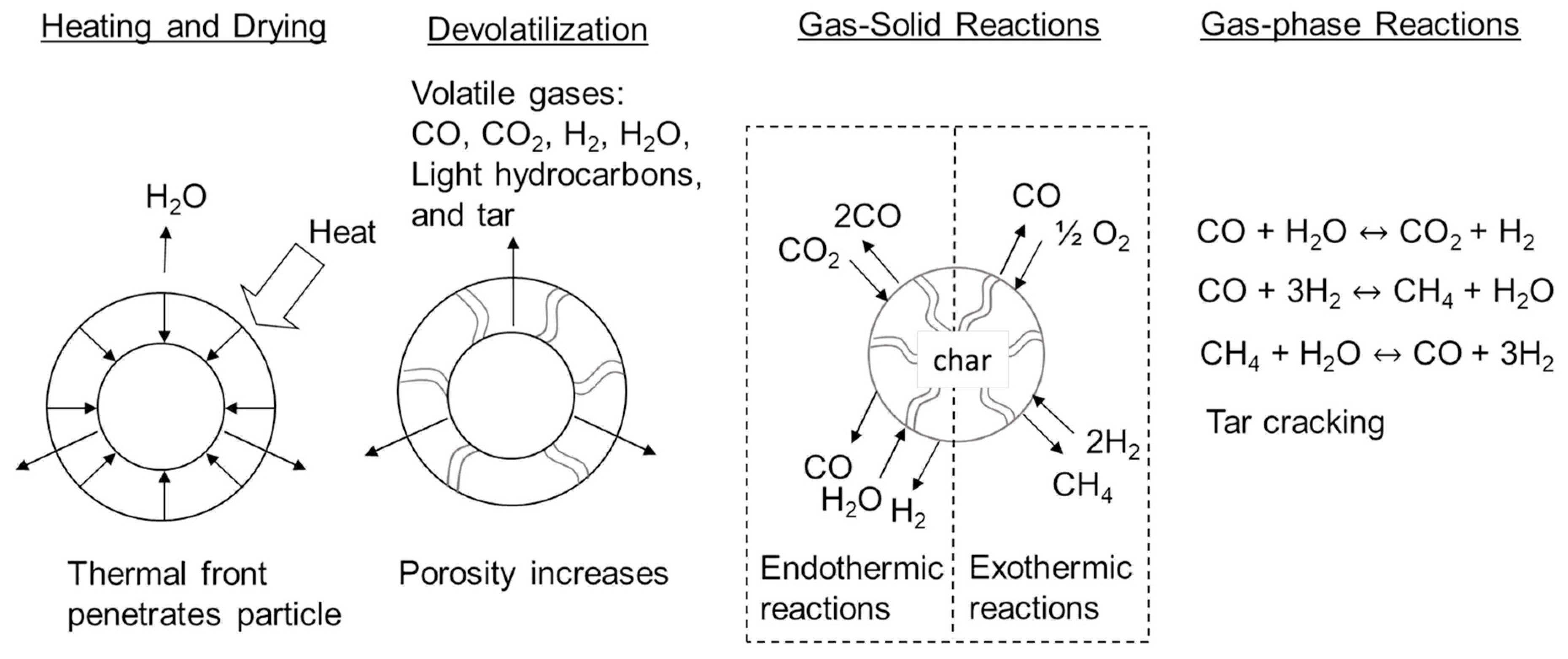
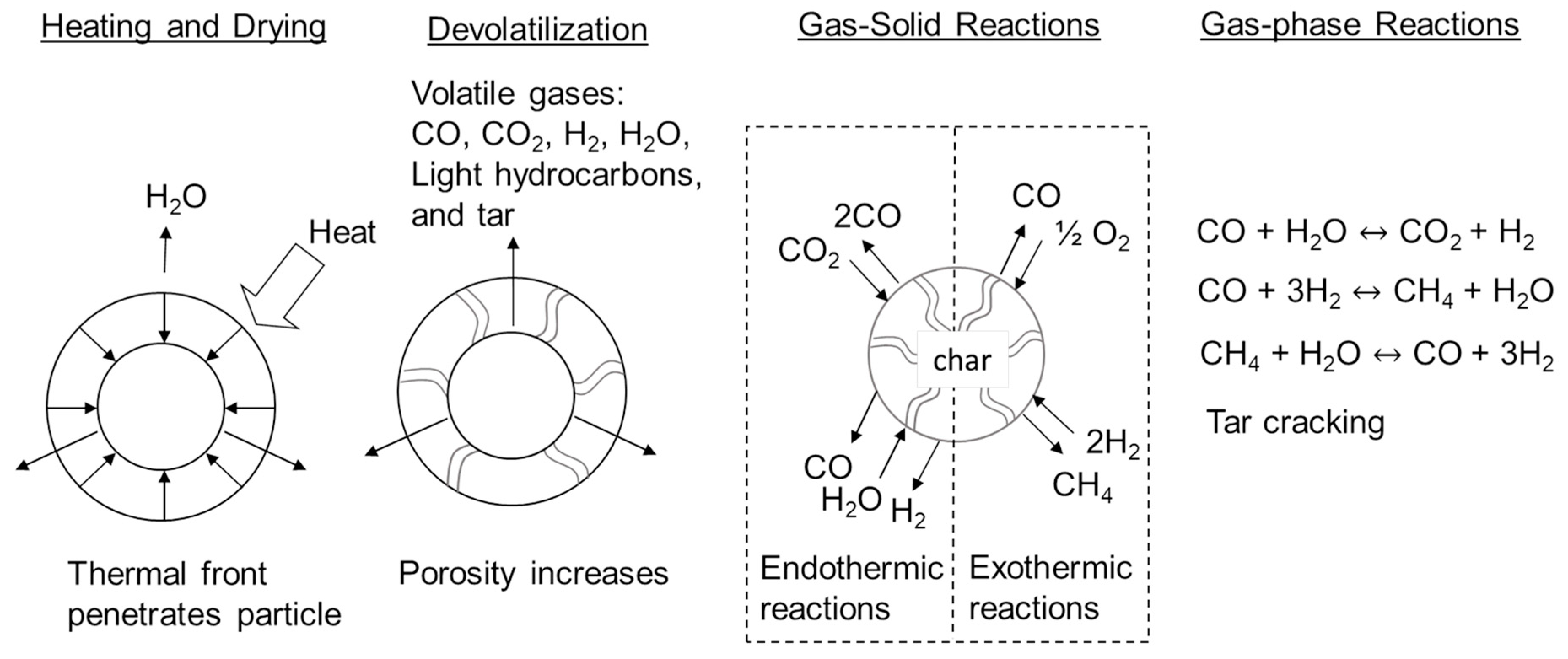
Figure 3 illustrates the fundamental processes responsible for slow pyrolysis, fast pyrolysis and gasification [
18]. All three processes begin by heating a particle of biomass or other solid organic material. A thermal front penetrates the particle, raising its temperature high enough to first drive off moisture after which the temperature continues to rise to the reactor temperature (300–600 °C for pyrolysis and 800–1500 °C for gasification). Light gases and vapors, including carbon monoxide CO, CO
2, produced water, hydrogen (H
2), light hydrocarbons and heavy organic compounds (known as tar) are released from the solid at temperatures as low as 250 °C and continuing up to 500 °C. Most commonly this process is endothermic although it can be exothermic under certain conditions as subsequently described.
The combination of heating, drying and devolatilization at modest temperatures (<600 °C) in the absence of oxygen is characterized as pyrolysis. Slow pyrolysis occurs when the heating of the particle is relatively slow and residence times of released vapors is relatively long, both of which encourage formation of char vs. liquid product and likely approaches chemical equilibrium in the distribution of products. Fast pyrolysis is a non-equilibrium process that strives for rapid particle heating and short vapor residence times for the purpose of maximizing liquid products at the expense of char. As temperature increases, chemical equilibrium favors the conversion of both char and condensable vapors (tar) into CO and H
2 [
19]. This increasing prevalence of permanent gases among products with increasing temperature, the result of the gas-solid and gas-phase reactions illustrated in
Figure 3, represents a transition to gasification.
Even though neither pyrolysis nor gasification requires oxygen to proceed, both require a source of energy to drive the predominately endothermic reactions of these processes [
20]. Occasionally for gasification and traditionally for pyrolysis, this energy is provided through direct or indirect heat exchange between the reactor and a furnace, the latter often fired with char produced from the pyrolyzer or gasifier. Most gasifiers, though, admit oxygen (O
2) or air to the reactor at equivalence ratios (defined as the actual oxygen/fuel ratio divided by the stoichiometric oxygen/fuel ratio) of around 0.20–0.25 to partially oxidize products of gasification and thus provide the energy demand for the process, achieving what is known as “autothermal operation” [
19]. In this scheme the gas product is diluted with the products of this oxidation (CO
2 and additionally nitrogen gas in the case of air-blown gasification). The resulting simplifications in reactor design and operation often justifies this autothermal approach to gasification. Recent research has shown that fast pyrolyzers also can be operated autothermally by admitting air at equivalence ratios of 0.06–0.12, accompanied by dramatic intensification of pyrolysis while suffering only minor losses in oil yield [
21].
If O
2 is present in the reactor, it is generally not able to diffuse to the surface of the particle until devolatilization is complete, which may be a few seconds for fast pyrolysis or several minutes or even longer for slow pyrolysis [
18]. Under conditions of high temperature and oxygen equivalence ratios, volatiles rapidly oxidize in a spherical shell immediately surrounding the particle, which is characterized as flaming combustion. At lower temperatures and equivalence ratios, as occurs in autothermal pyrolyzers and gasifiers, the volatiles burn more diffusely in the gaseous volume of the reactor. Once the particle has devolatilized to a porous carbonaceous solid (char), O
2 can diffuse to its surface or even into the particle’s pores and react to form CO. Other gas-solid reactions upon completion of devolatilization include reaction of biochar with CO, H
2 and water to form CO
2, methane (CH
4) and CO and H
2, respectively.
Some of the major characteristics that distinguish slow pyrolysis, fast pyrolysis and gasification are summarized in
Table 1. These are described in more detail in the sections that follow.
3. Pyrolysis as a Pathway to Carbon Negative Energy
Pyrolysis is traditionally defined as the thermal deconstruction of solid organic materials in the absence of oxygen [
22] although it may also occur in the presence of oxygen under conditions of low equivalence ratios [
21]. Slow pyrolysis, operating at temperatures comparable to fast pyrolysis, is distinguished by much lower heating rates [
23] resulting in char and gas rather than liquid being the primary products. As illustrated in
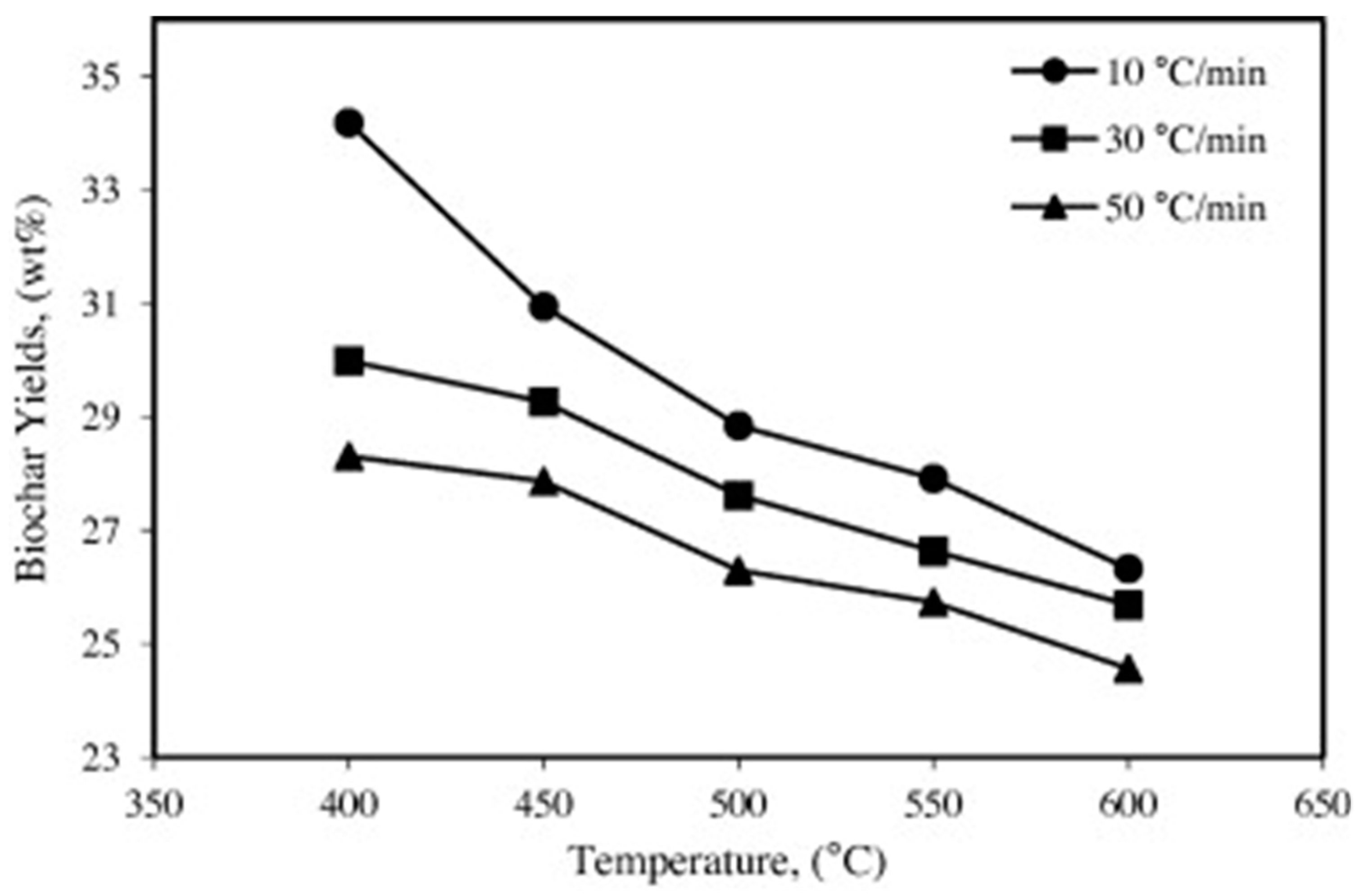
Figure 4, char yields increase with decreasing temperature and heating rate [
24]. Theoretical yields of char, based on proximate analysis, can be 50–70 wt% although actual yields are usually less than 80% of theoretical values [
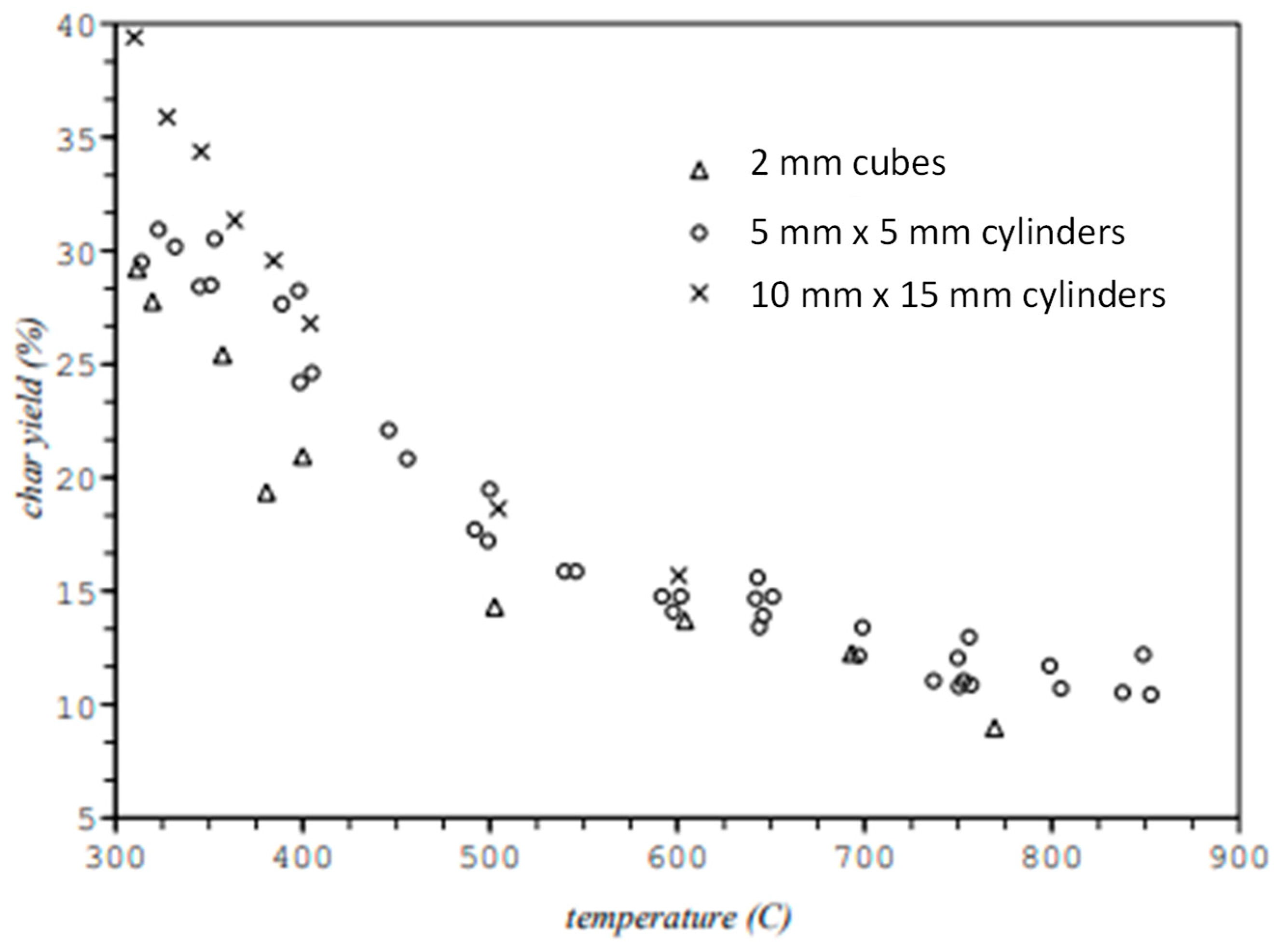
25]. A number of other operating conditions can influence char yields, including grind of feedstock, reactor ventilating rate and operating pressure. Large particles tend to produce higher char yields especially at lower pyrolysis temperatures (see
Figure 5) [
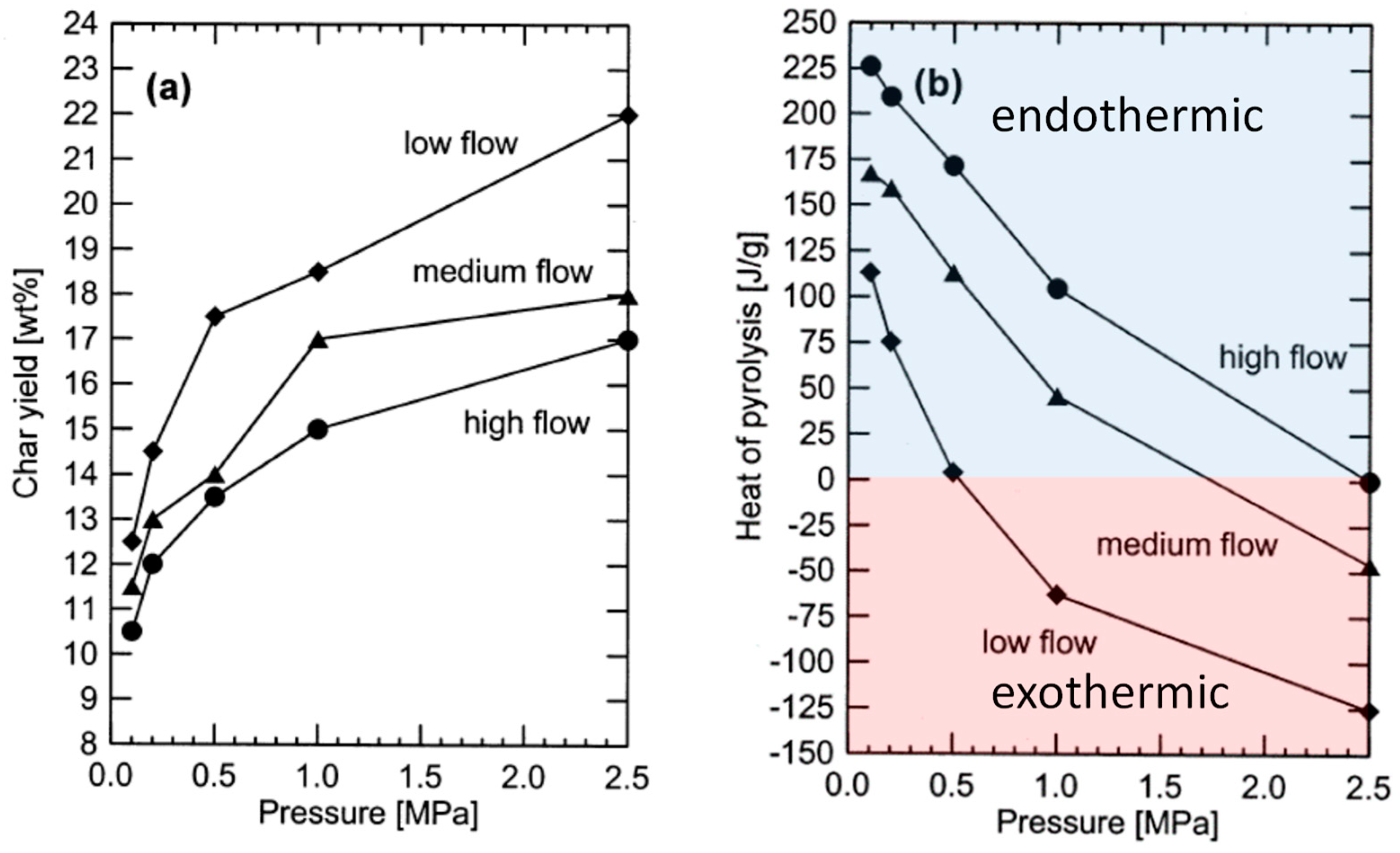
26]. Increasing the pressure of operation and decreasing the ventilation rate of gas through a pyrolysis reactor encourages volatiles to condense and dehydrate to form so-called secondary char to distinguish it from char formed directly from solid organic material. As shown in
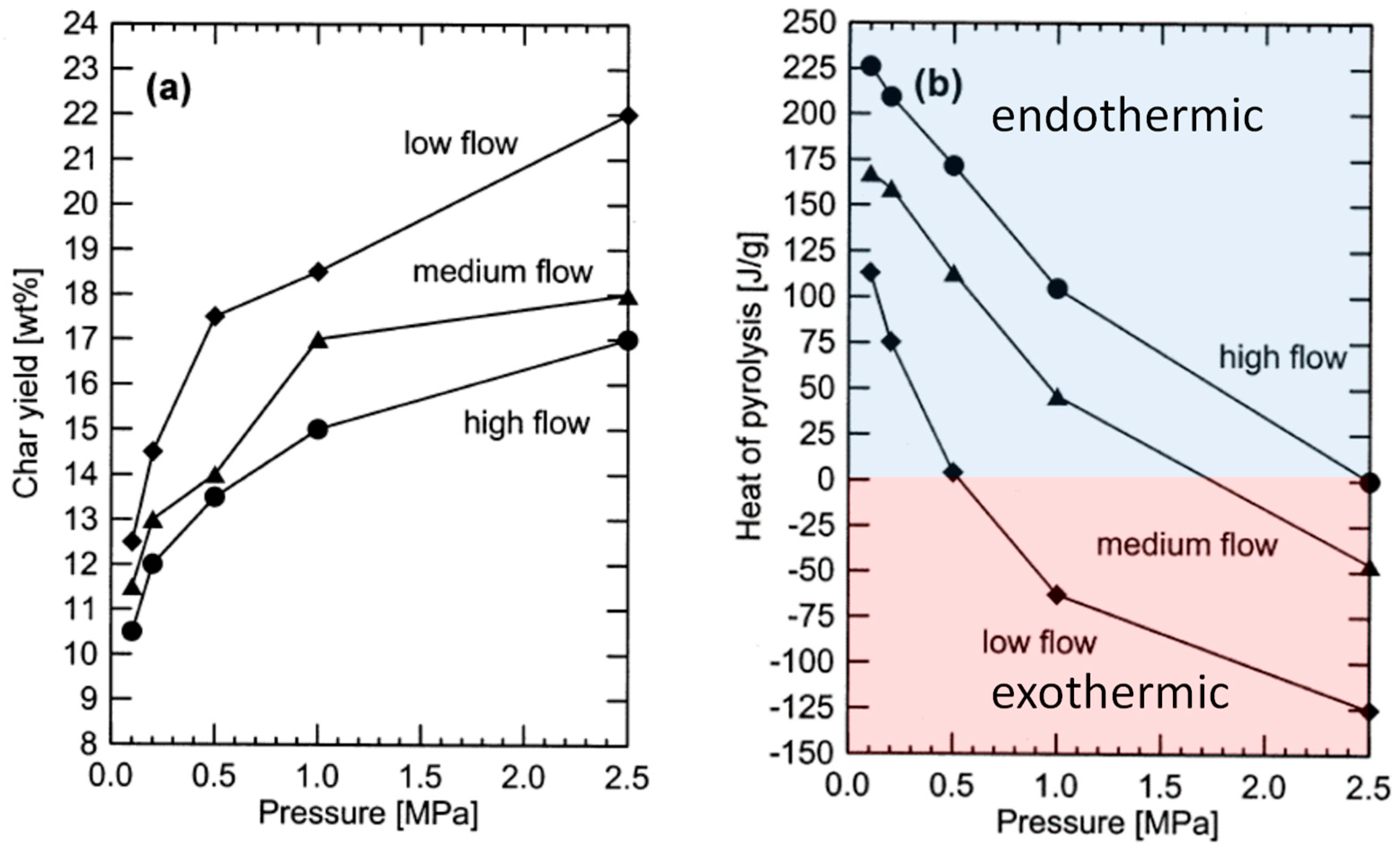
Figure 6a, char yields can increase 30% in moving from high to low ventilation rates and can increase by over 80% in increasing pressure from atmospheric to 2.5 MPa. The literature contains contradictory reports on the effect of moisture on char yields, [
27] although Antal et al. [
28] provide evidence that moisture improves yields in closed, pressurized kiln vessels.
A prominent advantage of slow pyrolysis for carbon negative energy is the simple construction and operation of slow pyrolysis reactors, making them suitable for small plants processing widely distributed biomass supplies. As illustrated in
Figure 6b, if operated at elevated pressure (above 0.5 MPa) and low ventilation rates, slow pyrolysis has prospects for being an exothermic process, requiring neither an external source of heat nor partial oxidation of products to drive the process [
29]. Even though pressurized “charcoal kilns” have been demonstrated, [
28] they bring additional cost of construction and complexity of operation that negates an important advantage of slow pyrolysis.
Fast pyrolysis is characterized by rapid heating of solid organic material followed by rapid cooling of the volatilized products to assure maximum yield of condensable organic vapors from this non-equilibrium process. If vapors released from devolatilizing biomass dwell too long at pyrolysis temperatures, they undergo secondary reactions that crack heavy molecules to light oxygenated compounds (especially in the presence of hot char particles) [
30] or repolymerize phenolic compounds to high molecular weight oligomers, [
31,
32] neither of which are particularly desirable liquid products for upgrading to fuels or chemicals. Under conditions of rapid heating, most volatiles are released in as little as 1–2 s, [
33] although devolatilization can continue for as long as 40 s [
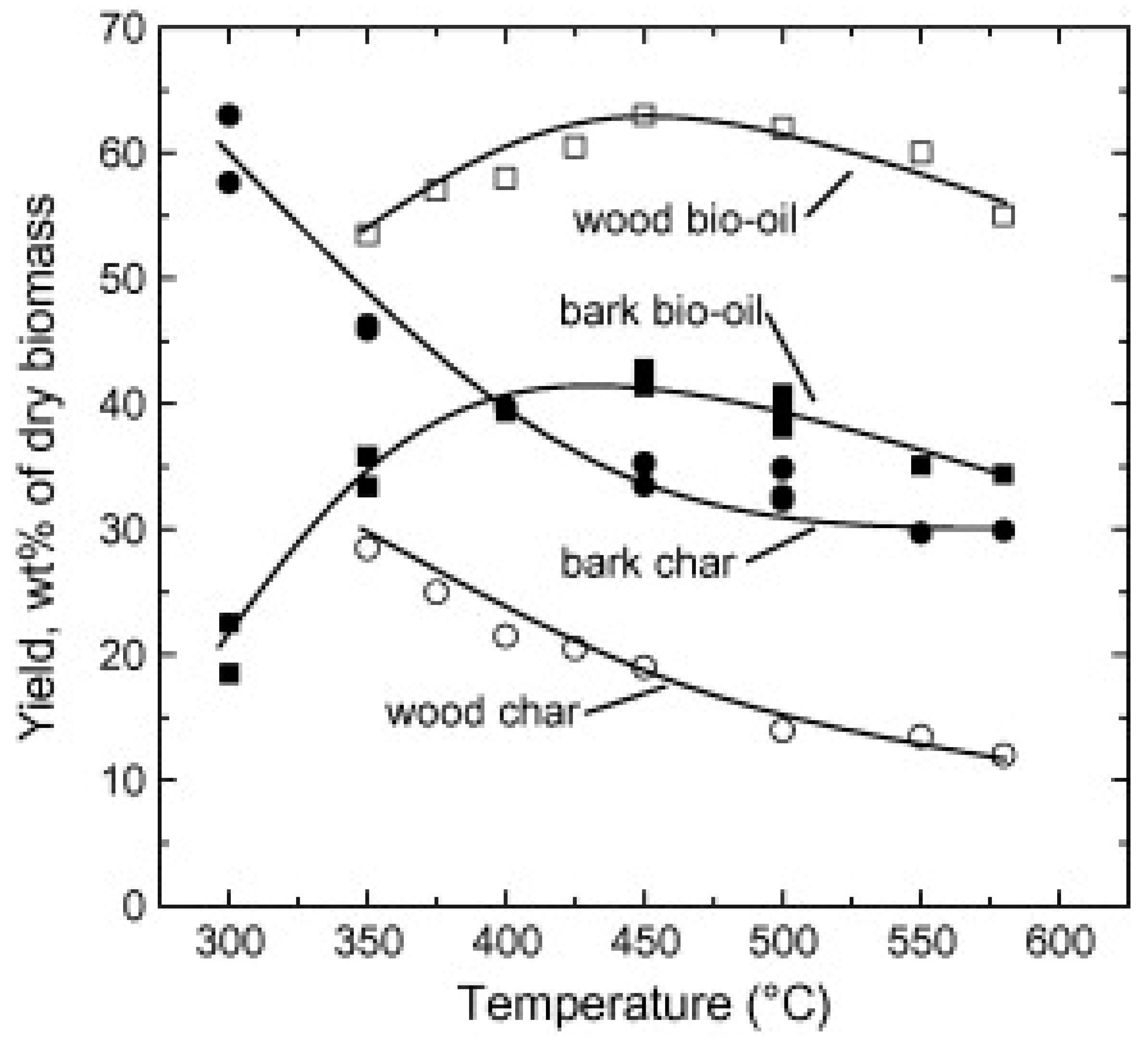
34]. Accordingly, solids residence times should be much longer than vapor residence times to minimize volatile content of char and maximize bio-oil yields from condensation of volatiles downstream of the pyrolysis reactor. The optimal temperature for maximum bio-oil yields occurs around 500 °C (see
Figure 7) [
35]. At higher temperatures both bio-oil and char yields decrease; that is the process shifts towards gasification of the solid organic reactant.
To favor liquid over char formation, both high heat fluxes at the surface of the biomass particles and rapid conduction of heat to the center of the particle are critical. Over the years, a wide range of reactor configurations have been evaluated to rapidly convey heat to the surface of particles while rapidly transporting vapors out of the reactor without overheating the particles [
22]. Due to their outstanding convection coefficients, fluidized beds have been widely employed for fast pyrolysis, either bubbling or circulating beds. In continuous operation, char particles, which tend to accumulate at the surface of the bed, can be removed with an overflow pipe. However, their low particle density and high friability compared to the starting biomass causes attrited char to readily elutriate from the bed with gases and vapors where they can be separated from the product stream with gas cyclones.
Pyrolysis reactors can be categorized according to the method that the energy for pyrolysis is provided to them: indirectly heated, directly heated and autothermal operation [
36]. Indirectly heated reactors either pass hot gases around the reactor perimeter or through heat exchange tubes within the reactor (for research reactors electrical heaters are sometimes used to provide this thermal energy). Achieving high heat fluxes at modest temperatures can be problematic in gaseous environments. Even though fluidized beds can achieve high heat fluxes at reactor walls or heat exchange surfaces, erosion can be a problem and immersed tubes can compromise the mixing of sand and gas that is responsible for the high convection coefficients of fluidized beds. Directly heated reactors contact heat transfer media with the biomass to improve heat transfer rates compared to indirect heat transfer. Even though the sensible enthalpy of gases may be adequate for the modest endothermicity for slow pyrolysis (<0.225 MJ/kg, see
Figure 6b), it is inadequate for providing the enthalpy for fast pyrolysis, which is on the order of 1 MJ/kg [
37]. For this reason, granular media is often employed to transport thermal energy from a heat source to the reactor, usually either a fluidized bed [
38] or an auger reactor [
39].
Autothermal operation provides the enthalpy for pyrolysis through partial combustion of either the reactant or products of pyrolysis [
40]. Traditional charcoal kilns operate on this principle, with a fire ignited in a pile of wood covered with earth except for a few vents at the bottom and a small flue at the top for the purpose of providing just enough air to meet the energy needs for slow pyrolysis of the wood [
41]. The possibility of autothermal operation of fast pyrolyzers received little attention until recently, probably because it seemed likely that oxygen would preferentially oxidize the vapors intended for bio-oil production. In fact, in a fluidized bed reactor, the oxygen preferentially oxidizes char with only a small penalty on bio-oil yield [
42,
43]. Not only does autothermal pyrolysis simplify reactor design and operation, it achieves several-fold process intensification in reactor throughput by overcoming the heat transfer bottleneck of conventional (non-oxidative) pyrolysis [
40].
Both slow and fast pyrolysis produce solid carbon sequestration products although slow pyrolysis produces as much as three times more char. Despite these advantages, an economic study by Brown et al. [
44] suggests that it would be a less profitable enterprise for the production of char than fast pyrolysis. The only co-product of slow pyrolysis is a low caloric value gas that today is only worth
$2–4/GJ while fast pyrolysis produces bio-oil and gas as co-products. The bio-oil can be upgraded to transportation fuel worth
$23/GJ or more.
Char has several advantages as a carbon sequestration agent. Marmiroli et al. [
45] estimate the bulk density of wood char to be around 450 kg/m
3 while Brewer et al. [
46] report carbon content of 80%, thus achieving 364 kg carbon storage per cubic centimeter. This is several fold greater than for CO
2 even when stored under tens of atmospheres of pressure [
47]. Char is also recalcitrant, with a half-life measured in hundreds of years or even longer even when stored in soils [
48]. Finally, char stored in soils, in which case it is referred to as biochar, provides ecosystem services beyond carbon sequestration including improving the water holding and nutrient retention capacities of soil [
49,
50,
51]. The annual carbon removal potential of biochar has been estimated to be as much as 1.2 trillion tons of carbon, representing 10%–15% of the total carbon removal potential of terrestrial ecosystems [
52].
One of the major challenges of biochar as carbon sequestration agent is lack of uniform properties among biochars, which are dependent on the feedstock and pyrolysis conditions. For example, the high ash content of herbaceous biomass results in biochars with lower fixed carbon content than woody biomass [
46], reducing its carbon sequestration potential per unit mass. Biochar produced at lower temperatures and shorter solids residence times contain more volatile matter (labile carbon) [
53], which very quickly mineralizes in soils and does not contribute toward long-term carbon sequestration [
54]. Biochar production often yields a fine powder (see
Figure 8), which can be difficult to apply on fields and can even be a fire hazard. However, this problem can be largely mitigated through prilling the biochar with a small amount of binder material into granules, as shown in
Figure 8.
Fast pyrolysis technology has undergone significant advancement since its emergence in the early 1980′s. Readers seeking more comprehensive information on these advances are directed to a recent review article [
21] and book [
55] on the subject prepared by the author.
4. Gasification as a Pathway to Carbon Negative Energy
Gasification can be broadly characterized according to the method by which thermal energy is provided to support devolatilization of biomass and other endothermic reactions including gas-solid reactions and gas-phase reactions (see
Figure 3) [
19]. These include indirect heating (allothermal gasification) using an external heat source and partial oxidation (autothermal gasification) in which combustion at equivalence ratios of 0.2–0.3 releases enough thermal energy to support the endothermic reactions of the process.
Most allothermal gasifiers are based on fluidized bed reactors with heat supplied by transporting hot granular bed media between a combustor and the gasification vessel [
19]. However, there are also chemical looping allothermal gasifiers that transfer chemical energy between a combustor and gasifier [
56]. These system have the advantage of producing high caloric-value gas with relatively little carbon dioxide (from combustion) or nitrogen (from air) that characterize partial oxidation gasification. However, they add an additional level of complexity in the design and operation of the gasifier.
Autothermal gasifiers can be either air-blown or oxygen-blown [
19]. Air-blown gasifiers are relatively simple but generally operate at lower temperatures than oxygen-blown systems and the product gas is heavily diluted with nitrogen from the air. Oxygen-blown gasifiers, by operating at higher temperatures, attain very high carbon conversion and produce gas that is mostly CO and H
2. Of course, the need for oxygen increases the capital and operating costs for these more efficient gasifiers.
The intended use of the product gas strongly influences whether the gasifier is designed to operate at elevated pressures, which can be 10 bar or higher. Atmospheric gasification is suitable for many process heat applications and gas fermentation [
57]. However, for use in gas turbine or gas-to-liquids synthesis, pressurized gas is required. Even though gas from an atmospheric pressure gasifier could be cooled and compressed to pressures appropriate for downstream applications, it is rarely deemed practical because of the large volume of gas generated in the gasifier. Generating the gas in a pressurized reactor is more energy efficient although the need for pressure vessels and feedstock lock hoppers adds to the complexity and capital cost of the installation.
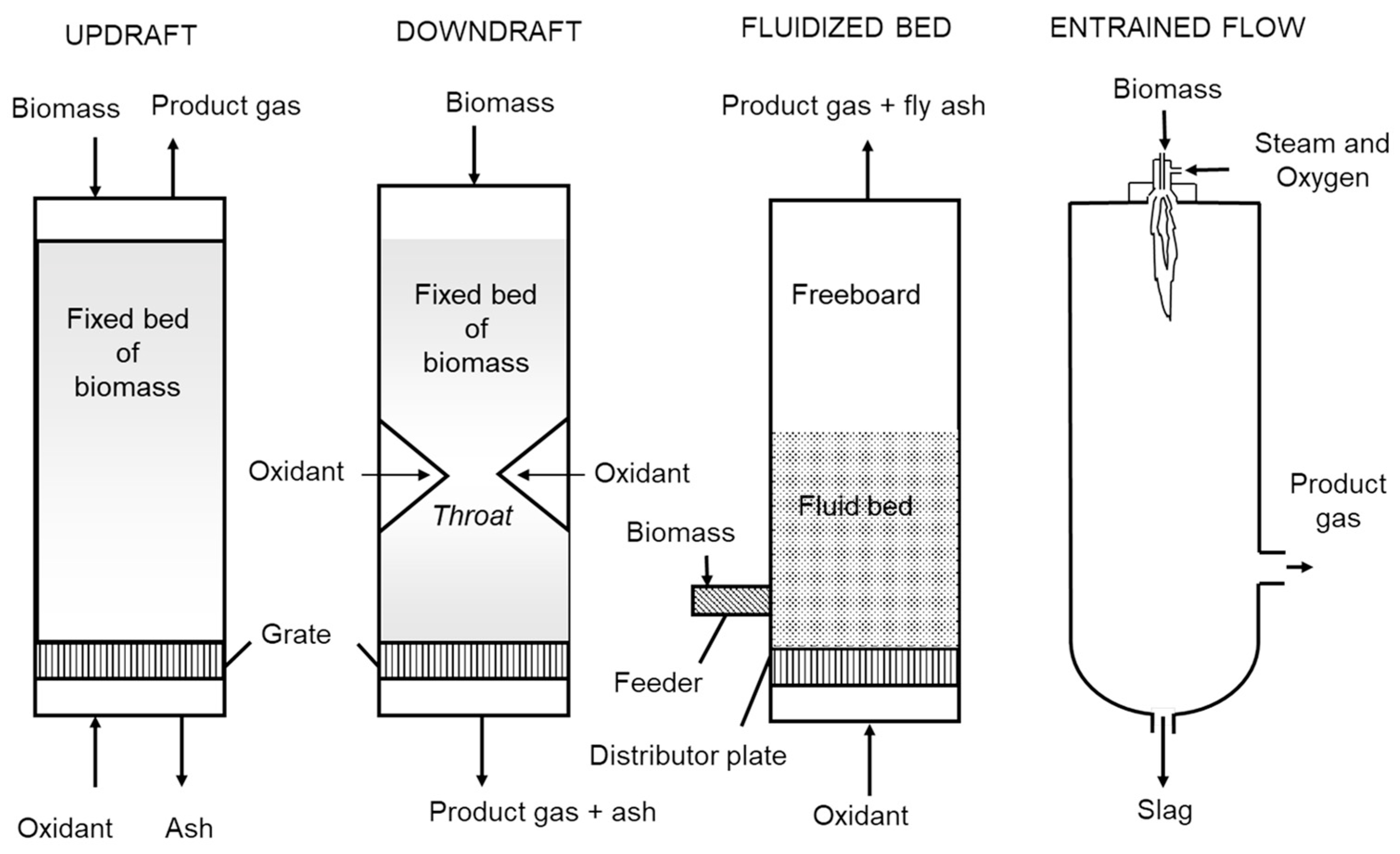
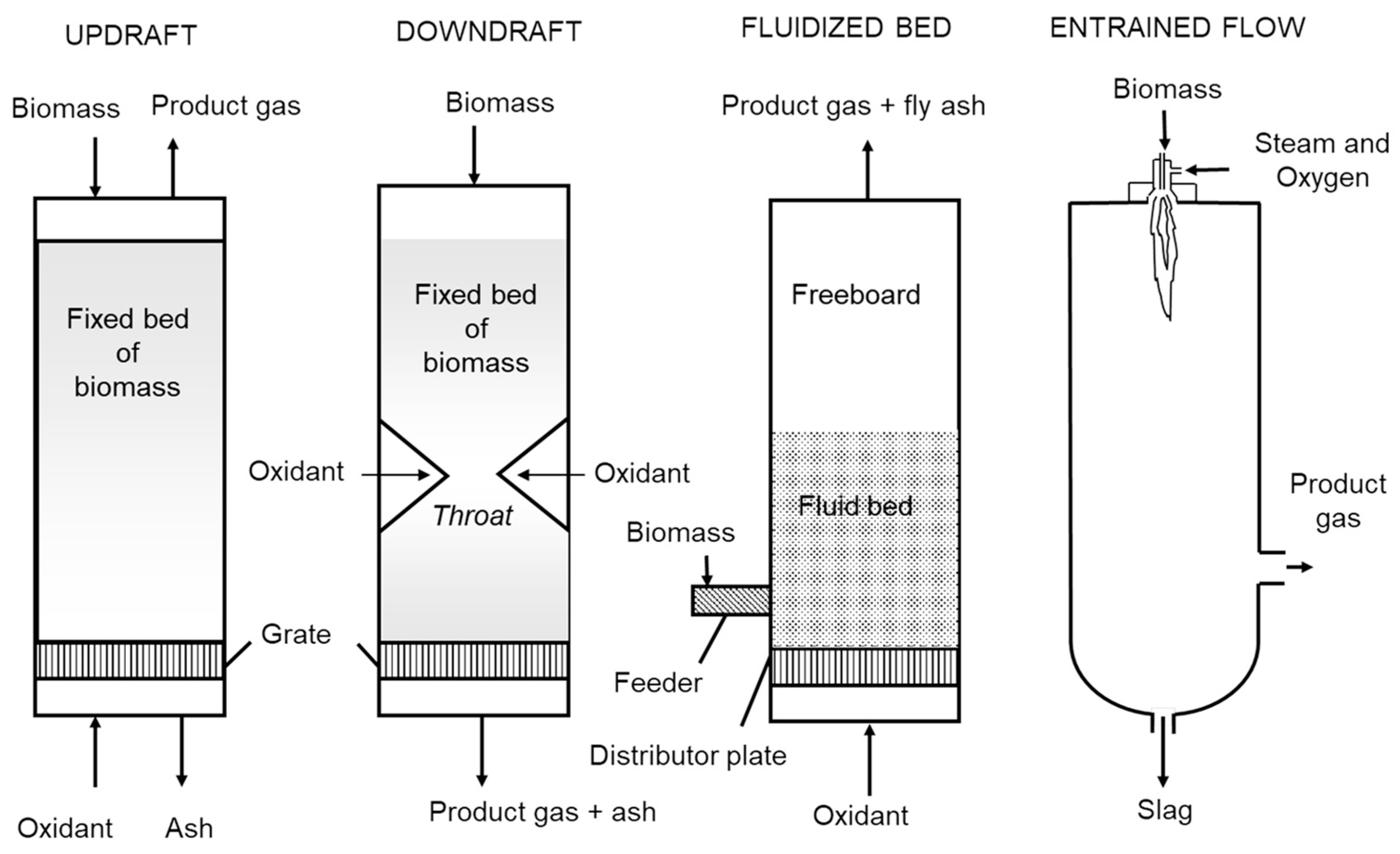
Biomass gasifiers can be further classified according to the methods used to contact solid feedstock and gasification agents (steam, oxygen and/or air). These include updraft, downdraft, fluidized beds and entrained flow (see
Figure 9) [
18]. Updraft and downdraft gasifiers are both fixed bed reactors with biomass moving downward through them by gravity. Updraft gasifiers were the basis of the earliest gasifiers. Biomass is fed from the top of the gasifier onto a grate through which air flows upward. Even though very simple in operation, the counterflow of biomass and air produces high concentrations of tar. It is generally not considered suitable for modern heat and power systems. The downdraft gasifier is a distinct improvement over updraft gasifiers. An induced draft fan draws gases downward through a grate, causing the concurrent downward flow of biomass and gases (air and product gas). Air is drawn into the gasifier at a constriction in the cross-section of the reactor known as the throat where charcoal is oxidized to high temperatures. Biomass approaching the hot charcoal is pyrolyzed to vapors and additional charcoal. The vapors flow through the hot charcoal where tars are cracked to light gases while the fresh charcoal replenishes the burned charcoal. This gasifier produces gas with very little tar, but is not readily scaled beyond 400 kg/h.
The fluidized bed gasifier is a multi-phase system of gas and particulate matter [
39]. A bubbling fluidized bed consists of an emulsion phase of particulate matter suspended in an upward flowing gas and a bubble phase consisting of large voids moving upward through the emulsion phase. A bubbling fluidized bed transitions to a circulating fluidized bed when sufficient gas is passed through the reactor to cause a dilute suspension of particulate matter with no discernible bubble phase. Particulate matter is continuously circulated between the reactor (riser) and the return line (standpipe). Both kinds of fluidized beds are characterized by high rates of heat transfer and solids mixing. The bubbling fluidized bed is of simpler design and is probably more suited for smaller scale applications. The circulating fluidized bed achieves higher carbon conversion because of the circulation of solids between riser and standpipe. It is more appropriate for larger scale applications.
Entrained flow gasifiers achieve very high carbon conversions and virtually tar-free product gas by reacting very fine particles of solid fuel in a stream of pure oxygen at temperatures of 1200–1500 °C [
19]. At these temperatures, ash in the feedstock melts and forms a slag that flows down the walls of the gasifier to a quench section. Due to their high gasification efficiency, entrained flow reactors have been favored in commercial-scale gasification of coal [
58]. However, entrained flow reactors face several limitations in biomass gasification. The requirement of feedstock being comminuted to sizes finer than 200 µm [
59] is a significant barrier to utilizing them for biomass gasification. While coals are friable materials that are easily crushed to fine particles, biomass fibers are inherently elastic and are more apt to bend than break under compressive forces. Thus, significant energy is consumed in comminuting biomass compared to coal and minerals [
60]. For pressurized gasification, coal particles are frequently mixed with water to form pumpable slurries containing as much as 50% coal that can be injected at high pressures into the gasifier. The hydrophilic nature of biomass makes it impossible to produced high solids content slurries. Grindability and hydrophobicity of biomass can be remarkably increased through torrefaction of biomass, although this adds significantly to production cost of producer gas [
61]. The slagging of ash in high temperature entrained flow gasifiers is a distinct disadvantage in the gasification of biomass since the ash includes the important plant nutrients of potassium and phosphorous. Experiments in gasification of biomass in entrained flow reactors have found that these nutrients react to form melts of potassium silicates or crystals of phosphates and diphosphates, [
62] which would ultimately report to the slag in vitrified form, likely making these nutrients no longer plant available. Considering the looming worldwide shortage of phosphorous, [
63] this is an important issue to resolve before adopting this technology for carbon negative energy.
The gaseous product of biomass gasification is ideally a mixture of non-condensable gases consisting primarily of CO and H
2 and smaller amounts of CO
2 and light alkanes and alkenes [
64]. In practice it can contain significant tar, hydrogen sulfide (H
2S), carbonyl sulfide (COS), ammonia (NH
3), hydrogen cyanide (HCN), hydrogen chloride (HCl) and alkali metals. Most of these represent serious air pollutants if the gas is combusted or catalyst poisons if the gas is used in chemical synthesis. Thus, they must be removed often to very low concentrations depending on the downstream application (see
Table 2) [
18]. In many cases, separate unit operations are required for each contaminant to achieve the very high levels of removal required for some applications, which adds considerably to the expense of gasification [
65].
Fluidized bed and entrained flow reactors are the two most likely candidates for carbon negative energy from biomass gasification because of their ability to scale, which will be important for operation at elevated pressure and attainment of comprehensive gas cleaning. It is useful to compare the economic performance in considering their prospects for carbon negative energy. Swanson et al. [
66] performed a technoeconomic comparison of low temperature (870 °C) fluidized bed gasification and high temperature (1300 °C) entrained flow gasification at 28 bar, both processing 2000 tons per day to produce Fisher-Tropsch liquids. Despite higher investment costs for the entrained flow gasifier system, its higher product output produced fuel that was 12% cheaper than from the fluidized bed gasifier. Unfortunately, the capital cost for these plants were estimated to be very high, between
$14.50 and
$15.40 per gallon of annual capacity.
Even though fluidized bed gasification produces as much as 10 wt% char, the primary opportunity for carbon negative energy via biomass gasification is through geological storage of CO
2. The amount of CO
2 that could be sequestered per metric ton of biomass gasified depends not only on the carbon conversion to gas, but the application of the gas. For example, oxygen-blown entrained flow gasification of wood (CH
1.4O
0.66) at 20% equivalence ratio might produce gas containing 93% of its carbon as CO and 7% as CO
2:
Sequestering this small fraction of the carbon in the biomass before burning the producer gas for heat or power would be significantly less than the carbon sequestered in biochar from pyrolysis (for fast pyrolysis and slow pyrolysis, respectively, about 20% and 56% of the biomass carbon). On the other hand, adding steam to the gasifier or reacting the product gas with steam over a catalyst to water-gas shift the products to a H
2:CO ratio of 2.0 for gas-to-liquid synthesis would increase the fraction of carbon in CO
2 to 53%:
In principle, all the carbon in the product gas could be water-gas shifted to CO
2 producing hydrogen gas as the energy product and sequestering 100% of the carbon as CO
2:
The coupling of biomass gasification with CO
2 sequestration is known as biomass energy with carbon capture and sequestration (BECCS) [
67]. Originally developed to reduce the net CO
2 emissions from coal- and gas-fired power plants to near zero, [
68] carbon capture and sequestration (CCS) has been expanded into the concept of carbon negative energy via gasification. Even though flue gas from combusting fuels could be scrubbed to remove CO
2, typically around 8–14 vol%, as the calculations in Equations (1)–(3) illustrate, gasification streams that were water-gas shifted for the purpose of fuels production or electric power generation would contain 27–38 vol% CO
2. These high concentrations would significantly reduce the cost of CO
2 capture using amines or alkaline aqueous solutions similar to those used by DAC to remove much lower concentrations. Several influential studies suggest that CCS will play an important role in decarbonizing society, [
69,
70,
71] contributing as much as 80% of global CO
2 emissions reductions by 2050 [
71]. Wide-scale deployment of BECCS to provide carbon negative power and fuels is estimated to be able to not simply avoid future GHG emissions but remove 3.5–5.2 Gt/y of CO
2 by 2050 [
4].
Clearly, gasification has more potential to sequester carbon from a given quantity of biomass than does pyrolysis. However, this important advantage must be balanced against its prominent disadvantages. First is the challenge of reliable and economical long-term storage of gaseous CO
2. The density of carbon dioxide stored as supercritical fluid in geological deposits or as dissolved gas in saline aquifers at depths of 800 m is only 260 kg/m
3 and 35 kg/m
3, respectively [
47]. Since carbon only accounts for 27% of this mass, the actual carbon storage density is 70 kg/m
3 and 9.5 kg/m
3. This compares poorly against biochar with carbon storage density of 364 kg/m
3.
Both sequestration in oceans and geological formations has been proposed. In principle, the high pressures and vast extent of deep ocean waters could sequester more CO
2 as dissolved gas or solid CO
2 hydrates than would be emitted from combusting the estimated global fossil fuel resources of 5000–10,000 GtC [
12]. Small scale, in-situ experiments have established the technical feasibility of ocean storage, [
72] but efforts to conduct large scale experiments in the ocean have met with public opposition over concerns about negative impacts on ocean ecosystems [
73].
Geological storage includes injecting CO
2 as compressed gas or supercritical fluid into porous geological formations that are capped by low permeability caprock (known as hydrodynamic trapping) or into liquids including petroleum reservoirs or saline aquifers (known as solubility trapping) [
12]. Hydrodynamic trapping is likely to be the most attractive near-term approach to geological storage while solubility trapping is thought to be more reliable in preventing CO
2 gas from returning to the atmosphere. Leakage around injection points or through cracks in the caprock are serious concerns for hydrodynamic cracking. Abrupt leakage through injection well failure or collapse of caprock could cause grievous harm to animals or people [
73]. Solubility trapping has the potential advantage of solubilized CO
2 reacting with minerals in the geological formation to form carbonates that would permanently sequester carbon, a process known as mineral trapping [
74]. Both hydrodynamic and solubility trapping will require site monitoring for very long periods of time.
The energy and cost penalties associated with capturing carbon vs. emitting it into the atmosphere is significant. An analysis in 2015 by Supekar and Skerlos [
75] found that adding carbon capture to the flue gas stream from pulverized coal (PC) boilers would decrease power plant thermal efficiency by 11%–23%-points increasing electricity costs from
$0.084/kWh to
$0.14–
$0.16/kWh. That same year Sanchez et al. [
76] reported that BECCS could produce carbon negative power for
$0.13–
$0.19/kWh. BECCS demonstration projects have yet to emerge although a number of integrated coal gasification/electric power generation/carbon sequestration demonstration plants have been planned and/or abandoned in the last several years, as detailed in an MIT report by Herzog [
77]. As stated by the author, “A major reason [for failure] is the recurring theme of the high cost of gasification. The current technologies just seem too expensive for the power sector”. However, the author goes on to note that many of these projects were developed during a period when gas and oil prices dramatically decreased, contributing to the unattractiveness of coal power with or without carbon sequestration.
The Herzog report observes that successful CCS projects have employed already pure streams of CO
2 requiring little more than compression, transportation and storage costs. Sanchez et al. [
13] reports that CO
2 from ethanol plants could be captured and compressed for pipeline transport to sequestration sites for around
$32/MT CO
2. An IPCC study [
73] indicates the cost of transporting CO
2 a distance of 250 km by pipeline and geologically sequestering it would add
$1.60–
$16.30/MT CO
2 resulting in the total cost to capture and sequester CO
2 from this “biochemical BECCS” system of less than
$50/MT CO
2. Not surprising, these appear to be the first BECCS systems to move toward commercialization [
78].
Finally, the scale of gasification might not be compatible with biomass resources, especially when integrated with gas turbine power cycles and gas-to-liquids synthesis. These energy products require pressurized gasifiers and extensive gas cleaning operations, which are only economical in large-scale deployments. Experience in the advanced biofuels industry suggest that the low bulk density of biomass and the distributed nature of this resource is incompatible with the 2000 MT/d scale originally envisioned by the U.S. DOE for advanced biorefineries [
79].




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}