
Comprehensive Analysis of Mutation-Based and Expressed Genes-Based Pathways in Head and Neck Squamous Cell Carcinoma

 , , , , ,
, , , , ,
Abstract
1. Introduction
2. Patients and Methods
2.1. Patients and Data Collection
2.2. Targeted Gene Sequencing and mRNA Expression Assay
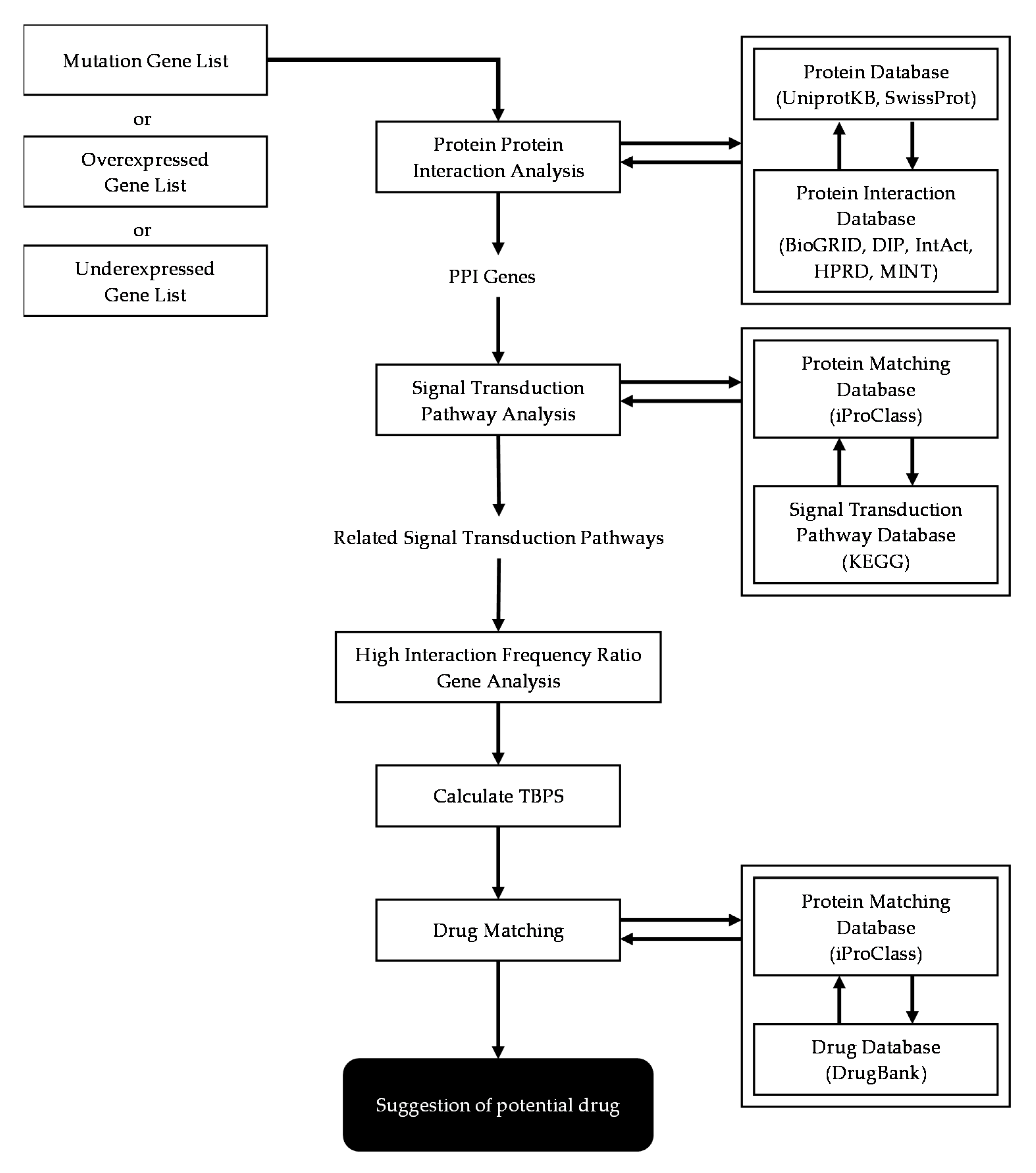
2.3. Basic Scheme of Protein-Protein Interaction Network Analysis
2.4. Gene List Enrichment
2.5. PPI Mapping of Mutated Genes and Over- or Under-Expressed Genes
2.6. Signal Transduction Pathway Analysis
2.7. High Interaction Frequency Ratio Genes Analysis
2.8. Treatment Benefit Prediction Score (TBPS) Calculation
2.9. Potential Treatment Recommendation for Patient
2.10. Comparison of Mutation-Based pathway and Over- or Under-Expressed Genes-Based Pathways
2.11. Validation of TBPS in Two HNSCC Patients Treated with Targeted Agents
3. Results
3.1. Clinical Characteristics
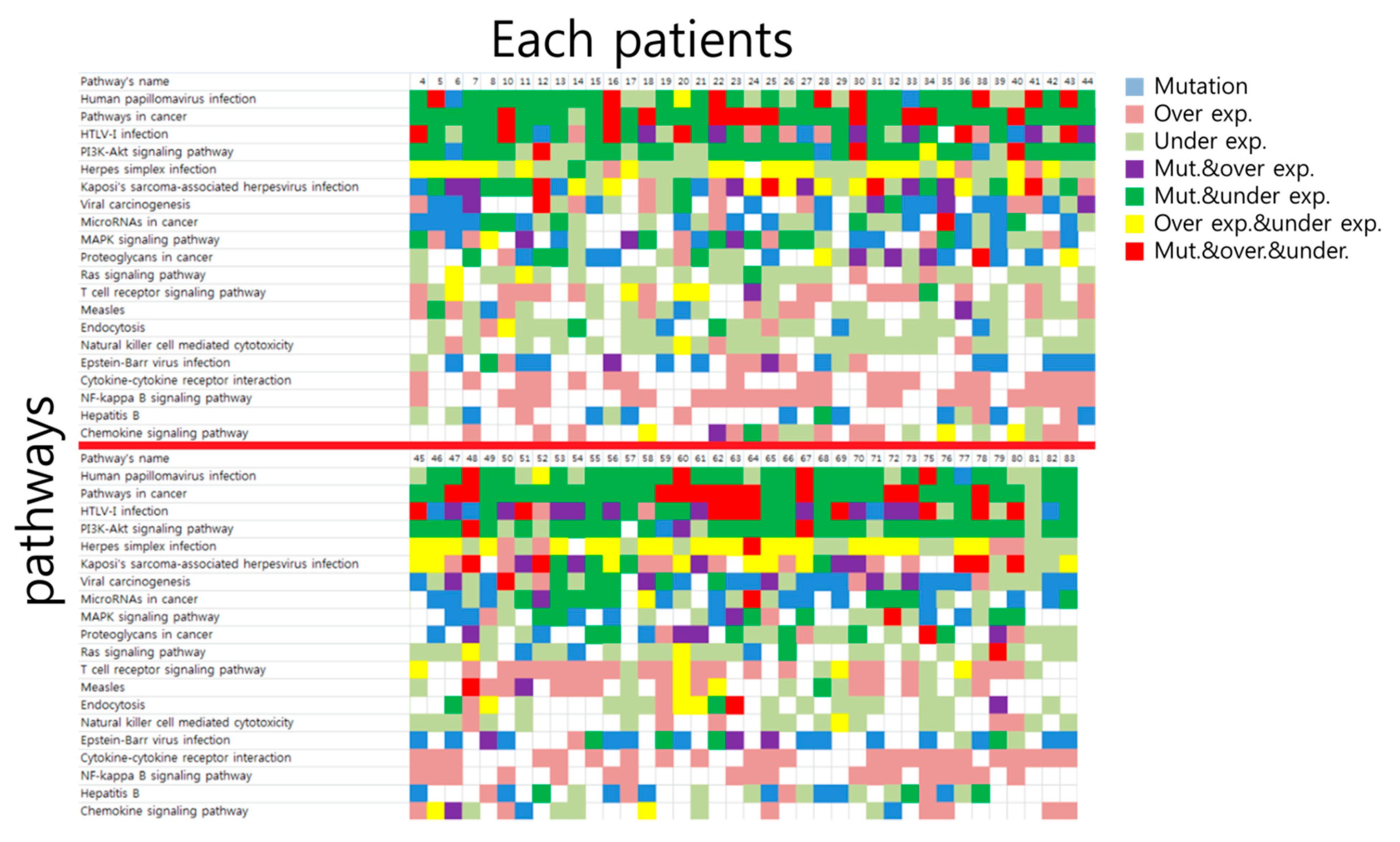
3.2. Top 10 Signaling Pathway Discovered by Mutation-Based Analysis and mRNA Expressed Genes-Based Analysis
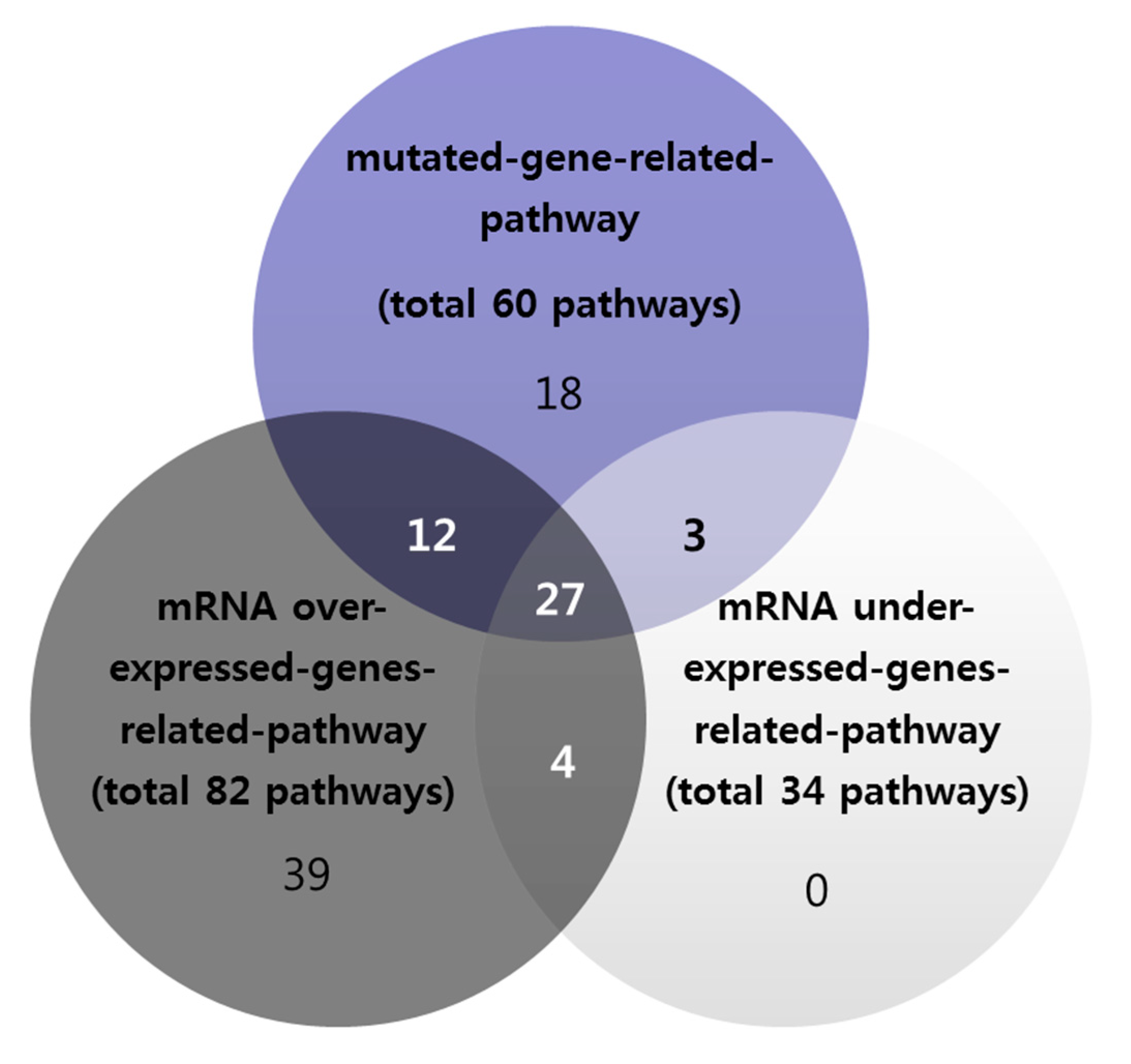
3.3. Overlapping of Mutation-Based Pathway and Over- or Under-Expressed Genes-Based Pathways
3.4. Calculation of Treatment Benefit Prediction Score (TBPS)
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Cancer Genome Atlas Network; Sheikine, Y.; Kuo, F.C.; Lindeman, N.I. Clinical and Technical Aspects of Genomic Diagnostics for Precision Oncology. J. Clin. Oncol. 2017, 35, 929–933. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Network. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature 2015, 517, 576–582. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, N.; Frederick, M.J.; Pickering, C.R.; Bettegowda, C.; Chang, K.; Li, R.J.; Fakhry, C.; Xie, T.X.; Zhang, J.; Wang, J.; et al. Exome sequencing of head and neck squamous cell carcinoma reveals inacti-vating mutations in NOTCH1. Science 2011, 333, 1154–1157. [Google Scholar] [CrossRef] [PubMed]
- Stransky, N.; Egloff, A.M.; Tward, A.D.; Kostic, A.D.; Cibulskis, K.; Sivachenko, A.; Kryukov, G.V.; Lawrence, M.S.; Sougnez, C.; McKenna, A.; et al. The Mutational Landscape of Head and Neck Squamous Cell Carcinoma. Science 2011, 333, 1157–1160. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.M.; Cho, S.H.; Hwang, I.G.; Choi, J.W.; Chang, H.; Ahn, M.-J.; Park, K.U.; Kim, J.-W.; Ko, Y.H.; Ahn, H.K.; et al. Investigating the Feasibility of Targeted Next-Generation Sequencing to Guide the Treatment of Head and Neck Squamous Cell Carcinoma. Cancer Res. Treat. 2019, 51, 300–312. [Google Scholar] [CrossRef] [PubMed]
- Keam, B.; Kim, H.R.; Yun, H.J. Triumph Trial: One Small Step Could Become One Giant Leap for Precision Oncology in Head and Neck Cancer. Cancer Res. Treat. 2019, 51, 413–414. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Ivanov, A.A.; Su, R.; Gonzalez-Pecchi, V.; Qi, Q.; Liu, S.; Webber, P.; McMillan, E.; Rusnak, L.; Pham, C.; et al. The OncoPPi network of cancer-focused protein–protein interactions to inform biological insights and therapeutic strategies. Nat. Commun. 2017, 8, 14356. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, A.A.; Revennaugh, B.; Rusnak, L.; Gonzalez-Pecchi, V.; Mo, X.; Johns, M.A.; Du, Y.; Cooper, L.A.; Moreno, C.S.; Khuri, F.R.; et al. The OncoPPi Portal: An integrative resource to explore and prioritize pro-tein-protein interactions for cancer target discovery. Bioinformatics 2018, 34, 1183–1191. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, A.A.; Khuri, F.R.; Fu, H. Targeting protein–protein interactions as an anticancer strategy. Trends Pharmacol. Sci. 2013, 34, 393–400. [Google Scholar] [CrossRef] [PubMed]
- Murakami, Y.; Tripathi, L.P.; Prathipati, P.; Mizuguchi, K. Network analysis and in silico prediction of protein-protein interac-tions with applications in drug discovery. Curr. Opin. Struct. Biol. 2017, 44, 134–142. [Google Scholar] [CrossRef] [PubMed]
- Park, I.J.; Yu, Y.S.; Mustafa, B.; Park, J.Y.; Seo, Y.B.; Kim, G.-D.; Kim, J.; Kim, C.M.; Noh, H.D.; Hong, S.-M.; et al. A Nine-Gene Signature for Predicting the Response to Preoperative Chemoradiotherapy in Patients with Locally Advanced Rectal Cancer. Cancers 2020, 12, 800. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.M.; Hwang, S.; Keam, B.; Yu, Y.S.; Kim, J.H.; Kim, D.S.; Bae, S.H.; Kim, G.D.; Lee, J.K.; Seo, Y.B.; et al. Gene Signature for Sorafenib Susceptibility in Hepatocellular Carcinoma: Different Ap-proach with a Predictive Biomarker. Liver Cancer 2020, 9, 182–192. [Google Scholar] [CrossRef] [PubMed]
- Costello, M.; Pugh, T.J.; Fennell, T.J.; Stewart, C.; Lichtenstein, L.; Meldrim, J.C.; Fostel, J.L.; Friedrich, D.C.; Perrin, D.; Dionne, D.; et al. Discovery and characterization of artifactual mutations in deep coverage targeted capture sequencing data due to oxidative DNA damage during sample preparation. Nucleic Acids Res. 2013, 41, e67. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Hoang, T.H.; Joshi, P.; Hong, S.-H.; Giardina, C.; Shin, N.-G. A route-based pathway analysis framework integrating mutation information and gene expression data. Methods 2017, 124, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Park, Y.; Kim, S. MIDAS: Mining differentially activated subpaths of KEGG pathways from multi-class RNA-seq data. Methods 2017, 124, 13–24. [Google Scholar] [CrossRef] [PubMed]
- The Mutation Consequences and Pathway Analysis working group of the International Cancer Genome Consortium; Creixell, P.; Reimand, J.; Haider, S.; Wu, G.; Shibata, T.; Vazquez, M.; Mustonen, V.; Gonzalez-Perez, A.; Pearson, J.; et al. Pathway and network analysis of cancer genomes. Nat. Methods 2015, 12, 615–621. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| n = 93 | n | % |
|---|---|---|
| Age, median (range) | 59 (28–80) | |
| Gender | ||
| Female | 18 | 19 |
| Male | 75 | 81 |
| Anatomic site | ||
| Oropharnx | 26 | 28 |
| Oral cavity | 35 | 38 |
| Hypopharynx | 15 | 16 |
| Glottic larynx | 9 | 10 |
| Supraglottic larynx | 3 | 3 |
| Maxillary sinus | 5 | 5 |
| Tobacco use | ||
| Never | 26 | 28 |
| Former | 49 | 53 |
| Current | 18 | 19 |
| Alcohol use | ||
| Never | 34 | 37 |
| Former | 33 | 35 |
| Current | 26 | 28 |
| Initial clinical stage | ||
| I–III | 54 | 58 |
| IV | 39 | 42 |
| HPV status | ||
| Positive | 20 | 22 |
| Negative | 56 | 60 |
| Unknown | 17 | 18 |
| Mutated Based Analysis | mRNA Over-Expressed Genes-Based Analysis | mRNA Under-Expressed Genes-Based Analysis | ||||
|---|---|---|---|---|---|---|
| Rank | Number of Patients (n = 77) | Pathway Name | Number of Patients | Pathway Name | Number of Patients | Pathway Name |
| 1 | 74 | Pathways in cancer | 47 | Herpes simplex infection | 77 | Pathways in cancer |
| 2 | 63 | PI3K-Akt signaling pathway | 47 | Kaposi’s sarcoma-associated herpesvirus infection | 74 | Human papillomavirus infection |
| 3 | 62 | HTLV-I infection | 43 | T cell receptor signaling pathway | 71 | PI3K-Akt signaling pathway |
| 4 | 61 | Human papillomavirus infection | 42 | NF-kappa B signaling pathway | 68 | Herpes simplex infection |
| 5 | 41 | MicroRNAs in cancer | 42 | Cytokine-cytokine receptor interaction | 48 | Ras signaling pathway |
| 6 | 41 | Viral carcinogenesis | 38 | HTLV-I infection | 46 | HTLV-I infection |
| 7 | 35 | Kaposi’s sarcoma-associated herpesvirus infection | 36 | Cell adhesion molecules (CAMs) | 46 | Kaposi’s sarcoma-associated herpesvirus infection |
| 8 | 34 | Epstein–Barr virus infection | 31 | Influenza A | 40 | Natural killer cell mediated cytotoxicity |
| 9 | 33 | MAPK signaling pathway | 31 | Toll-like receptor signaling pathway | 40 | Endocytosis |
| 10 | 29 | Proteoglycans in cancer | 25 | Measles | 34 | MicroRNAs in cancer |
| Matching Rate Comparison | |
|---|---|
| Comparison of mutation-based pathways | Number of patients |
| Over-expressed genes-based pathways > Under-expressed genes-based pathways | 6 |
| Over-expressed genes-based pathways < Under-expressed genes-based pathways | 64 |
| Over-expressed genes-based pathways = Under-expressed genes-based pathways | 7 |
| Average Matching Rate | |
| Average matching rate with mutation-based pathways | Average matching rate |
| Over-expressed genes-based pathways | 19.09% |
| Under-expressed genes-based pathways | 42.73% |
| (A) Over-Expression Genes-Related Analysis | ||||
|---|---|---|---|---|
| Patient No. | Druggable Pathway | Druggable Gene | TBPS | Matched Drug |
| 4 | Measles, Hematopoietic cell lineage, Chagas disease (American trypanosomiasis), T cell receptor signaling pathway, HTLV-I infection | CD3E | 72.7 | Muromonab |
| 5 | RIG-I-like receptor signaling pathway, Hepatitis C, IL-17 signaling pathway, MAPK signaling pathway, Toll-like receptor signaling pathway, Herpes simplex infection, Influenza A | TNF | 135.3 | Adalimumab |
| Golimumab | ||||
| Infliximab | ||||
| Toll-like receptor signaling pathway, Cell adhesion molecules (CAMs) | CD80 | 23.4 | Durvalumab | |
| Cell adhesion molecules (CAMs) | CD274 | 9.1 | Atezolizumab | |
| Avelumab | ||||
| Durvalumab | ||||
| Cell adhesion molecules (CAMs) | PDCD1 | 9.1 | Nivolumab | |
| Pembrolizumab | ||||
| 16 | Influenza A, Epstein–Barr virus infection, Kaposi’s sarcoma-associated herpesvirus infection, Human papillomavirus infection, Pathways in cancer, Tuberculosis, Herpes simplex infection, HTLV-I infection | JAK1 | 41.4 | Ruxolitinib |
| (B) Under-Expression Genes-Related Analysis | ||||
| Patient No. | Druggable Pathway | Druggable Gene | TBPS | Matched Drug |
| 57 | Axon guidance, T cell receptor signaling pathway, Measles, Natural killer cell mediated cytotoxicity | FYN | 30.2 | Dasatinib |
| T cell receptor signaling pathway, Pathways in cancer, Human papillomavirus infection, Natural killer cell mediated cytotoxicity | GRB2 | 18.9 | Pegademase bovine | |
| Kaposi’s sarcoma-associated herpesvirus infection, Pathways in cancer, Human papillomavirus infection, HTLV-I infection | PIK3R1 | 16.9 | Isoprenaline | |
| T cell receptor signaling pathway, HTLV-I infection, Natural killer cell mediated cytotoxicity | LCK | 16 | Dasatinib | |
| Nintedanib | ||||
| Ponatinib | ||||
| (C) Mutation Genes-Related Analysis | ||||
| Patient No. | Druggable Pathway | Druggable Gene | TBPS | Matched Drug |
| 3 | Pathways in cancer, PI3K-Akt signaling pathway, HTLV-I infection, Human papillomavirus infection, MicroRNAs in cancer, Kaposi’s sarcoma-associated herpesvirus infection, Epstein–Barr virus infection, Breast cancer, Prostate cancer | TP53 | 42.5 | Acetylsalicylic acid |
| Pathways in cancer, PI3K-Akt signaling pathway, Human papillomavirus infection, MicroRNAs in cancer, Focal adhesion, Breast cancer, Prostate cancer | GRB2 | 27.7 | Pegademase bovine | |
| Mutated Based Analysis | ||||
|---|---|---|---|---|
| Pathway Name | High Frequency Gene | Frequency Ratio | TBPS | Maching_Drug_Names |
| Ras signaling pathway | AKT1 | 100.0 | 4.8 | Enzastaurin |
| Pathways in cancer | AKT1 | 100.0 | 3.9 | Arsenic trioxide, Enzastaurin |
| Melanoma | RAF1 | 50.0 | 3.7 | Dabrafenib |
| Proteoglycans in cancer | EGFR | 50.0 | 3.6 | Dacomitinib |
| Regulation of actin cytoskeleton | PDGFRB | 100.0 | 3.3 | Becaplermin |
| Breast cancer | EGFR | 50.0 | 3.3 | Lapatinib, Neratinib, Trastuzumab |
| Regulation of actin cytoskeleton | FGFR1 | 100.0 | 3.3 | Palifermin |
| Regulation of actin cytoskeleton | FGFR2 | 100.0 | 3.3 | Palifermin |
| Breast cancer | ESR2 | 50.0 | 3.3 | Tamoxifen |
| Gastric cancer | EGFR | 50.0 | 3.1 | Trastuzumab |
| PI3K-Akt signaling pathway | HSP90AA1 | 50.0 | 2.6 | Alvespimycin, Tanespimycin |
| PI3K-Akt signaling pathway | FGFR1 | 50.0 | 2.6 | Erdafitinib |
| PI3K-Akt signaling pathway | FGFR2 | 50.0 | 2.6 | Erdafitinib |
| PI3K-Akt signaling pathway | PDGFRB | 50.0 | 2.6 | Erdafitinib, Midostaurin |
| PI3K-Akt signaling pathway | HSP90AB1 | 50.0 | 2.6 | Tanespimycin |
| MAPK signaling pathway | EGFR | 50.0 | 2.5 | Afatinib, Canertinib, Cetuximab, Erlotinib, Gefitinib, Lapatinib, Necitumumab, Olmutinib, Osimertinib, Panitumumab, Pelitinib, Rindopepimut, Vandetanib, Zalutumumab |
| MAPK signaling pathway | PDGFRB | 50.0 | 2.5 | Becaplermin, Dasatinib, Imatinib, Midostaurin, Pazopanib, Regorafenib, Sorafenib, Sunitinib |
| MAPK signaling pathway | RAF1 | 50.0 | 2.5 | Dabrafenib, Regorafenib, Sorafenib |
| MAPK signaling pathway | EPHA2 | 50.0 | 2.5 | Dasatinib, Regorafenib |
| MAPK signaling pathway | FGFR2 | 50.0 | 2.5 | Lenvatinib, Nintedanib, Regorafenib |
| MAPK signaling pathway | FGFR1 | 50.0 | 2.5 | Lenvatinib, Nintedanib, Regorafenib, Sorafenib |
| mRNA Based Analysis | ||||
| Pathway Name | High Frequency Gene | Frequency Ratio | TBPS | Maching_Drug_Names |
| Proteoglycans in cancer | EGFR | 40.0 | 2.9 | Dacomitinib |
| MAPK signaling pathway | EGFR | 80.0 | 2.6 | Afatinib, Canertinib, Cetuximab, Erlotinib, Gefitinib, Lapatinib, Necitumumab, Olmutinib, Osimertinib, Panitumumab, Pelitinib, Rindopepimut, Vandetanib, Zalutumumab |
| MAPK signaling pathway | FGFR3 | 80.0 | 2.6 | Lenvatinib, Nintedanib, Pazopanib |
| MAPK signaling pathway | FGFR2 | 80.0 | 2.6 | Lenvatinib, Nintedanib, Regorafenib |
| MAPK signaling pathway | FGFR1 | 80.0 | 2.6 | Lenvatinib, Nintedanib, Regorafenib, Sorafenib |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Keam, B.; Park, J.-Y.; Kim, J.-P.; Kim, G.-D.; Yu, Y.-S.; Cho, S.-H.; Kim, S.; Ahn, H.-K.; Chun, S.-H.; Kwon, J.-H.; et al. Comprehensive Analysis of Mutation-Based and Expressed Genes-Based Pathways in Head and Neck Squamous Cell Carcinoma. Processes 2021, 9, 792. https://doi.org/10.3390/pr9050792
Keam B, Park J-Y, Kim J-P, Kim G-D, Yu Y-S, Cho S-H, Kim S, Ahn H-K, Chun S-H, Kwon J-H, et al. Comprehensive Analysis of Mutation-Based and Expressed Genes-Based Pathways in Head and Neck Squamous Cell Carcinoma. Processes. 2021; 9(5):792. https://doi.org/10.3390/pr9050792
Chicago/Turabian StyleKeam, Bhumsuk, Jin-Young Park, Jin-Pyo Kim, Gun-Do Kim, Yun-Suk Yu, Sang-Hee Cho, Sangwoo Kim, Hee-Kyung Ahn, Sang-Hoon Chun, Jung-Hye Kwon, and et al. 2021. "Comprehensive Analysis of Mutation-Based and Expressed Genes-Based Pathways in Head and Neck Squamous Cell Carcinoma" Processes 9, no. 5: 792. https://doi.org/10.3390/pr9050792
APA StyleKeam, B., Park, J.-Y., Kim, J.-P., Kim, G.-D., Yu, Y.-S., Cho, S.-H., Kim, S., Ahn, H.-K., Chun, S.-H., Kwon, J.-H., Yun, T., Kim, J.-W., Kim, J.-E., Ahn, M.-J., Kim, J.-H., & Yun, H.-J. (2021). Comprehensive Analysis of Mutation-Based and Expressed Genes-Based Pathways in Head and Neck Squamous Cell Carcinoma. Processes, 9(5), 792. https://doi.org/10.3390/pr9050792