
Modelling Cell Metabolism: A Review on Constraint-Based Steady-State and Kinetic Approaches



Abstract
1. Introduction
2. Topological Representation of Metabolic Networks
2.1. Representation Based on the Graph-Theory
2.2. Representation Based on the Petri net Theory
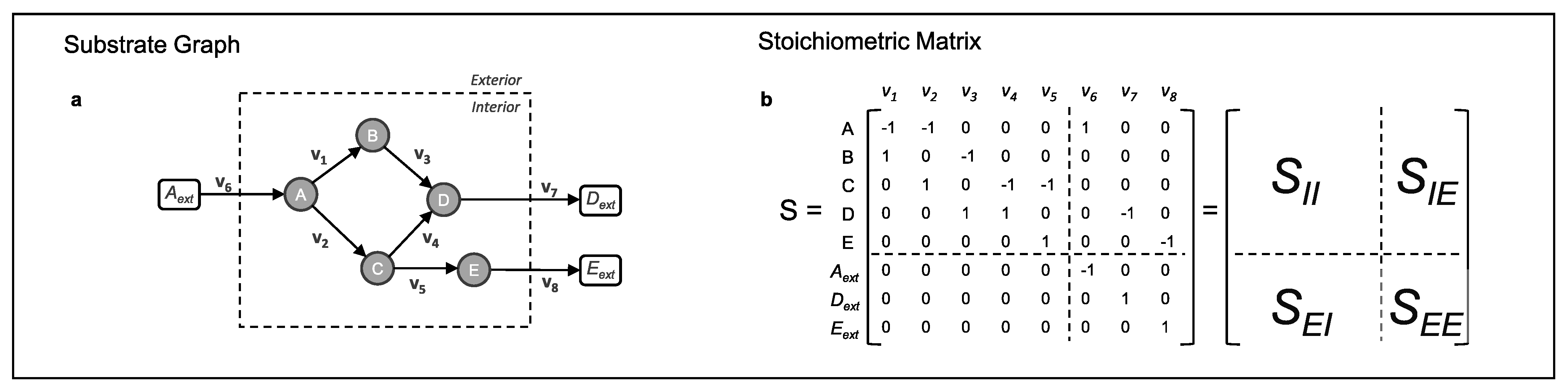
2.3. The Stoichiometric Matrix of a Metabolic Network
3. Metabolism Is a Constrained System
3.1. The Null Space of Stoichiometric Matrix
3.2. Metabolic Pathway Analysis (MPA)
3.3. Metabolic Flux Analysis (MFA): Admissible Metabolic Fluxes
13C-Metabolic Flux Analysis
4. Constraints Augmented with a Hypothesized Objective Function for the Cell
- To curate metabolic reactions from the annotated genome data.
- To identify the topology and the structure of a network.
- To identify the flux vector solution space at steady state.
- To impose constraints and bounds on fluxes.
- To define hypothesized objective function of interest for a biosystem.
- To realize the vector space of the solution and identify the optimum solution (i.e., the flux map).
5. Thermodynamics-Based Constraints
The general struggle for existence of animate beings is therefore not a struggle for raw materials, nor for energy, but a struggle for entropy which becomes available through the transition of heat from the hot sun to the cold earth.(Boltzman, 1886)
5.1. Estimation of the Gibbs Free Energy Change
Group Contribution Methods (GCMs)
5.2. Modelling Approaches Complying to the Thermodynamics-Based Constraints
- Assignment of thermodynamically feasible directions to all reactions in a network.
- Elimination of futile cycles (infeasible closed cycles).
- Consideration of thermodynamically coupled reactions.
5.3. Extensions of Constraint-Based Flux Balance Models Simulate Growth Dynamics
6. Kinetic Modelling
6.1. Approximate Kinetic Formats and the Quantification of Metabolic Regulation
6.1.1. Metabolic Control Analysis (MCA)
6.1.2. Biochemical System Theory (BST)
- In the domain where the underlying assumptions of a specific rate law approximation remain valid and not substantially violated throughout simulations;
- The rate laws are not the most important single factor in determination of dynamic behaviour of the network.
6.2. Regulated Kinetic Metabolic Models
6.3. Michaelis-Menten Kinetic Expression for Enzymatic Reactions
- Metabolic flux quantification: Carbon labelling experiments, uptake rate of nutrients, secretion rate of products.
- Enzyme concentrations measurement: MS/MS technique, absolute quantification by standard peptides or label-free methods in general, quantitative and qualitative proteomics.
- Kinetic parameters: database values for in vitro assays, existing models in literature with similar settings, estimation techniques.
- Substrate concentrations: Metabolomics.
6.4. Convenience Kinetics (CK)
6.5. Cybernetic Metabolic Models
7. Parameter Estimation Formulation
- Objective function formulation: enhancements can be introduced by collection of the data in several duplicates to then adjust the relative weight of the error in states experimental values.
- Constraints definition: bounds on parameter values should be introduced to keep them in the feasible biological ranges.
- Algorithm and solver selection: nature of the optimization problem and available data play a significant role in this step.
- Optimization option assignment: a set of options must be assigned for the solver. These options are descriptive of optimization algorithm’s details and accuracy.
- To impose a reference state and estimate the biosystem parameters around this state [221].
- To introduce thermodynamic limitations and therefore limit the parameter solution space [222].
- To introduce local stability constraints [223].
- To reduce the model directly through the model reduction techniques (listed in [146]).
8. Perspectives and Challenges Ahead
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- DiStefano, J.D., III. Dynamic Systems Biology Modeling and Simulation, 1st ed.; Academic Press: Cambridge, MA, USA, 2013; p. 884. [Google Scholar]
- Ingalls, B.P. Mathematical Modeling in Systems Biology: An Introduction; The MIT Press: Cambridge, MA, USA, 2013; p. 424. [Google Scholar]
- Palsson, B.O. Systems Biology: Properties of Reconstructed Networks; Cambridge University Press: Cambridge, UK, 2006. [Google Scholar]
- Jolicoeur, M. Modeling cell behavior: Moving beyond intuition. AIMS Bioeng. 2014, 1, 1–12. [Google Scholar] [CrossRef]
- Zhou, W.; Rehm, J.; Europa, A.; Hu, W.S. Alteration of mammalian cell metabolism by dynamic nutrient feeding. Cytotechnology 1997, 24, 99–108. [Google Scholar] [CrossRef] [PubMed]
- Reinhart Heinrich, S.S. The Regulation of Cellular Systems; Springer: Berlin/Heidelberg, Germany, 1996. [Google Scholar]
- Liebermeister, W.; Klipp, E. Bringing metabolic networks to life: Convenience rate law and thermodynamic constraints. Theor. Biol. Med Model. 2006, 3, 41. [Google Scholar] [CrossRef] [PubMed]
- Du, B.; Zielinski, D.C.; Kavvas, E.S.; Drager, A.; Tan, J.; Zhang, Z.; Ruggiero, K.E.; Arzumanyan, G.A.; Palsson, B.O. Evaluation of rate law approximations in bottom-up kinetic models of metabolism. BMC Syst. Biol. 2016, 10, 40. [Google Scholar] [CrossRef]
- Schilling, C.H.; Schuster, S.; Palsson, B.O.; Heinrich, R. Metabolic pathway analysis: Basic concepts and scientific applications in the post-genomic era. Biotechnol. Prog. 1999, 15, 296–303. [Google Scholar] [CrossRef]
- Ghorbaniaghdam, A.; Henry, O.; Jolicoeur, M. A kinetic-metabolic model based on cell energetic state: Study of CHO cell behavior under Na-butyrate stimulation. Bioprocess Biosyst. Eng. 2013, 36, 469–487. [Google Scholar] [CrossRef]
- Goffaux, G.; Hammami, I.; Jolicoeur, M. A Dynamic Metabolic Flux Analysis of Myeloid-Derived Suppressor Cells Confirms Immunosuppression-Related Metabolic Plasticity. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef]
- Calmels, C.; McCann, A.; Malphettes, L.; Andersen, M.R. Application of a curated genome-scale metabolic model of CHO DG44 to an industrial fed-batch process. Metab. Eng. 2019, 51, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Hefzi, H.; Ang, K.S.; Hanscho, M.; Bordbar, A.; Ruckerbauer, D.; Lakshmanan, M.; Orellana, C.A.; Baycin-Hizal, D.; Huang, Y.; Ley, D.; et al. A Consensus Genome-scale Reconstruction of Chinese Hamster Ovary Cell Metabolism. Cell Syst. 2016, 3. [Google Scholar] [CrossRef]
- Da Veiga Moreira, J.; Hamraz, M.; Abolhassani, M.; Schwartz, L.; Jolicoeur, M.; Peres, S. Metabolic therapies inhibit tumor growth in vivo and in silico. Sci. Rep. 2019, 9, 3153. [Google Scholar] [CrossRef]
- Katzir, R.; Polat, I.H.; Harel, M.; Katz, S.; Foguet, C.; Selivanov, V.A.; Sabatier, P.; Cascante, M.; Geiger, T.; Ruppin, E. The landscape of tiered regulation of breast cancer cell metabolism. Sci. Rep. 2019, 9, 17760. [Google Scholar] [CrossRef] [PubMed]
- Thiele, I.; Hyduke, D.R.; Steeb, B.; Fankam, G.; Allen, D.K.; Bazzani, S.; Charusanti, P.; Chen, F.C.; Fleming, R.M.; Hsiung, C.A.; et al. A community effort towards a knowledge-base and mathematical model of the human pathogen Salmonella Typhimurium LT2. BMC Syst. Biol. 2011, 5, 8. [Google Scholar] [CrossRef]
- Bailey, J.E. Mathematical modeling and analysis in biochemical engineering: Past accomplishments and future opportunities. Biotechnol. Prog. 1998, 14, 8–20. [Google Scholar] [CrossRef]
- Cloutier, M.; Chen, J.; De Dobbeleer, C.; Perrier, M.; Jolicoeur, M. A systems approach to plant bioprocess optimization. Plant Biotechnol. J. 2009, 7, 939–951. [Google Scholar] [CrossRef]
- Montegut, L.; Martinez-Basilio, P.C.; da Veiga Moreira, J.; Schwartz, L.; Jolicoeur, M. Combining lipoic acid to methylene blue reduces the Warburg effect in CHO cells: From TCA cycle activation to enhancing monoclonal antibody production. PLoS ONE 2020, 15, e0231770. [Google Scholar] [CrossRef]
- Fredrickson, A.G.; Megee, R.D.; Tsuchiya, H.M. Mathematical Models for Fermentation Processes**During preparation of this review, the authors were supported in part by USDA Grant No. 12-14-100-9178 and USDPH Grant No. GM 16692. In Advances in Applied Microbiology; Academic Press: Cambridge, MA, USA, 1970; Volume 13, pp. 419–465. [Google Scholar] [CrossRef]
- Jeong, H.; Tombor, B.; Albert, R.; Oltvai, Z.N.; Barabási, A.L. The large-scale organization of metabolic networks. Nature 2000, 407, 651–654. [Google Scholar] [CrossRef]
- Karp, P.D.; Krummenacker, M.; Paley, S.; Wagg, J. Integrated pathway-genome databases and their role in drug discovery. Trends Biotechnol. 1999, 17, 275–281. [Google Scholar] [CrossRef]
- Ma, H.; Zeng, A.P. Reconstruction of metabolic networks from genome data and analysis of their global structure for various organisms. Bioinformatics 2003, 19, 270–277. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.W.; Zeng, A.P. The connectivity structure, giant strong component and centrality of metabolic networks. Bioinformatics 2003, 19, 1423–1430. [Google Scholar] [CrossRef] [PubMed]
- Fell, D.A.; Wagner, A. The small world of metabolism. Nat. Biotechnol. 2000, 18, 1121–1122. [Google Scholar] [CrossRef]
- Wagner, A.; Fell, D.A. The small world inside large metabolic networks. Proc. R. Soc. London. Ser. B Biol. Sci. 2001, 268, 1803–1810. [Google Scholar] [CrossRef] [PubMed]
- Koschützki, D.; Schwöbbermeyer, H.; Schreiber, F. Ranking of network elements based on functional substructures. J. Theor. Biol. 2007, 248, 471–479. [Google Scholar] [CrossRef] [PubMed]
- Meiss, M.R.; Menczer, F.; Vespignani, A. Structural analysis of behavioral networks from the Internet. J. Phys. A Math. Theor. 1993, 41, 224022. [Google Scholar] [CrossRef]
- Edwards, J.S.; Palsson, B.O. The Escherichia coli MG1655 in silico metabolic genotype: Its definition, characteristics, and capabilities. Proc. Natl. Acad. Sci. USA 2000, 97, 5528–5533. [Google Scholar] [CrossRef]
- Koschützki, D.; Schreiber, F. Centrality analysis methods for biological networks and their application to gene regulatory networks. Gene Regul. Syst. Biol. 2008, 2, 193. [Google Scholar] [CrossRef] [PubMed]
- Wuchty, S.; Stadler, P.F. Centers of complex networks. J. Theor. Biol. 2003, 223, 45–53. [Google Scholar] [CrossRef]
- Reddy, V.N.; Mavrovouniotis, M.L.; Liebman, M.N. Petri Net Representations in Metabolic Pathways. In Proceedings of the 1st International Conference on Intelligent Systems for Molecular Biology, Bethesda, MD, USA, 6–9 July 1993; Volume 93, pp. 328–336. [Google Scholar]
- Chen, M.; Hu, M.; Hofestadt, R. A systematic petri net approach for multiple-scale modeling and simulation of biochemical processes. Appl. Biochem. Biotechnol. 2011, 164, 338–352. [Google Scholar] [CrossRef] [PubMed]
- Zevedei-Oancea, I.; Schuster, S. Topological analysis of metabolic networks based on petri net theory. Stud. Health Technol. Inf. 2011, 162, 17–37. [Google Scholar]
- Chaouiya, C. Petri net modelling of biological networks. Briefings Bioinform. 2007, 8, 210–219. [Google Scholar] [CrossRef]
- Marsan, M.A.; Balbo, G.; Conte, G.; Donatelli, S.; Franceschinis, G. Modelling with Generalized Stochastic Petri Nets. ACM Sigmetrics Perform. Eval. Rev. 1998, 26. [Google Scholar] [CrossRef]
- Goss, P.J.E.; Peccoud, J. Quantitative modeling of stochastic systems in molecular biology by using stochastic Petri nets. Proc. Natl. Acad. Sci. USA 1998, 95, 6750. [Google Scholar] [CrossRef]
- Hofestadt, R.; Thelen, S. Quantitative modeling of biochemical networks. In Silico Biol. 1998, 1, 39–53. [Google Scholar] [PubMed]
- Valk, R. Self-Modifying Nets, a Natural Extension of Petri Nets; Automata, Languages and Programming; Ausiello, G., Böhm, C., Eds.; Springer: Berlin/Heidelberg, Germany, 1978; pp. 464–476. [Google Scholar]
- Varma, A.; Palsson, B.O. Metabolic flux balancing: Basic concepts, scientific and practical use. Nat. Biotechnol. 1994, 12, 994–998. [Google Scholar] [CrossRef]
- Ravasz, E.; Somera, A.L.; Mongru, D.A.; Oltvai, Z.N.; Barabási, A.L. Hierarchical organization of modularity in metabolic networks. Science 2002, 297, 1551–1555. [Google Scholar] [CrossRef] [PubMed]
- Bilke, S.; Peterson, C. Topological properties of citation and metabolic networks. Phys. Rev. E 2001, 64, 036106. [Google Scholar] [CrossRef] [PubMed]
- Fell, D.A.; Small, J.R. Fat synthesis in adipose tissue. An examination of stoichiometric constraints. Biochem. J. 1986, 238, 781–786. [Google Scholar] [CrossRef]
- Majewski, R.; Domach, M. Simple constrained-optimization view of acetate overflow in E. coli. Biotechnol. Bioeng. 1990, 35, 732–738. [Google Scholar] [CrossRef]
- Papoutsakis, E.T. Equations and calculations for fermentations of butyric acid bacteria. Biotechnol. Bioeng. 1984, 26, 174–187. [Google Scholar] [CrossRef]
- Savinell, J.M.; Palsson, B.O. Network analysis of intermediary metabolism using linear optimization. I. Development of mathematical formalism. J. Theor. Biol. 1992, 154, 421–454. [Google Scholar] [CrossRef]
- Fernandes, S.; Robitaille, J.; Bastin, G.; Jolicoeur, M.; Wouwer, A.V. Dynamic metabolic flux analysis of underdetermined and overdetermined metabolic networks. IFAC PapersOnLine 2016, 49, 318–323. [Google Scholar] [CrossRef]
- Zamorano, F.; Wouwer, A.V.; Bastin, G. A detailed metabolic flux analysis of an underdetermined network of CHO cells. J. Biotechnol. 2010, 150, 497–508. [Google Scholar] [CrossRef]
- Robert, W.L.; Maciek, R.A. Dynamic metabolic flux analysis (DMFA): A framework for determining fluxes at metabolic non-steady state. Metab. Eng. 2011, 13, 745–755. [Google Scholar] [CrossRef]
- Ahn, W.S.; Antoniewicz, M.R. Towards dynamic metabolic flux analysis in CHO cell cultures. Biotechnol. J. 2012, 7, 61–74. [Google Scholar] [CrossRef] [PubMed]
- Niklas, J.; Heinzle, E. Metabolic flux analysis in systems biology of mammalian cells. Adv. Biochem. Eng. 2012, 127, 109–132. [Google Scholar] [CrossRef]
- Ahn, W.S.; Antoniewicz, M.R. Metabolic flux analysis of CHO cells at growth and non-growth phases using isotopic tracers and mass spectrometry. Metab. Eng. 2011, 13, 598–609. [Google Scholar] [CrossRef] [PubMed]
- Wagner, C.; Urbanczik, R. The geometry of the flux cone of a metabolic network. Biophys. J. 2005, 89, 3837–3845. [Google Scholar] [CrossRef]
- Schauer, M.; Heinrich, R. Quasi-steady-state approximation in the mathematical modeling of biochemical reaction networks. Math. Biosci. 1983, 65, 155–170. [Google Scholar] [CrossRef]
- Stephanopoulos, G.; Aristidou, A.A.; Nielsen, J. Metabolic Engineering: Principles and Methodologies; Academic Press: Cambridge, MA, USA, 1998. [Google Scholar]
- Niklas, J.; Schräder, E.; Sandig, V.; Noll, T.; Heinzle, E. Quantitative characterization of metabolism and metabolic shifts during growth of the new human cell line AGE1.HN using time resolved metabolic flux analysis. Bioprocess Biosyst. Eng. 2011, 34, 533–545. [Google Scholar] [CrossRef] [PubMed]
- Smallbone, K.; Simeonidis, E.; Broomhead, D.S.; Kell, D.B. Something from nothing: Bridging the gap between constraint-based and kinetic modelling. FEBS J. 2007, 274, 5576–5585. [Google Scholar] [CrossRef]
- Famili, I.; Palsson, B.O. The Convex Basis of the Left Null Space of the Stoichiometric Matrix Leads to the Definition of Metabolically Meaningful Pools. Biophys. J. 2003, 85, 16–26. [Google Scholar] [CrossRef]
- Llaneras, F.; Pico, J. Which Metabolic Pathways Generate and Characterize the Flux Space? A Comparison among Elementary Modes, Extreme Pathways and Minimal Generators. J. Biomed. Biotechnol. 2010. [Google Scholar] [CrossRef] [PubMed]
- Klamt, S.; Regensburger, G.; Gerstl, M.P.; Jungreuthmayer, C.; Schuster, S.; Mahadevan, R.; Zanghellini, J.; Müller, S. From elementary flux modes to elementary flux vectors: Metabolic pathway analysis with arbitrary linear flux constraints. PLoS Comput. Biol. 2017, 13, e1005409. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, S.; Cluett, W.R.; Mahadevan, R. Constructing kinetic models of metabolism at genome-scales: A review. Biotechnol. J. 2015, 10, 1345–1359. [Google Scholar] [CrossRef] [PubMed]
- Kitano, H. Biological robustness. Nat. Rev. Genet. 2004, 5, 826–837. [Google Scholar] [CrossRef] [PubMed]
- Schuster, S.; Hilgetag, C. On Elementary Flux Modes in Biochemical Reaction Systems at Steady State. J. Biol. Syst. 1994, 2, 165–182. [Google Scholar] [CrossRef]
- Schuster, S.; Fell, D.A.; Dandekar, T. A general definition of metabolic pathways useful for systematic organization and analysis of complex metabolic networks. Nat. Biotechnol. 2000, 18, 326–332. [Google Scholar] [CrossRef] [PubMed]
- Schuster, S.; Hilgetag, C.; Woods, J.H.; Fell, D.A. Reaction routes in biochemical reaction systems: Algebraic properties, validated calculation procedure and example from nucleotide metabolism. J. Math. Biol. 2002, 45, 153–181. [Google Scholar] [CrossRef]
- Trinh, C.T.; Wlaschin, A.; Srienc, F. Elementary mode analysis: A useful metabolic pathway analysis tool for characterizing cellular metabolism. Appl. Microbiol. Biotechnol. 2009, 81, 813–826. [Google Scholar] [CrossRef]
- Henry, C.S.; Broadbelt, L.J.; Hatzimanikatis, V. Discovery and analysis of novel metabolic pathways for the biosynthesis of industrial chemicals: 3-hydroxypropanoate. Biotechnol. Bioeng. 2010. [Google Scholar] [CrossRef]
- von Kamp, A.; Klamt, S. Growth-coupled overproduction is feasible for almost all metabolites in five major production organisms. Nat. Commun. 2017, 8, 15956. [Google Scholar] [CrossRef]
- Pfeiffer, T.; Sanchez, V.; Nuno, J.; Montero, F.; Schuster, S. METATOOL: For studying metabolic networks. Bioinformatics 1999, 15, 251–257. [Google Scholar] [CrossRef] [PubMed]
- Zanghellini, J.; Ruckerbauer, D.E.; Hanscho, M.; Jungreuthmayer, C. Elementary flux modes in a nutshell: Properties, calculation and applications. Biotechnol. J. 2013, 8, 1009–1016. [Google Scholar] [CrossRef]
- Peres, S.; Schuster, S.; Dague, P. Thermodynamic constraints for identifying elementary flux modes. Biochem Soc Trans 2018, 46, 641–647. [Google Scholar] [CrossRef] [PubMed]
- Urbanczik, R. Enumerating constrained elementary flux vectors of metabolic networks. IET Syst. Biol. 2007, 1, 274–279. [Google Scholar] [CrossRef]
- Papin, J.; Stelling, J.; Price, N.; Klamt, S.; Schuster, S.; Palsson, B. Comparison of network-based pathway analysis methods. Trends Biotechnol. 2004, 22, 400–405. [Google Scholar] [CrossRef]
- Papin, J.A.; Price, N.D.; Wiback, S.J.; Fell, D.A.; Palsson, B.O. Metabolic pathways in the post-genome era. Trends Biochem. Sci. 2003, 28, 250–258. [Google Scholar] [CrossRef]
- Gagneur, J.; Klamt, S. Computation of elementary modes: A unifying framework and the new binary approach. BMC Bioinform. 2004, 5, 175. [Google Scholar] [CrossRef]
- Klamt, S.; Stelling, J. Combinatorial complexity of pathway analysis in metabolic networks. Mol. Biol. Rep. 2002, 29, 233–236. [Google Scholar] [CrossRef] [PubMed]
- Klamt, S.; Stelling, J. Two approaches for metabolic pathway analysis? Trends Biotechnol. 2003, 21, 64–69. [Google Scholar] [CrossRef]
- Klamt, S.; Gilles, E.D. Minimal cut sets in biochemical reaction networks. Bioinformatics 2004, 20, 226–234. [Google Scholar] [CrossRef]
- Hadicke, O.; Klamt, S. Computing complex metabolic intervention strategies using constrained minimal cut sets. Metab. Eng. 2011, 13, 204–213. [Google Scholar] [CrossRef] [PubMed]
- Schilling, C.H.; Palsson, B.Ø. Assessment of the Metabolic Capabilities of Haemophilus influenzae Rd through a Genome-scale Pathway Analysis. J. Theor. Biol. 2000, 203, 249–283. [Google Scholar] [CrossRef]
- Carlson, R.; Srienc, F. Fundamental Escherichia coli biochemical pathways for biomass and energy production: Identification of reactions. Biotechnol. Bioeng. 2004, 85, 1–19. [Google Scholar] [CrossRef]
- Trinh, C.T.; Thompson, R.A. Elementary mode analysis: A useful metabolic pathway analysis tool for reprograming microbial metabolic pathways. In Reprogramming Microbial Metabolic Pathways; Springer: Berlin/Heidelberg, Germany, 2012; pp. 21–42. [Google Scholar]
- Banerjee, D.; Eng, T.; Lau, A.K.; Sasaki, Y.; Wang, B.; Chen, Y.; Prahl, J.P.; Singan, V.R.; Herbert, R.A.; Liu, Y.; et al. Genome-scale metabolic rewiring improves titers rates and yields of the non-native product indigoidine at scale. Nat. Commun. 2020, 11, 5385. [Google Scholar] [CrossRef]
- Vernardis, S.I.; Goudar, C.T.; Klapa, M.I. Metabolic profiling reveals that time related physiological changes in mammalian cell perfusion cultures are bioreactor scale independent. Metab. Eng. 2013, 19, 1–9. [Google Scholar] [CrossRef]
- Sheikholeslami, Z.; Jolicoeur, M.; Henry, O. Elucidating the effects of postinduction glutamine feeding on the growth and productivity of CHO cells. Biotechnol. Prog. 2014, 30, 535–546. [Google Scholar] [CrossRef]
- Sheikholeslami, Z.; Jolicoeur, M.; Henry, O. The impact of the timing of induction on the metabolism and productivity of CHO cells in culture. Biochem. Eng. J. 2013, 79, 162–171. [Google Scholar] [CrossRef]
- Orth, J.D.; Thiele, I.; Palsson, B.Ø. What is flux balance analysis? Nat. Biotechnol. 2010, 28, 245–248. [Google Scholar] [CrossRef] [PubMed]
- Kruger, N.J.; Ratcliffe, R.G. Fluxes through plant metabolic networks: Measurements, predictions, insights and challenges. Biochem. J. 2015, 465, 27–38. [Google Scholar] [CrossRef] [PubMed]
- Raman, K.; Chandra, N. Flux balance analysis of biological systems: Applications and challenges. Briefings Bioinform. 2009, 10, 435–449. [Google Scholar] [CrossRef] [PubMed]
- Antoniewicz, M.R. A guide to metabolic flux analysis in metabolic engineering: Methods, tools and applications. Metab. Eng. 2020. [Google Scholar] [CrossRef] [PubMed]
- Schügerl, K.; Bellgardt, K.H. Bioreaction Engineering—Modeling and Control; Springer: Berlin/Heidelberg, Germany, 2000. [Google Scholar] [CrossRef]
- Villadsen, J.; Nielsen, J.; Lidén, G. Bioreaction Engineering Principles, 3rd ed.; Springer: Berlin/Heidelberg, Germany, 2011. [Google Scholar] [CrossRef]
- Bonarius, H.P.J.; Hatzimanikatis, V.; Meesters, K.P.H.; de Gooijer, C.D.; Schmid, G.; Tramper, J. Metabolic flux analysis of hybridoma cells in different culture media using mass balances. Biotechnol. Bioeng. 1996, 50, 299–318. [Google Scholar] [CrossRef]
- Klapa, M.I.; Park, S.M.; Sinskey, A.J.; Stephanopoulos, G. Metabolite and isotopomer balancing in the analysis of metabolic cycles: I. Theory. Biotechnol. Bioeng. 1999, 62, 375–391. [Google Scholar] [CrossRef]
- Park, S.M.; Klapa, M.I.; Sinskey, A.J.; Stephanopoulos, G. Metabolite and isotopomer balancing in the analysis of metabolic cycles: II. Applications. Biotechnol. Bioeng. 1999, 62, 392–401. [Google Scholar] [CrossRef]
- Varma, A.; Palsson, B.O. Stoichiometric flux balance models quantitatively predict growth and metabolic by-product secretion in wild-type Escherichia coli W3110. Appl. Environ. Microbiol. 1994, 60, 3724–3731. [Google Scholar] [CrossRef] [PubMed]
- Antoniewicz, M.R.; Kelleher, J.K.; Stephanopoulos, G. Determination of confidence intervals of metabolic fluxes estimated from stable isotope measurements. Metab. Eng. 2006, 8, 324–337. [Google Scholar] [CrossRef] [PubMed]
- Zupke, C.; Stephanopoulos, G. Intracellular flux analysis in hybridomas using mass balances and in vitro 13C nmr. Biotechnol. Bioeng. 1995, 45, 292–303. [Google Scholar] [CrossRef]
- Antoniewicz, M.R.; Kelleher, J.K.; Stephanopoulos, G. Elementary metabolite units (EMU): A novel framework for modeling isotopic distributions. Metab. Eng. 2007, 9. [Google Scholar] [CrossRef]
- Antoniewicz, M.R. Parallel labeling experiments for pathway elucidation and 13C metabolic flux analysis. Curr. Opin. Biotechnol. 2015, 36, 91–97. [Google Scholar] [CrossRef]
- Szyperski, T. Biosynthetically Directed Fractional 13C-labeling of Proteinogenic Amino Acids. Eur. J. Biochem. 1995, 232, 433–448. [Google Scholar] [CrossRef]
- Long, C.P.; Antoniewicz, M.R. High-resolution (13)C metabolic flux analysis. Nat. Protoc. 2019, 14, 2856–2877. [Google Scholar] [CrossRef]
- Spagou, K.; Theodoridis, G.; Wilson, I.; Raikos, N.; Greaves, P.; Edwards, R.; Nolan, B.; Klapa, M.I. A GC-MS metabolic profiling study of plasma samples from mice on low- and high-fat diets. J. Chromatogr. B 2011, 879, 1467–1475. [Google Scholar] [CrossRef]
- Kanani, H.; Chrysanthopoulos, P.K.; Klapa, M.I. Standardizing GC-MS metabolomics. J. Chromatogr. B 2008, 871, 191–201. [Google Scholar] [CrossRef]
- Kanani, H.H.; Klapa, M.I. Data correction strategy for metabolomics analysis using gas chromatography-mass spectrometry. Metab. Eng. 2007, 9, 39–51. [Google Scholar] [CrossRef] [PubMed]
- Papadimitropoulos, M.P.; Vasilopoulou, C.G.; Maga-Nteve, C.; Klapa, M.I. Untargeted GC-MS Metabolomics. Methods Mol. Biol. 2018, 1738, 133–147. [Google Scholar] [CrossRef]
- Choi, J.; Antoniewicz, M.R. Tandem mass spectrometry: A novel approach for metabolic flux analysis. Metab. Eng. 2011, 13, 225–233. [Google Scholar] [CrossRef] [PubMed]
- Millard, P.; Schmitt, U.; Kiefer, P.; Vorholt, J.A.; Heux, S.; Portais, J.C. ScalaFlux: A scalable approach to quantify fluxes in metabolic subnetworks. PLoS Comput. Biol. 2020, 16, e1007799. [Google Scholar] [CrossRef]
- Edwards, J.S.; Palsson, B.O. How will bioinformatics influence metabolic engineering? Biotechnol. Bioeng. 1998, 58, 162–169. [Google Scholar] [CrossRef]
- Jol, S.J.; Kümmel, A.; Terzer, M.; Stelling, J.; Heinemann, M. System-Level Insights into Yeast Metabolism by Thermodynamic Analysis of Elementary Flux Modes. PLoS Comput. Biol. 2012, 8, e1002415. [Google Scholar] [CrossRef] [PubMed]
- Covert, M.W.; Schilling, C.H.; Famili, I.; Edwards, J.S.; Goryanin, I.; Selkov, E.; Palsson, B.O. Metabolic modeling of microbial strains in silico. Trends Biochem. Sci. 2001, 26, 179–186. [Google Scholar] [CrossRef]
- Lewis, N.E.; Hixson, K.K.; Conrad, T.M.; Lerman, J.A.; Charusanti, P.; Polpitiya, A.D.; Adkins, J.N.; Schramm, G.; Purvine, S.O.; Lopez-Ferrer, D.; et al. Omic data from evolved E. coli are consistent with computed optimal growth from genome-scale models. Mol. Syst. Biol. 2010, 6, 390. [Google Scholar] [CrossRef] [PubMed]
- Kibele, A.; Granacher, U.; Muehlbauer, T.; Behm, D.G. Stable, Unstable and Metastable States of Equilibrium: Definitions and Applications to Human Movement. J. Sport. Sci. Med. 2015, 14, 885–887. [Google Scholar]
- Qian, H.; Beard, D.A.; Liang, S.D. Stoichiometric network theory for nonequilibrium biochemical systems. Eur. J. Biochem. 2003, 270, 415–421. [Google Scholar] [CrossRef]
- Demirel, Y.; Sandler, S.I. Thermodynamics and bioenergetics. Biophys. Chem. 2002, 97, 87–111. [Google Scholar] [CrossRef]
- Schoepp-Cothenet, B.; van Lis, R.; Atteia, A.; Baymann, F.; Capowiez, L.; Ducluzeau, A.L.; Duval, S.; ten Brink, F.; Russell, M.J.; Nitschke, W. On the universal core of bioenergetics. Biochim. Biophys. Acta 2013, 1827, 79–93. [Google Scholar] [CrossRef] [PubMed]
- Branscomb, E.; Russell, M.J. Turnstiles and bifurcators: The disequilibrium converting engines that put metabolism on the road. Biochim. Biophys. Acta 2013, 1827, 62–78. [Google Scholar] [CrossRef]
- Qian, H. Entropy production and excess entropy in a nonequilibrium steady-state of single macromolecules. Phys. Rev. E 2002, 65, 021111. [Google Scholar] [CrossRef]
- Ataman, M.; Hatzimanikatis, V. Heading in the right direction: Thermodynamics-based network analysis and pathway engineering. Curr. Opin. Biotechnol. 2015, 36, 176–182. [Google Scholar] [CrossRef]
- Jankowski, M.D.; Henry, C.S.; Broadbelt, L.J.; Hatzimanikatis, V. Group contribution method for thermodynamic analysis of complex metabolic networks. Biophys. J. 2008, 95, 1487–1499. [Google Scholar] [CrossRef] [PubMed]
- Mavrovouniotis, M.L. Group contributions for estimating standard gibbs energies of formation of biochemical compounds in aqueous solution. Biotechnol. Bioeng. 1990, 36, 1070–1082. [Google Scholar] [CrossRef]
- Du, B.; Zielinski, D.C.; Palsson, B.O. Estimating Metabolic Equilibrium Constants: Progress and Future Challenges. Trends Biochem. Sci. 2018, 43, 960–969. [Google Scholar] [CrossRef] [PubMed]
- Alberty, R.A. Thermodynamics of Biochemical Reactions; John Wiley & Sons: Hoboken, NJ, USA, 2003. [Google Scholar] [CrossRef]
- Alberty, R.A. Calculation of Standard Transformed Formation Properties of Biochemical Reactants and Standard Apparent Reduction Potentials of Half Reactions. Arch. Biochem. Biophys. 1998, 358, 25–39. [Google Scholar] [CrossRef] [PubMed]
- Thauer, R.K. Biochemistry of methanogenesis: A tribute to Marjory Stephenson. 1998 Marjory Stephenson Prize Lecture. Microbiology 1998, 144 Pt 9, 2377–2406. [Google Scholar] [CrossRef]
- Thauer, R.K.; Jungermann, K.; Decker, K. Energy conservation in chemotrophic anaerobic bacteria. Bacteriol. Rev. 1977, 41, 100–180. [Google Scholar] [CrossRef]
- Dolfing, J.; Harrison, B.K. Gibbs free energy of formation of halogenated aromatic compounds and their potential role as electron acceptors in anaerobic environments. Environ. Sci. Technol. 1992, 26, 2213–2218. [Google Scholar] [CrossRef]
- Dolfing, J.; Janssen, D.B. Estimates of Gibbs free energies of formation of chlorinated aliphatic compounds. Biodegradation 1994, 5, 21–28. [Google Scholar] [CrossRef]
- Henry, C.S.; Broadbelt, L.J.; Hatzimanikatis, V. Thermodynamics-based metabolic flux analysis. Biophys. J. 2007, 92, 1792–1805. [Google Scholar] [CrossRef]
- Alberty, R.A. Equilibrium compositions of solutions of biochemical species and heats of biochemical reactions. Proc. Natl. Acad. Sci. USA 1991, 88, 3268–3271. [Google Scholar] [CrossRef] [PubMed]
- Zerfass, C.; Asally, M.; Soyer, O.S. Interrogating metabolism as an electron flow system. Curr. Opin. Syst. Biol. 2019, 13, 59–67. [Google Scholar] [CrossRef]
- Hamilton, J.J.; Dwivedi, V.; Reed, J.L. Quantitative assessment of thermodynamic constraints on the solution space of genome-scale metabolic models. Biophys. J. 2013, 105, 512–522. [Google Scholar] [CrossRef]
- Du, B.; Zhang, Z.; Grubner, S.; Yurkovich, J.T.; Palsson, B.O.; Zielinski, D.C. Temperature-Dependent Estimation of Gibbs Energies Using an Updated Group-Contribution Method. Biophys. J. 2018, 114, 2691–2702. [Google Scholar] [CrossRef]
- Noor, E.; Bar-Even, A.; Flamholz, A.; Lubling, Y.; Davidi, D.; Milo, R. An integrated open framework for thermodynamics of reactions that combines accuracy and coverage. Bioinformatics 2012, 28, 2037–2044. [Google Scholar] [CrossRef] [PubMed]
- Beard, D.A.; Liang, S.d.; Qian, H. Energy Balance for Analysis of Complex Metabolic Networks. Biophys. J. 2002, 83, 79–86. [Google Scholar] [CrossRef]
- Kummel, A.; Panke, S.; Heinemann, M. Putative regulatory sites unraveled by network-embedded thermodynamic analysis of metabolome data. Mol. Syst. Biol. 2006, 2, 2006.0034. [Google Scholar] [CrossRef]
- Reed, J.L.; Vo, T.D.; Schilling, C.H.; Palsson, B.O. An expanded genome-scale model of Escherichia coli K-12 (iJR904 GSM/GPR). Genome Biol. 2003, 4, R54. [Google Scholar] [CrossRef] [PubMed]
- Peres, S.; Jolicoeur, M.; Moulin, C.; Dague, P.; Schuster, S. How important is thermodynamics for identifying elementary flux modes? PLoS ONE 2017, 12, e0171440. [Google Scholar] [CrossRef]
- Gerstl, M.P.; Jungreuthmayer, C.; Zanghellini, J. tEFMA: Computing thermodynamically feasible elementary flux modes in metabolic networks. Bioinformatics 2015, 31, 2232–2234. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Ley, D.; Andersen, M.R. Fed-Batch CHO Cell Culture for Lab-Scale Antibody Production. Methods Mol. Biol. 2018, 1674, 147–161. [Google Scholar] [CrossRef]
- Mahadevan, R.; Edwards, J.S.; Doyle, F.J. Dynamic flux balance analysis of diauxic growth in Escherichia coli. Biophys. J. 2002, 83, 1331–1340. [Google Scholar] [CrossRef]
- Kyriakopoulos, S.; Ang, K.S.; Lakshmanan, M.; Huang, Z.; Yoon, S.; Gunawan, R.; Lee, D.Y. Kinetic Modeling of Mammalian Cell Culture Bioprocessing: The Quest to Advance Biomanufacturing. Biotechnol. J. 2018, 13, 1700229. [Google Scholar] [CrossRef]
- Sauro, H.M. Enzyme Kinetics for Systems Biology; Ambrosius Publishing: Leipzig, Germany, 2011. [Google Scholar]
- Liu, L.; Bockmayr, A. Formalizing Metabolic-Regulatory Networks by Hybrid Automata. Acta Biotheor. 2020, 68, 73–85. [Google Scholar] [CrossRef]
- Liu, L.; Bockmayr, A. Regulatory dynamic enzyme-cost flux balance analysis: A unifying framework for constraint-based modeling. J. Theor. Biol. 2020, 501, 110317. [Google Scholar] [CrossRef]
- Strutz, J.; Martin, J.; Greene, J.; Broadbelt, L.; Tyo, K. Metabolic kinetic modeling provides insight into complex biological questions, but hurdles remain. Curr. Opin. Biotechnol. 2019, 59, 24–30. [Google Scholar] [CrossRef]
- Ederer, M.; Gilles, E.D. Thermodynamically Feasible Kinetic Models of Reaction Networks. Biophys. J. 2007, 92, 1846–1857. [Google Scholar] [CrossRef]
- Cloutier, M.; Chen, J.; Tatge, F.; McMurray-Beaulieu, V.; Perrier, M.; Jolicoeur, M. Kinetic metabolic modelling for the control of plant cells cytoplasmic phosphate. J. Theor. Biol. 2009, 259, 118–131. [Google Scholar] [CrossRef] [PubMed]
- Julien, R.; Jingkui, C.; Mario, J.; Néstor, V.T. A Single Dynamic Metabolic Model Can Describe mAb Producing CHO Cell Batch and Fed-Batch Cultures on Different Culture Media. PLoS ONE 2015, 10, e0136815. [Google Scholar] [CrossRef]
- Henry, O.; Kamen, A.; Perrier, M. Monitoring the physiological state of mammalian cell perfusion processes by on-line estimation of intracellular fluxes. J. Process Control 2007, 17, 241–251. [Google Scholar] [CrossRef]
- Atefeh, G.; Olivier, H.; Mario, J. An in-silico study of the regulation of CHO cells glycolysis. J. Theor. Biol. 2014, 357, 112–122. [Google Scholar] [CrossRef]
- Almquist, J.; Cvijovic, M.; Hatzimanikatis, V.; Nielsen, J.; Jirstrand, M. Kinetic models in industrial biotechnology—Improving cell factory performance. Metab. Eng. 2014, 24, 38–60. [Google Scholar] [CrossRef] [PubMed]
- Tummler, K.; Lubitz, T.; Schelker, M.; Klipp, E. New types of experimental data shape the use of enzyme kinetics for dynamic network modeling. FEBS J. 2014, 281, 549–571. [Google Scholar] [CrossRef]
- Visser, D.; Heijnen, J.J. Dynamic simulation and metabolic re-design of a branched pathway using linlog kinetics. Metab. Eng. 2003, 5, 164–176. [Google Scholar] [CrossRef]
- Heijnen, J.J. Approximative kinetic formats used in metabolic network modeling. Biotechnol. Bioeng. 2005, 91, 534–545. [Google Scholar] [CrossRef] [PubMed]
- Sriyudthsak, K.; Shiraishi, F.; Hirai, M.Y. Mathematical Modeling and Dynamic Simulation of Metabolic Reaction Systems Using Metabolome Time Series Data. Front. Mol. Biosci. U6 2016, 3. [Google Scholar] [CrossRef]
- Horn, F.; Jackson, R. General mass action kinetics. Arch. Ration. Mech. Anal. 1972, 47, 81–116. [Google Scholar] [CrossRef]
- Chellaboina, V.; Bhat, S.P.; Haddad, W.M.; Bernstein, D.S. Modeling and analysis of mass-action kinetics. IEEE Control Syst. 2009, 29, 69–78. [Google Scholar]
- Dräger, A.; Kronfeld, M.; Ziller, M.J.; Supper, J.; Planatscher, H.; Magnus, J.B.; Oldiges, M.; Kohlbacher, O.; Zell, A. Modeling metabolic networks in C. glutamicum: A comparison of rate laws in combination with various parameter optimization strategies. BMC Syst. Biol. 2009, 3. [Google Scholar] [CrossRef]
- Hamby, D. A review of techniques for parameter sensitivity analysis of environmental models. Environ. Monit. Assess. 1994, 32, 135–154. [Google Scholar] [CrossRef]
- Cazzaniga, P.; Damiani, C.; Besozzi, D.; Colombo, R.; Nobile, M.S.; Gaglio, D.; Pescini, D.; Molinari, S.; Mauri, G.; Alberghina, L.; et al. Computational Strategies for a System-Level Understanding of Metabolism. Metabolites 2014, 4, 1034–1087. [Google Scholar] [CrossRef]
- Di Maggio, J.; Diaz Ricci, J.C.; Diaz, M.S. Global sensitivity analysis in dynamic metabolic networks. Comput. Chem. Eng. 2010, 34, 770–781. [Google Scholar] [CrossRef]
- Chan, K.; Scott, E.M.; Saltelli, A. Sensitivity Analysis: Edited by Andrea Saltelli, Karen Chan, E. Marian Scott; Wiley: Chichester, UK; Toronto, ON, Canada, 2000. [Google Scholar]
- Degenring, D.; Froemel, C.; Dikta, G.; Takors, R. Sensitivity analysis for the reduction of complex metabolism models. J. Process Control 2004, 14, 729–745. [Google Scholar] [CrossRef]
- Kacser, H.; Burns, J.A. The control of flux. Symp. Soc. Exp. Biol. 1973, 27, 65–104. [Google Scholar] [CrossRef]
- Heinrich, R.; Rapoport, T.A. A Linear Steady-State Treatment of Enzymatic Chains. Eur. J. Biochem. 1974, 42, 97–105. [Google Scholar] [CrossRef]
- Visser, D.; Heijnen, J.J. The Mathematics of Metabolic Control Analysis Revisited. Metab. Eng. 2002, 4, 114–123. [Google Scholar] [CrossRef]
- Fell, D.A. Metabolic control analysis: A survey of its theoretical and experimental development. Biochem. J. 1992, 286, 313. [Google Scholar] [CrossRef] [PubMed]
- Hatzimanikatis, V.; Floudas, C.A.; Bailey, J.E. Analysis and design of metabolic reaction networks via mixed-integer linear optimization. AIChE J. 1996, 42, 1277–1292. [Google Scholar] [CrossRef]
- Savageau, M.A. Biochemical systems analysis. II. The steady-state solutions for an n-pool system using a power-law approximation. J. Theor. Biol. 1969, 25, 370. [Google Scholar] [CrossRef]
- Savageau, M.A. Biochemical systems analysis: I. Some mathematical properties of the rate law for the component enzymatic reactions. J. Theor. Biol. 1969, 25, 365–369. [Google Scholar] [CrossRef]
- Voit, E.O. Biochemical Systems Theory: A Review. ISRN Biomath. 2013, 2013, 1–53. [Google Scholar] [CrossRef]
- Wang, F.S.; Ko, C.L.; Voit, E.O. Kinetic modeling using S-systems and lin-log approaches. Biochem. Eng. J. 2007, 33, 238–247. [Google Scholar] [CrossRef]
- Hatzimanikatis, V.; Bailey, J.E. MCA has more to say. J. Theor. Biol. 1996, 182, 233–242. [Google Scholar] [CrossRef]
- Voit, E.O. The best models of metabolism. Wiley Interdiscip. Reviews. Syst. Biol. Med. 2017, 9. [Google Scholar] [CrossRef] [PubMed]
- Shuler, M.; Kargı, F. Bioprocess Engineering: Basic Concepts; Prentice-Hall International Series in the Physical and Chemical Engineering Sciences; Prentice Hall: Upper Saddle River, NJ, USA, 1992; p. 91. [Google Scholar]
- Audagnotto, M.; Dal Peraro, M. Protein post-translational modifications: In silico prediction tools and molecular modeling. Comput. Struct. Biotechnol. J. 2017, 15, 307–319. [Google Scholar] [CrossRef]
- Kim, O.D.; Rocha, M.; Maia, P. A Review of Dynamic Modeling Approaches and Their Application in Computational Strain Optimization for Metabolic Engineering. Front. Microbiol. 2018, 9. [Google Scholar] [CrossRef]
- Nolan, R.P.; Lee, K. Dynamic model of CHO cell metabolism. Metab. Eng. 2011, 13, 108–124. [Google Scholar] [CrossRef]
- Bozovic, O.; Zanobini, C.; Gulzar, A.; Jankovic, B.; Buhrke, D.; Post, M.; Wolf, S.; Stock, G.; Hamm, P. Real-time observation of ligand-induced allosteric transitions in a PDZ domain. Proc. Natl. Acad. Sci. USA 2020, 117, 26031–26039. [Google Scholar] [CrossRef]
- Goldbeter, A.; Caplan, S.R. Oscillatory Enzymes. Annu. Rev. Biophys. Bioeng. 1976, 5, 449–476. [Google Scholar] [CrossRef]
- Palsson, B.O. Mathematical Modelling Of Dynamics And Control In Metabolic Networks. PhD Thesis, University of Wisconsin-Madison, Madison, WI, USA, 1984. [Google Scholar]
- Alaka, M.; Yan, X.; René, W.; Maria, K.; Claire, G.; Sophie, B.; Félix, M.; Lucie, B.; Linda, L.; Rita, L.; et al. The cumate gene-switch: A system for regulated expression in mammalian cells. BMC Biotechnol. 2006, 6. [Google Scholar] [CrossRef]
- Korzeniewski, B.; Liguzinski, P. Theoretical studies on the regulation of anaerobic glycolysis and its influence on oxidative phosphorylation in skeletal muscle. Biophys. Chem. 2004, 110, 147–169. [Google Scholar] [CrossRef] [PubMed]
- Covert, M.W.; Xiao, N.; Chen, T.J.; Karr, J.R. Integrating metabolic, transcriptional regulatory and signal transduction models in Escherichia coli. Bioinformatics 2008, 24, 2044–2050. [Google Scholar] [CrossRef] [PubMed]
- Fendt, S.M.; Buescher, J.M.; Rudroff, F.; Picotti, P.; Zamboni, N.; Sauer, U. Tradeoff between enzyme and metabolite efficiency maintains metabolic homeostasis upon perturbations in enzyme capacity. Mol. Syst. Biol. 2010, 6, 356. [Google Scholar] [CrossRef]
- Westerhoff, H.V.; Jensen, P.R.; Snoep, J.L.; Kholodenko, B.N. Thermodynamics of complexity—The live cell. Thermochim. Acta 1998, 309, 111–120. [Google Scholar] [CrossRef]
- Arkun, Y.; Yasemi, M. Dynamics and control of the ERK signaling pathway: Sensitivity, bistability, and oscillations. PLoS ONE 2018, 13, e0195513. [Google Scholar] [CrossRef]
- Johnson, K.A.; Goody, R.S. The Original Michaelis Constant: Translation of the 1913 Michaelis-Menten Paper. Biochemistry 2011, 50, 8264–8269. [Google Scholar] [CrossRef]
- Henri, V. Théorie générale de l’action de quelques diastases par Victor Henri [C. R. Acad. Sci. Paris 135 (1902) 916-919]. Comptes Rendus Biol. 2006, 329, 47–50. [Google Scholar] [CrossRef]
- Cornish-Bowden, A. One hundred years of Michaelis-Menten kinetics. Perspect. Sci. 2015, 4, 3–9. [Google Scholar] [CrossRef]
- Bajzer, Ž.; Strehler, E.E. About and beyond the Henri-Michaelis-Menten rate equation for single-substrate enzyme kinetics. Biochem. Biophys. Res. Commun. 2012, 417, 982–985. [Google Scholar] [CrossRef]
- Ren, X.; Deschênes, J.S.; Tremblay, R.; Peres, S.; Jolicoeur, M. A kinetic metabolic study of lipid production in Chlorella protothecoides under heterotrophic condition. Microb. Cell Factories 2019, 18, 113. [Google Scholar] [CrossRef]
- Kompala, D.S.; Ramkrishna, D.; Tsao, G.T. Cybernetic modeling of microbial growth on multiple substrates. Biotechnol. Bioeng. 1984, 26, 1272–1281. [Google Scholar] [CrossRef]
- Ramakrishna, R.; Ramkrishna, D.; Konopka, A.E. Cybernetic modeling of growth in mixed, substitutable substrate environments: Preferential and simultaneous utilization. Biotechnol. Bioeng. 1996, 52, 141–151. [Google Scholar] [CrossRef]
- Ramkrishna, D.; Song, H.S. Dynamic models of metabolism: Review of the cybernetic approach. AIChE J. 2012, 58, 986–997. [Google Scholar] [CrossRef]
- Guardia, M.J.; Gambhir, A.; Europa, A.F.; Ramkrishna, D.; Hu, W.S. Cybernetic Modeling and Regulation of Metabolic Pathways in Multiple Steady States of Hybridoma Cells. Biotechnol. Prog. 2000, 16, 847–853. [Google Scholar] [CrossRef] [PubMed]
- Young, J.D.; Henne, K.L.; Morgan, J.A.; Konopka, A.E.; Ramkrishna, D. Integrating cybernetic modeling with pathway analysis provides a dynamic, systems-level description of metabolic control. Biotechnol. Bioeng. 2008, 100, 542–559. [Google Scholar] [CrossRef] [PubMed]
- Kompala, D.S.; Ramkrishna, D.; Jansen, N.B.; Tsao, G.T. Investigation of bacterial growth on mixed substrates: Experimental evaluation of cybernetic models. Biotechnol. Bioeng. 1986, 28, 1044–1055. [Google Scholar] [CrossRef]
- Kim, J.I.; Varner, J.D.; Ramkrishna, D. A hybrid model of anaerobic E. coli GJT001: Combination of elementary flux modes and cybernetic variables. Biotechnol. Prog. 2008, 24, 993–1006. [Google Scholar] [CrossRef]
- Song, H.S.; Morgan, J.A.; Ramkrishna, D. Systematic development of hybrid cybernetic models: Application to recombinant yeast co-consuming glucose and xylose. Biotechnol. Bioeng. 2009, 103, 984–1002. [Google Scholar] [CrossRef]
- Aboulmouna, L.; Raja, R.; Khanum, S.; Gupta, S.; Maurya, M.R.; Grama, A.; Subramaniam, S.; Ramkrishna, D. Cybernetic modeling of biological processes in mammalian systems. Curr. Opin. Chem. Eng. 2020. [Google Scholar] [CrossRef]
- Schomburg, I.; Chang, A.; Ebeling, C.; Gremse, M.; Heldt, C.; Huhn, G.; Schomburg, D. BRENDA, the enzyme database: Updates and major new developments. Nucleic Acids Res. 2004, 32, D431–D433. [Google Scholar] [CrossRef]
- Goldberg, R.N.; Tewari, Y.B.; Bhat, T.N. Thermodynamics of enzyme-catalyzed reactions- a database for quantitative biochemistry. Bioinformatics 2004, 20, 2874–2877. [Google Scholar] [CrossRef]
- Caspi, R.; Foerster, H.; Fulcher, C.A.; Hopkinson, R.; Ingraham, J.; Kaipa, P.; Krummenacker, M.; Paley, S.; Pick, J.; Rhee, S.Y.; et al. MetaCyc: A multiorganism database of metabolic pathways and enzymes. Nucleic Acids Res. 2006, 34, D511–D516. [Google Scholar] [CrossRef]
- Khim Chong, C.; Saberi Mohamad, M.; Deris, S.; Shahir Shamsir, M.; Wen Choon, Y.; En Chai, L. A Review on Modelling Methods, Pathway Simulation Software and Recent Development on Differential Evolution Algorithms for Metabolic Pathways in Systems Biology. Curr. Bioinform. 2014, 9, 509–521. [Google Scholar] [CrossRef]
- Polisetty, P.K.; Voit, E.O.; Gatzke, E.P. Identification of metabolic system parameters using global optimization methods. Theor. Biol. Med Model. 2006, 3, 4. [Google Scholar] [CrossRef]
- Reali, F.; Priami, C.; Marchetti, L. Optimization Algorithms for Computational Systems Biology. Front. Appl. Math. Stat. 2017, 3. [Google Scholar] [CrossRef]
- Cavazzuti, M. Optimization Methods: From Theory to Design Scientific and Technological Aspects in Mechanics; Springer Science & Business Media: Berlin/Heidelberg, Germany, 2012. [Google Scholar]
- Floudas, C.A. Deterministic Global Optimization: Theory, Methods and Applications; Springer Science & Business Media: Berlin/Heidelberg, Germany, 2013; Volume 37. [Google Scholar]
- Dominique, V.; Filip, L.; Jan Van, I. Dynamic estimation of specific fluxes in metabolic networks using non-linear dynamic optimization. BMC Syst. Biol. 2014, 8. [Google Scholar] [CrossRef]
- Gopalakrishnan, S.; Dash, S.; Maranas, C. K-FIT: An accelerated kinetic parameterization algorithm using steady-state fluxomic data. Metab. Eng. 2020, 61, 197–205. [Google Scholar] [CrossRef]
- Lubitz, T.; Liebermeister, W. Parameter balancing: Consistent parameter sets for kinetic metabolic models. Bioinformatics 2019, 35, 3857–3858. [Google Scholar] [CrossRef]
- Horst, R.; Tuy, H. Global Optimization: Deterministic Approaches; Springer Science & Business Media: Berlin/Heidelberg, Germany, 2013. [Google Scholar]
- Dutton, R.L.; Scharer, J.M.; Moo-Young, M. Descriptive parameter evaluation in mammalian cell culture. Cytotechnology 1998, 26, 139–152. [Google Scholar] [CrossRef]
- Chou, I.C.; Voit, E.O. Recent developments in parameter estimation and structure identification of biochemical and genomic systems. Math. Biosci. 2009, 219, 57–83. [Google Scholar] [CrossRef] [PubMed]
- Kutalik, Z.; Moulton, V.; Tucker, W. S-system parameter estimation for noisy metabolic profiles using Newton-flow analysis. IET Syst. Biol. 2007, 1, 174–180. [Google Scholar] [CrossRef] [PubMed]
- Voit, E.O.; Goel, G.; Chou, I.; Fonseca, L.L. Estimation of metabolic pathway systems from different data sources. IET Syst. Biol. 2009, 3, 513–522. [Google Scholar] [CrossRef]
- Penas, D.R.; González, P.; Egea, J.A.; Doallo, R.; Banga, J.R. Parameter estimation in large-scale systems biology models: A parallel and self-adaptive cooperative strategy. BMC Bioinform. 2017, 18, 52. [Google Scholar] [CrossRef] [PubMed]
- Khodayari, A.; Maranas, C.D. A genome-scale Escherichia coli kinetic metabolic model k-ecoli457 satisfying flux data for multiple mutant strains. Nat. Commun. 2016, 7, 13806. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.; Lafontaine Rivera, J.G.; Contador, C.A.; Asenjo, J.A.; Liao, J.C. Reducing the allowable kinetic space by constructing ensemble of dynamic models with the same steady-state flux. Metab. Eng. 2011, 13, 60–75. [Google Scholar] [CrossRef]
- Henry, C.S.; Jankowski, M.D.; Broadbelt, L.J.; Hatzimanikatis, V. Genome-scale thermodynamic analysis of Escherichia coli metabolism. Biophys. J. 2006, 90, 1453–1461. [Google Scholar] [CrossRef]
- Greene, J.L.; Wäechter, A.; Tyo, K.E.J.; Broadbelt, L.J. Acceleration Strategies to Enhance Metabolic Ensemble Modeling Performance. Biophys. J. 2017, 113, 1150–1162. [Google Scholar] [CrossRef]
- Dräger, A.; Kronfeld, M.; Supper, J.; Planatscher, H.; Magnus, J.B.; Oldiges, M.; Zell, A. Benchmarking evolutionary algorithms on convenience kinetics models of the Valine and Leucine Biosynthesis in C. glutamicum. In Proceedings of the 2007 IEEE Congress on Evolutionary Computation, Singapore, 25–28 September 2007; IEEE: Piscataway, NJ, USA, 2007; pp. 896–903. [Google Scholar]
- Spieth, C.; Hassis, N.; Streichert, F. Comparing mathematical models on the problem of network inference. In Proceedings of the 8th Annual Conference on Genetic and Evolutionary Computation, Washington, DC, USA, 8–12 July 2016; ACM: New York, NY, USA, 2006; pp. 279–286. [Google Scholar]
- Salman, A.; Engelbrecht, A.P.; Omran, M.G. Empirical analysis of self-adaptive differential evolution. Eur. J. Oper. Res. 2007, 183, 785–804. [Google Scholar] [CrossRef]
- Thangaraj, R.; Pant, M.; Abraham, A. A simple adaptive differential evolution algorithm. In Proceedings of the 2009 World Congress on Nature & Biologically Inspired Computing (NaBIC), Coimbatore, India, 9–11 December 2009; pp. 457–462. [Google Scholar]
- Feng, L.; Yang, Y.F.; Wang, Y.X. A new approach to adapting control parameters in differential evolution algorithm. In Simulated Evolution and LEARNING, Proceedings of the 7th International Conference on Simulated Evolution and Learning, Melbourne, Australia, 7–10 December 2008; Springer: Berlin/Heidelberg, Germany, 2008; pp. 21–30. [Google Scholar]
- Long, C.P.; Antoniewicz, M.R. Metabolic flux responses to deletion of 20 core enzymes reveal flexibility and limits of E. coli metabolism. Metab. Eng. 2019, 55, 249–257. [Google Scholar] [CrossRef]
- Covert, M.W.; Knight, E.M.; Reed, J.L.; Herrgard, M.J.; Palsson, B.O. Integrating high-throughput and computational data elucidates bacterial networks. Nature 2004, 429, 92–96. [Google Scholar] [CrossRef]
- Herrgard, M.J.; Lee, B.S.; Portnoy, V.; Palsson, B.Ø. Integrated analysis of regulatory and metabolic networks reveals novel regulatory mechanisms in Saccharomyces cerevisiae. Genome Res. 2006, 16, 627–635. [Google Scholar] [CrossRef] [PubMed]
- Yeo, H.C.; Hong, J.; Lakshmanan, M.; Lee, D.Y. Enzyme capacity-based genome scale modelling of CHO cells. Metab. Eng. 2020, 60, 138–147. [Google Scholar] [CrossRef]
- Klamt, S.; Mahadevan, R.; von Kamp, A. Speeding up the core algorithm for the dual calculation of minimal cut sets in large metabolic networks. BMC Bioinform. 2020, 21, 510. [Google Scholar] [CrossRef] [PubMed]
- Schneider, P.; von Kamp, A.; Klamt, S. An extended and generalized framework for the calculation of metabolic intervention strategies based on minimal cut sets. PLoS Comput. Biol. 2020, 16, e1008110. [Google Scholar] [CrossRef]
- Raj, K.; Venayak, N.; Mahadevan, R. Novel two-stage processes for optimal chemical production in microbes. Metab. Eng. 2020, 62, 186–197. [Google Scholar] [CrossRef]
- Machado, D.; Herrgård, M.J. Co-evolution of strain design methods based on flux balance and elementary mode analysis. Metab. Eng. Commun. 2015, 2, 85–92. [Google Scholar] [CrossRef]
- Zampieri, M.; Sauer, U. Model-based media selection to minimize the cost of metabolic cooperation in microbial ecosystems. Bioinformatics 2016, 32, 1733–1739. [Google Scholar] [CrossRef]
- Jarrett, A.M.; Hormuth, D.A.; Wu, C.; Kazerouni, A.S.; Ekrut, D.A.; Virostko, J.; Sorace, A.G.; DiCarlo, J.C.; Kowalski, J.; Patt, D.; et al. Evaluating patient-specific neoadjuvant regimens for breast cancer via a mathematical model constrained by quantitative magnetic resonance imaging data. Neoplasia 2020, 22, 820–830. [Google Scholar] [CrossRef] [PubMed]
- Pappalardo, F.; Russo, G.; Tshinanu, F.M.; Viceconti, M. In silico clinical trials: Concepts and early adoptions. Brief. Bioinform. 2019, 20, 1699–1708. [Google Scholar] [CrossRef] [PubMed]
- Ho, D.; Quake, S.R.; McCabe, E.R.B.; Chng, W.J.; Chow, E.K.; Ding, X.; Gelb, B.D.; Ginsburg, G.S.; Hassenstab, J.; Ho, C.M.; et al. Enabling Technologies for Personalized and Precision Medicine. Trends Biotechnol. 2020, 38, 497–518. [Google Scholar] [CrossRef] [PubMed]
- Chiappino-Pepe, A.; Pandey, V.; Ataman, M.; Hatzimanikatis, V. Integration of metabolic, regulatory and signaling networks towards analysis of perturbation and dynamic responses. Curr. Opin. Syst. Biol. 2017, 2, 59–66. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Thermodynamics-Based Constrained Model | ||
|---|---|---|
| Theory Name | Specification | Reference |
| Energy balance analysis (EBA) | Identifies reactions’ direction and flux limits based on the Gibbs free energy value of reactions. | [135] |
| Network-embedded thermodynamic analysis (NET) | Identifies thermodynamically feasible flux range or metabolite concentration range, based on the predefined standard Gibbs free energy and predetermined flux directions. | [136] |
| Thermodynamics-based metabolic flux analysis (TMFA) | Identifies flux direction, allowable flux range and also concentration ranges. Incorporates a MILP optimization. Based on the predefined standard Gibbs free energy. | [129] |
| MCA Coefficients Table | ||
|---|---|---|
| Name | Mathematical Formulation | Description |
| Control coefficients | Flux: | The coefficients are a measure of |
| Intracellular metabolite: | the relative change in a flux or concentration upon a relative change of an enzyme activity level | |
| Response coefficients | Flux: | The coefficients are a measure of |
| Intracellular metabolite: | the effect of a change of an external parameter, on intracellular fluxes and concentrations | |
| Elasticity coefficient | Intracellular reaction rate: | The elasticity is a local measure quantifying the relative change in reaction rate upon a relative change in metabolite concentration, while maintaining other concentrations and parameters constant |
| Approximate Kinetic Formats Based on MCA and BST | |
|---|---|
| Type of Approximate Rate Law | Mathematical Formulation |
| Log-lin | |
| Lin-log | |
| S-system | |
| General mass action (GMA) | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yasemi, M.; Jolicoeur, M. Modelling Cell Metabolism: A Review on Constraint-Based Steady-State and Kinetic Approaches. Processes 2021, 9, 322. https://doi.org/10.3390/pr9020322
Yasemi M, Jolicoeur M. Modelling Cell Metabolism: A Review on Constraint-Based Steady-State and Kinetic Approaches. Processes. 2021; 9(2):322. https://doi.org/10.3390/pr9020322
Chicago/Turabian StyleYasemi, Mohammadreza, and Mario Jolicoeur. 2021. "Modelling Cell Metabolism: A Review on Constraint-Based Steady-State and Kinetic Approaches" Processes 9, no. 2: 322. https://doi.org/10.3390/pr9020322
APA StyleYasemi, M., & Jolicoeur, M. (2021). Modelling Cell Metabolism: A Review on Constraint-Based Steady-State and Kinetic Approaches. Processes, 9(2), 322. https://doi.org/10.3390/pr9020322