1. Introduction
SARS-CoV-2 virus was first detected in Wuhan, China in December 2019. The SARS-CoV-2 disease also known as COVID-19 has become a pandemic and continues to be a global burden, impacting all levels of health care systems and economies. As of 3 November 2021, over 251.2 million people have been infected worldwide with more than 5 million deaths (
https://covid19.who.int/ (accessed on 3 November 2021)). In sub-Saharan Africa, COVID-19 cases are relatively low compared to other parts of the world even though the reported numbers might be underestimated.
In Mali, the first case of COVID-19 was detected on 26 March 2020. By November 2021, 16,527 cases were reported with 581 deaths according the WHO report. The Malian government implemented protection and control measures of the disease. The country has experienced three waves of the outbreak.
Global efforts to control the disease included genomic surveillance as a key component of control strategies. The genomic epidemiology of COVID 19 has provided a framework for tracing the origins of the circulating virus, identifying clusters or hotspots of transmission, route of transmission, and detecting new variants [
1,
2,
3]. For instance, new variants reported in the United Kingdom, South Africa, and Brazil have been identified through genomic surveillance [
4,
5,
6,
7].
However, Africa has been lagging behind with providing sequences of the virus. The first African SARS-CoV-2 was provided by Nigeria [
8]. To date, only a few laboratories in Africa are able to sequence the genome of the virus. Most of the samples are sent abroad in institutions with sequencing capacities. For instance, the first sequences from Mali deposited in GISAID were sequenced in Germany. However, shipping samples overseas is expensive. In addition, ethical and regulatory issues constitute major challenges. Therefore, developing local capacity in sequencing seems an optimal option for developing countries such as Mali.
As the pandemic progresses, more sequences are needed to monitor the evolution of the virus. Until recently, Malian researchers did not have access to next-generation sequencing locally. The Malaria Research and Training Center at the University of Science, Techniques and Technology of Bamako has recently acquired a MiSeq instrument for its malaria genomics research. However, the urgency of the COVID-19 pandemic prompted the use of the existing resource to sequence SARS-CoV-2 genome. Indeed, a combination of factors including lack of preparedness, lack of specific funding, and difficulty in shipping samples abroad led to the necessity of using malaria research resources to sequence SARS-CoV-2 genomes. We describe here the first genome of SARS-CoV-2 sequenced locally and highlight the importance of implementing genomic surveillance and discuss some challenges related to access to reagents during a pandemic.
2. Materials and Methods
2.1. Samples
Two samples were collected in June 2020 by the Centre d’Infectiologie Charles Merieux’s mobile laboratory, which was dispatched by the Ministry of Health to handle an outbreak of SARS-CoV-2 in Timbuktu, more than 1000 km North of Bamako, which is the capital city of Mali. A nasopharyngeal specimen served as starting material for RNA extraction. Total RNA was extracted using a Qiamp viral RNA mini kit (Qiagen Cat. 57704) according to the manufacturer’s instructions. Real-time transcriptase polymerase chain reaction (RT-PCR) was used for the detection of positive samples. RT-PCR tests were based on the N gene and the RNA-dependent RNA polymerase (RdRp) gene located on the ORF1ab.
2.2. Library Preparation
RNA was quantified with a fluorometer (Denovix QFX) using Denovix RNA quantification kit. Total RNA was depleted of ribosomal RNA prior to library preparation. Briefly, 200 ng of input RNA was subjected to rRNA depletion according to the Ribo-Zero Gold rRNA depletion protocol (Illumina, 96 samples, Cat no. 20020599). Libraries were prepared from the depleted RNA using the Illumina TruSeq Stranded Total RNA Library Prep Kit (Cat no. 20020599). The protocol included the following steps: RNA fragmentation, cDNA synthesis, adenylation of the 3′-end, adapter ligation, and cDNA enrichment (TruSeq Stranded Total RNA protocol). For cDNA synthesis, because the transcriptase Superscript II was not available, we slightly modified the Illumina protocol by replacing it with the SuperScript IV (Introgen cat. 12594025). In addition, PCR was performed at 50 °C instead of 42 °C. cDNA was quantified with the fluorometer using DeNovix dsDNA Broad Range Quantitation Reagent according to the manufacturer’s instructions. We adapted a QuadroMACS Separation Unit (Miltenyi Biotec Ltd., Cat no.130-090-976, Tokyo, Japan) for all bead-based purification steps described in the TruSeq protocol (
Figure 1).
2.3. Library SIZE Estimation of the cDNA Library Using an Agarose Gel
During this pandemic, access to reagents was a real challenge. While our order for Bioanalyzer had been pending for months, we decided to estimate our library size using 1% agarose gel. Briefly, 2 uL of the library was loaded using NEB loading dye (Cat: NEB #B7025), which was selected because it did not make any shadow on the gel, as did the bromophenol blue dye. Gel image was visualized and saved as a .jpeg file. The image was subsequently analyzed using ImageJ software, which is a gel analysis tool [
9].
2.4. Sequencing
Libraries were quantified and normalized with nuclease-free ultrapure water, spiked with 5% of Phi X control, and run onto the Illumina MiSeq instrument (Illumina, San Diego, CA, USA) for sequencing. A MiSeq reagent kit v3 (150-cycle) was used according to the manufacturer’s instructions. Twenty (20) pM of the normalized library were loaded onto the MiSeq instrument.
2.5. Data Analysis
All data were processed locally using a recently built bioinformatics infrastructure located at the Malaria Research and Training Center. Raw data (FastQ files) were transferred from the MiSeq instrument (via BaseSpace account) to a local server and analyzed according to the workflow in
Supplemental Figure S1. Quality control was performed using
FastQC and
Multi-FastQC tools. The QC plot showed that all reads were of high quality and did not require any trimming. Subsequently, reads were mapped onto the SARS-CoV-2 reference genome (accession number: MN908947.3) using the Burrows Wheeler aligner (
BWA) version 0.7.17 [
10] with default parameters. The post-mapping file generated as a bam file was processed with
samtools [
11] and the
vcfR package to determine the percent of aligned reads, the depth of coverage, and mapping quality. Library estimation was performed using the
CollectInsertSizeMetrics tool version 2.8.1 implemented in Picard. The alignment was visualized with the
IGV (Interactive Genome Viewer) tool [
12]. A consensus sequence algorithm implemented in
IGV was also used to generate the consensus sequence from the alignment [
13].
Vcftools was used to generate a variant call file (vcf).
SnpEff implemented on the Galaxy server as well as Nextclade were used to annotate variants.
We performed de novo assembly of the reads using MEGAHIT software [
14]. Shorter contigs (<2000 bp) were filtered out, and the remaining contigs were queried with BLASTN on the NCBI database to recover the SARS-CoV-2 genome. Consensus sequence from the alignment approach was compared to the contigs from de novo assembly using
nucmer aligner [
15] and BLASTN.
3. Results
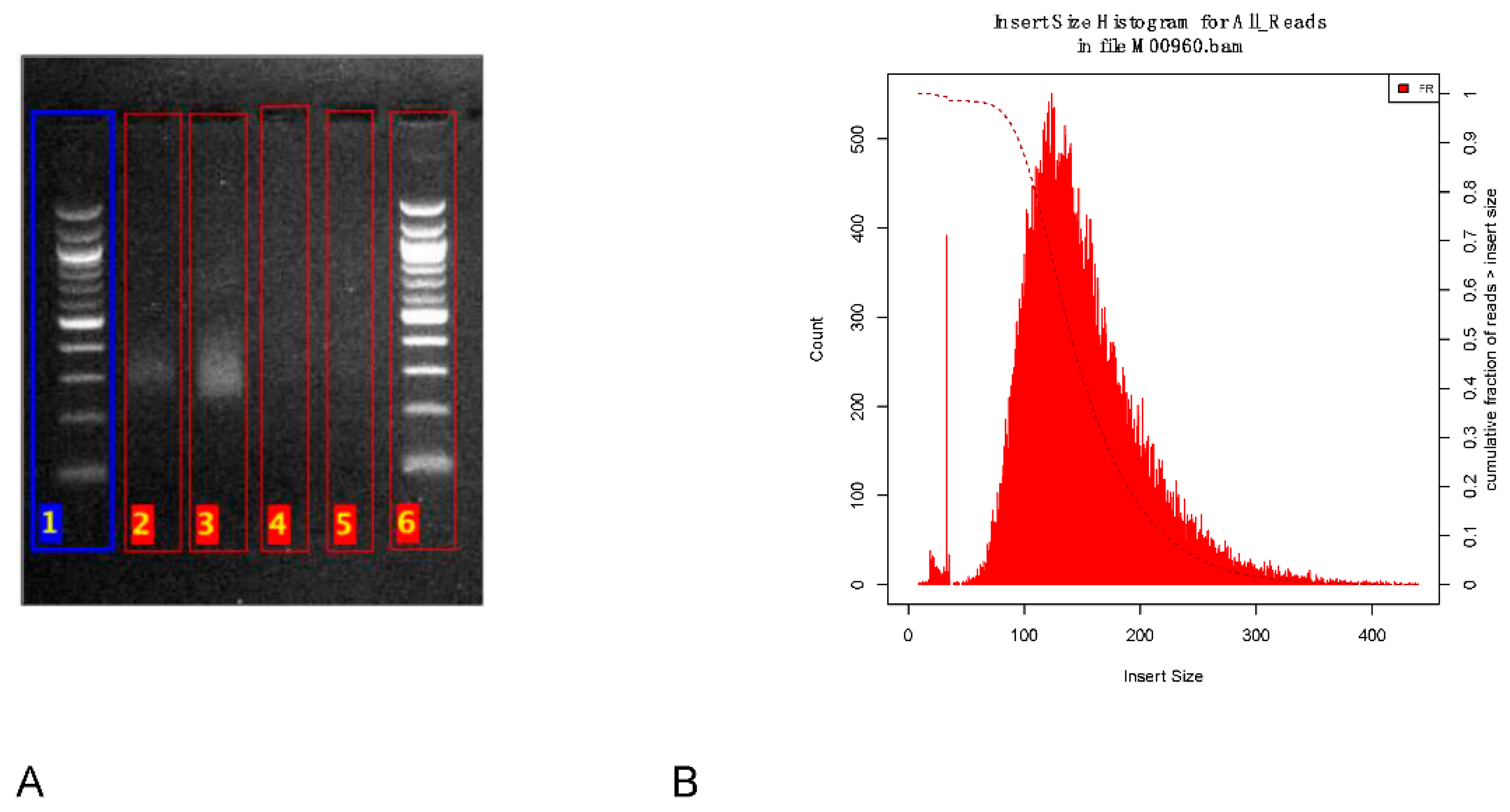
The library size was estimated to around 325 bp on the agarose gel (
Figure 2A). After sequencing, the average insert size was estimated from the reads to around 150 bp (
Figure 2B).
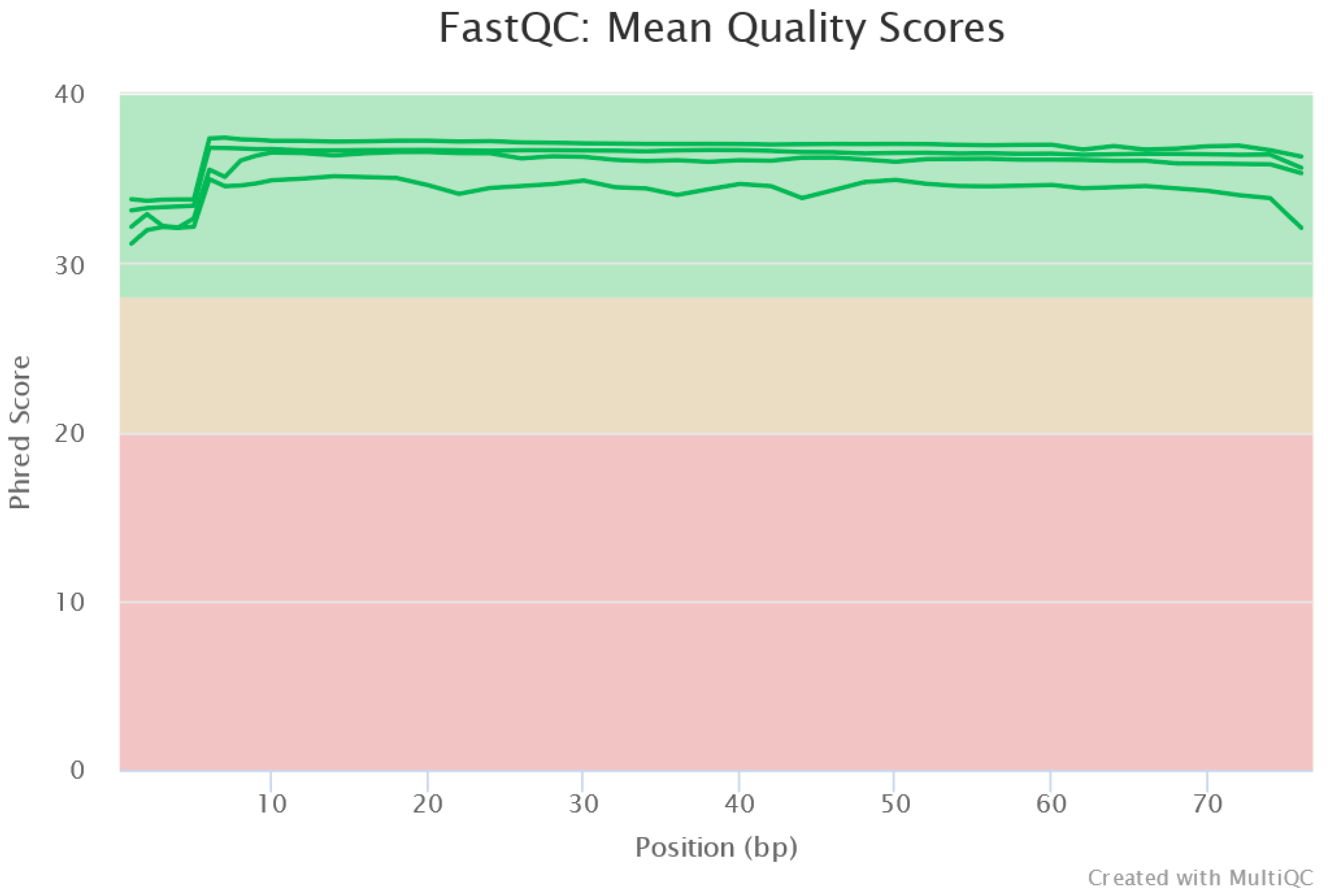
The MiSeq run output generated raw data with 96.22% of reads having a quality score Q > 30 (less than 1 error in every 1000 bp). The average quality measured as the Phred-score of the raw data was ≈35. Pre-processing of the raw data showed all reads were of high quality (
Figure 3). The reads did not require any end trimming.
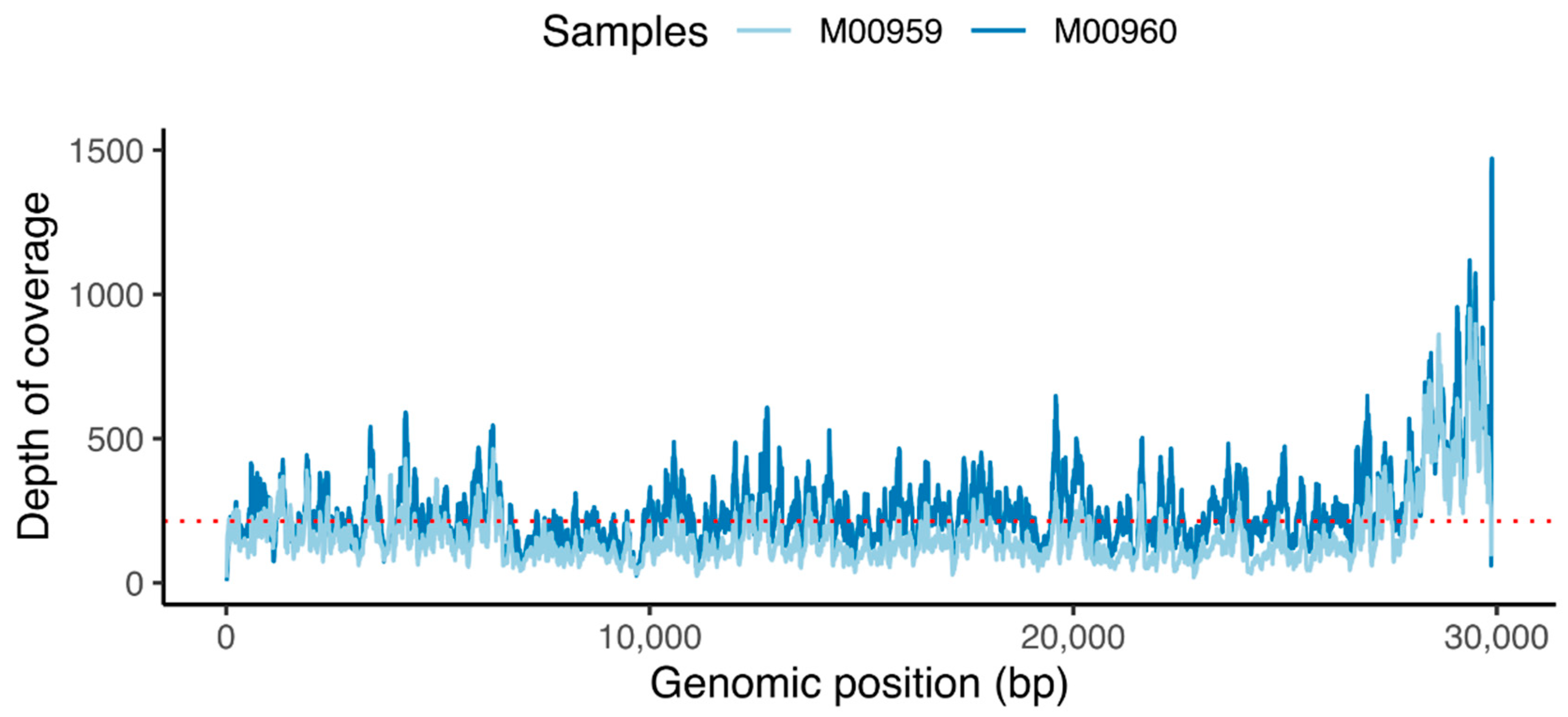
After pre-processing the data, to confirm that the reads contain SARS-CoV-2 sequences, we mapped the raw reads of sample 1 (M00960) and sample 2 (M00959) to the reference Wuhan genome (accession number: NC_045512). Post-alignment processing of the BAM files resulted in 0.13% and 0.97% of the total reads (104479 reads for sample 1 and 84,244 reads for sample 2) aligned to the reference genome covering 99% of genome. An average read depth of 214 (
Figure 4) and a mapping quality >50 were obtained (
Figure S2). Both the forward and the reverse sequences aligned well to the reference genome (
Figure S3). We generated a consensus genome of 29,903 base pairs from the bam file using a consensus mode implemented in the IGV tool. BLAST analysis showed top hits with 99.95% and 99.96% identity covering 100% of the query sequences respectively for sample 1 (
Figure S4) and sample 2 (data not shown).
In addition, we de novo assembled the reads with MEGAHIT software. Then, we performed a BLASTN analysis of the contigs using the NBCI non-redundant database. BLAST output showed both genomes hit the SARS-CoV-2 genome with a high percent identity. The hits corresponded to the SARS-CoV-2 genome with 100% coverage of the query and a percent identity of 99%.
To compare the consensus sequences of the resequencing versus de novo assembled genomes, we performed an alignment of the two sequences using BLASTN. The two sequences were identical with only one ambiguous nucleotide (Y: C or T) at the position 847 in the reference-based consensus sequence for sample 1. When sample 1 was compared to the reference Wuhan Hu-1 genome, 19 SNPs were identified, of which 2 were in the 5′ untranslated region (UTR) region, 6 were synonymous mutations, and 11 were non-synonymous (amino acid substitutions). Of the 11 non-synonymous mutations, two were located on the gene coding for the spike protein respectively at positions 23403 (amino acid D614G) and 23587 (amino acid Q675H) on the genome. We also noted one mutation at the position 28833 (C to T), which falls in the “Charité_N_R” primer target (
Table 1). Sample 2 contained 14 mutations, of which nine were non-synonymous. Interestingly, the spike protein had one mutation at the amino acid position 614 where D replaced N (D614N) (data not shown).
Twenty-one (21) Malian sequences provided by a German laboratory and deposited on GISAID were retrieved, and multiple alignments were performed with MAFFT. Subsequently, a phylogenetic tree was constructed with a neighbor-joining method implemented in the adegenet package in R. Our newly sequenced genomes clustered with the existing Malian samples sequenced abroad (
Figure 5).
To determine the lineage of the first new genome, we used the nextclade web application implemented on the Nextstrain website (
https://nextstrain.org/sars-cov-2/ (accessed on 3 November 2021)) and the pangolin lineage assigner. The analysis assigned the first genome to the B.1 lineage (pangolin classification), which is most common in the USA, UK, France, and it belonged to the 20A clade according to Nextstrain classification. The second genome was assigned to the A.21 lineage (pangolin classification).
Figure 1.
“Improvised device” used for ribosomal RNA depletion, RNA purification, and cDNA cleanup. The recommended plate magnetic separator is shown on the left.
Figure 1.
“Improvised device” used for ribosomal RNA depletion, RNA purification, and cDNA cleanup. The recommended plate magnetic separator is shown on the left.
Figure 2.
Library size estimation. (A) Agarose gel image showing the library size (smear at ~325 bp). Lanes 1, 6 represent 100 bp DNA ladders. Lanes 2–3 are undiluted libraries and lanes 4–5 are diluted libraries. (B) Library insert size estimated from the reads with Picard tool.
Figure 2.
Library size estimation. (A) Agarose gel image showing the library size (smear at ~325 bp). Lanes 1, 6 represent 100 bp DNA ladders. Lanes 2–3 are undiluted libraries and lanes 4–5 are diluted libraries. (B) Library insert size estimated from the reads with Picard tool.
Figure 3.
Read quality control showing high-quality sequences.
Figure 3.
Read quality control showing high-quality sequences.
Figure 4.
Depth of coverage. Red dotted line indicates the average read coverage (214×).
Figure 4.
Depth of coverage. Red dotted line indicates the average read coverage (214×).
Table 1.
SNPs detected in the newly sequenced genome using the VCFtools.
Table 1.
SNPs detected in the newly sequenced genome using the VCFtools.
| #CHROM | POS | REF | ALT | Gene | Mutation nt | Mutation aa |
|---|
| NC_045512 | 41 | T | A | ORF1ab | c.-225T>A | UTR |
| NC_045512 | 241 | C | T | ORF1ab | c.-25C>T | UTR |
| NC_045512 | 1783 | T | C | ORF1ab | c.1518T>C | p.Cys506Cys |
| NC_045512 | 2416 | C | T | ORF1ab | c.2151C>T | p.Tyr717Tyr |
| NC_045512 | 3037 | C | T | ORF1ab | c.2772C>T | p.Phe924Phe |
| NC_045512 | 5100 | C | T | ORF1ab | c.4835C>T | p.Ser1612Leu |
| NC_045512 | 10138 | C | T | ORF1ab | c.9873C>T | p.Asn3291Asn |
| NC_045512 | 11128 | G | A | ORF1ab | c.10863G>A | p.Met3621Ile |
| NC_045512 | 14322 | C | T | ORF1ab | c.14073C>T | p.Tyr4691Tyr |
| NC_045512 | 14408 | C | T | ORF1ab | c.14159C>T | p.Pro4720Leu |
| NC_045512 | 15654 | C | T | ORF1ab | c.15405C>T | p.Asp5135Asp |
| NC_045512 | 23403 | A | G | S | c.1841A>G | p.Asp614Gly |
| NC_045512 | 23587 | G | C | S | c.2025G>C | p.Gln675His |
| NC_045512 | 25433 | C | T | ORF3a | c.41C>T | p.Thr14Ile |
| NC_045512 | 25563 | G | T | ORF3a | c.171G>T | p.Gln57His |
| NC_045512 | 26038 | T | C | ORF3a | c.646T>C | p.Ser216Pro |
| NC_045512 | 28482 | A | G | N | c.209A>G | p.Gln70Arg |
| NC_045512 | 28600 | T | C | N | c.327T>C | p.Tyr109Tyr |
| NC_045512 | 28833 | C | T | N | c.560C>T | p.Ser187Leu |
Figure 5.
Neighbor-joining phylogenetic tree.
Figure 5.
Neighbor-joining phylogenetic tree.
4. Discussion
We report the first full genomes of SARS-CoV-2 sequenced and analyzed in Mali by Malian scientists. We have shown the feasibility of sequencing in a resource-limited setting. Genomic data are not only important for the development and surveillance of vaccines and diagnostic tests but also essential for monitoring the evolution of the virus and the transmission of the disease. Thus, our work has important implications for epidemiological tracing of the outbreak and could provide valuable information on routes of introduction of the virus and the natural history of the SARS-CoV-2 circulating in Mali. Genomic data could help in identifying the source and sink of the virus and guide interventions.
One of the major challenges during this pandemic is the availability of reagents. Therefore, we adapted an alternative solution using an agarose gel to estimate the size of our library. There was an overestimation of the size using the agarose gel. Even though the difference in library size estimation could be due to the adapters or to a clustering bias of shorter fragments, the library size estimation on agarose gel is less accurate. Nevertheless, the agarose gel electrophoresis could be useful for laboratories in resource-limited settings with no access to expensive and sophisticated instruments for accurate library size analysis such as a Bioanalyzer or a TapeStation.
When the pandemic started, the laboratory was missing some small equipment. For example, the magnetic separator recommended during DNA library preparation was missing. We modified the protocol by adapting another device used for cell separation for ribosomal RNA depletion as well as RNA and cDNA purification. This device is normally used in our laboratory for Plasmodium-infected cell separation. The sequencing platform was initially set up for malaria research. However, the COVID-19 pandemic showed the versality of its use and underscores the importance of developing local sequencing capacity. The study demonstrates how an existing research infrastructure for a parasitic disease can be leveraged to tackle a global health problem.
The recovered genomes contained common SNPs along with some private mutations. Interestingly, the D614G mutation on the spike protein associated with increased infectivity and virion spike density [
16,
17] was observed in one sample. The second sample has the D614N mutation on the spike protein. The average number of cases in Timbuktu in June 2020 was around 50 per day as compared to a national average case of 20 per day [
18]. The presence of these mutations in Timbuktu might explain the sharp increase in cases in Timbuktu in June 2020, which prompted the Malian Ministry of Health to send a team with a mobile laboratory to detect cases, trace contacts, and contain the local outbreak.
The viruses were likely introduced in Mali from Europe, as the lineage tracing showed that it belongs to the B1 and A.21 lineages, which were common in Europe and the USA. This is not surprising because of frequent travels between Mali and Europe.
Our study has some limitations. One of these was the lack of quality control of total RNA before library preparation. For instance, we could not check the quality of the total RNA and did not know if the RNA was degraded or not because we did not have all the reagents required to do so. The decision to sequence the sample was solely based on the quantification of total RNA. Nonetheless, during this pandemic period, access to reagents remains a real challenge, and quality control of the total RNA may be bypassed. Indeed, as indicated above, we successfully repeated the experiment with a second and a third specimen (data not shown). Another issue was the library size estimation accuracy. Even though an agarose gel analysis represents an alternative solution to overcome fragment size determination with expensive equipment, it may overestimate the fragment size. In addition, the data presented here are very limited as only two samples were analyzed. Nevertheless, we demonstrate the feasibility of the approach. These methods could be used to estimate the size of DNA libraries that can be multiplexed (pooled) to generate higher numbers of sequences. As genomic epidemiology has become essential in the control and diagnostic of the COVID-19 pandemic, the approach described here would represent an important starting point in the genomic surveillance of the disease in resource-limited laboratories.
We provide the first genomes of SARS-CoV-2 sequenced locally in Mali by Malian scientists. We show that a laboratory set for malaria research can be readily adapted to sequencing other pathogens. Our results set a precedent for the genomic surveillance of SARS-CoV-2 strains circulating in the country. The platform could be used to provide insights in tracking the SAR-CoV-2 transmission dynamics, including the presence and evolution of variants. Such local data could inform health policy decisions for better control of the disease.
Supplementary Materials
The following are available online at
https://www.mdpi.com/article/10.3390/pr9122169/s1, Figure S1: Bioinformatics analysis workflow, Figure S2: Alignment quality metrics, Figure S3: Visualization of forward and reverse read alignment with IGV showing mutations supported by both reads, Figure S4: Blast hits.
Author Contributions
Conceptualization, A.D. (Antoine Dara) and A.A.D.; methodology, A.D. (Antoine Dara), B.K., A.D. (Amadou Daou), A.K.S., D.K. and C.D.; validation, B.K. and A.A.D.; formal analysis, A.D. (Antoine Dara); writing—original draft preparation, A.D. (Antoine Dara); writing—review and editing, A.D. (Antoine Dara), A.A.D. and B.K.; supervision, A.A.D. All authors have read and agreed to the published version of the manuscript.
Funding
We are thankful to Africa CDC and Illumina for providing us with reagents. This work was supported through the DELTAS Africa Initiative [DELGEME grant 107740/Z/15/Z]. The DELTAS Africa Initiative is an independent funding scheme of the African Academy of Sciences (AAS)’s Alliance for Accelerating Excellence in Science in Africa (AESA) and supported by the New Partnership for Africa’s Development Planning and Coordinating Agency (NEPAD Agency) with funding from the Wellcome Trust [DELGEME grant 107740/Z/15/Z] and the UK government. The views expressed in this publication are those of the author(s) and not necessarily those of AAS, NEPAD Agency, Wellcome Trust, or the UK government.
Institutional Review Board Statement
The use of the sample was approved by the ethical committee of the Faculty of Medicine and Odonto-Stomatology and of Pharmacy (N°2020/115/CE/FMOS/FAPH) of the University of Science, Techniques and Technologies of Bamako, Mali.
Informed Consent Statement
Patient consent was waived because this is part of the COVID-19 surveillance activities.
Data Availability Statement
Genome sequence has been deposited on the GISAID repository under the accession numbers EPI_ISL_683835 and EPI_ISL_2683873 (
https://www.gisaid.org/ (accessed on 3 November 2021)). The sequences are also available on NCBI under the GenBank accession numbers OL336607 and OL448870.
Acknowledgments
We are thankful French Development Agency (AFD) supported the acquisition of the Illumina MiSeq instrument.
Conflicts of Interest
Authors declared no conflict of interest.
References
- Pater, A.A.; Bosmeny, M.S.; Barkau, C.L.; Ovington, K.N.; Chilamkurthy, R.; Parasrampuria, M.; Eddington, S.B.; Yinusa, A.O.; White, A.A.; Metz, P.E.; et al. Emergence and Evolution of a Prevalent New SARS-CoV-2 Variant in the United States. BioRxiv 2021. Available online: https://www.biorxiv.org/content/10.1101/2021.01.11.426287v2 (accessed on 23 November 2021).
- Makoni, M. South Africa responds to new SARS-CoV-2 variant. Lancet 2021, 397, 267. [Google Scholar] [CrossRef]
- Tang, J.W.; Tambyah, P.A.; Hui, D.S. Emergence of a new SARS-CoV-2 variant in the UK. J. Infect. 2021, 82, e27–e28. [Google Scholar] [CrossRef] [PubMed]
- Rambaut, A.; Loman, N.; Pybus, O.; Barclay, W.; Barrett, J.; Carabelli, A.; Connor, T.; Peacock, T.; Robertson, D.L.; Volz, E. Preliminary Genomic Characterisation of an Emergent SARS-CoV-2 Lineage in the UK Defined by a Novel Set of Spike Mutations. Genome Epidemiol. 2020. Available online: https://virological.org/t/preliminary-genomic-characterisation-of-an-emergent-sars-cov-2-lineage-in-the-uk-defined-by-a-novel-set-of-spike-mutations/563 (accessed on 23 November 2021).
- Happi, A.N.; Ugwu, C.A.; Happi, C.T. Tracking the emergence of new SARS-CoV-2 variants in South Africa. Nat. Med. 2021, 27, 372–373. [Google Scholar] [CrossRef] [PubMed]
- Naveca, F.; Nascimento, V.; Souza, V.; Corado, A.; Nascimento, F.; Silva, G.; Costa, Á.; Duarte, D.; Pessoa, K.; Mejía, M.; et al. COVID-19 in Amazonas, Brazil, was driven by the persistence of endemic lineages and P.1 emergence. Nat. Med. 2021, 27, 1230–1238. [Google Scholar] [CrossRef] [PubMed]
- Resende, P.C.; Bezerra, J.F.; Vasconcelos, R.; Arantes, I.; Appolinario, L.; Mendonça, A.C.; Paixao, A.C.; Rodrigues, A.C.D.; Silva, T.; Rocha, A.S.; et al. Spike E484K Mutation in the First SARS-CoV-2 Reinfection Case Confirmed in Brazil, 2020. Virological 2021. Available online: https://virological.org/t/spike-e484k-mutation-in-the-first-sars-cov-2-reinfection-case-confirmed-in-brazil-2020/584 (accessed on 23 November 2021).
- Okwuraiwe, A.; Onwuamah, C.; Amoo, O.; Salako, B. First African SARS-CoV-2 Genome Sequence from Nigerian COVID-19 Case. Res. Gate 2020. Available online: https://virological.org/t/first-african-sars-cov-2-genome-sequence-from-nigerian-covid-19-case/421 (accessed on 23 November 2021).
- Ziraldo, R.; Shoura, M.J.; Fire, A.Z.; Levene, S.D. Deconvolution of nucleic-acid length distributions: A gel electrophoresis analysis tool and applications. Nucleic Acids Res. 2019, 47, e92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thorvaldsdóttir, H.; Robinson, J.T.; Mesirov, J.P. Integrative Genomics Viewer (IGV): High-performance genomics data visualization and exploration. Brief. Bioinform. 2013, 14, 178–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cavener, D.R. Comparison of the consensus sequence flanking translational start sites in Drosophila and vertebrates. Nucleic Acids Res. 1987, 15, 1353–1361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, D.; Liu, C.-M.; Luo, R.; Sadakane, K.; Lam, T.-W. MEGAHIT: An ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 2015, 31, 1674–1676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marçais, G.; Delcher, A.L.; Phillippy, A.; Coston, R.; Salzberg, S.; Zimin, A. MUMmer4: A fast and versatile genome alignment system. PLoS Comput. Biol. 2018, 14, e1005944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yurkovetskiy, L.; Wang, X.; Pascal, K.E.; Tomkins-Tinch, C.; Nyalile, T.P.; Wang, Y.; Baum, A.; Diehl, W.E.; Dauphin, A.; Carbone, C.; et al. Structural and Functional Analysis of the D614G SARS-CoV-2 Spike Protein Variant. Cell 2020, 183, 739–751.e8. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Jackson, C.B.; Mou, H.; Ojha, A.; Peng, H.; Quinlan, B.D.; Rangarajan, E.S.; Pan, A.; Vanderheiden, A.; Suthar, M.S.; et al. SARS-CoV-2 spike-protein D614G mutation increases virion spike density and infectivity. Nat. Commun. 2020, 11, 6013. [Google Scholar] [CrossRef] [PubMed]
- Institut National de Sante Publique. 2020. Available online: https://insp.ml/covid-ml/ (accessed on 3 November 2021).
| Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).


 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
