
In Silico Assessment and Molecular Docking Studies of Some Phyto-Triterpenoid for Potential Disruption of Mortalin-p53 Interaction

 ,
,  and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Ligand Preparation
2.2. Protein Preparation
2.3. Validation Docking
2.4. Molecular Docking Using AutoDock 4.2.6
3. Results and Discussion
3.1. Validation Docking
3.2. Drug-Like and Pharmacokinetic Properties Assessment
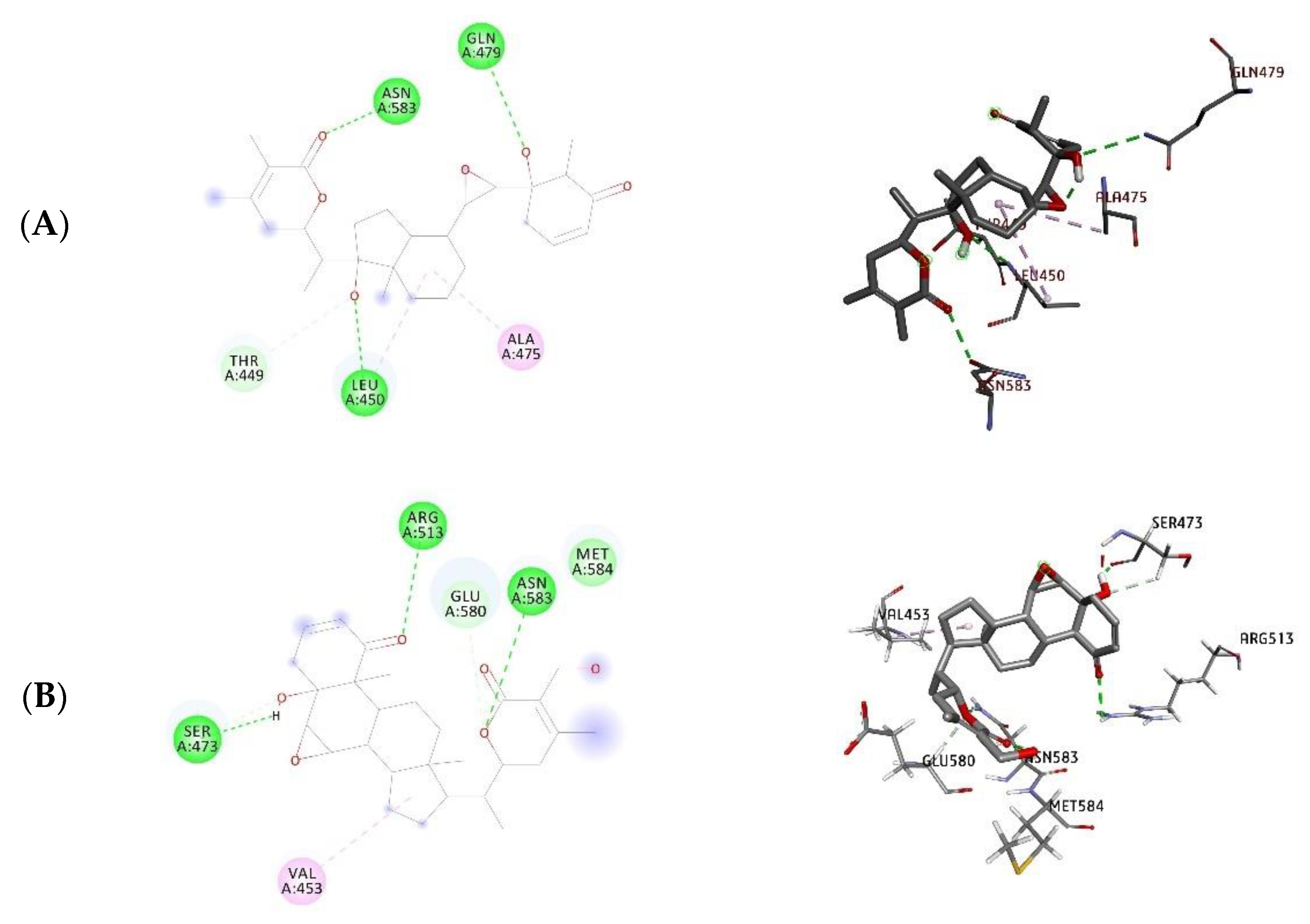
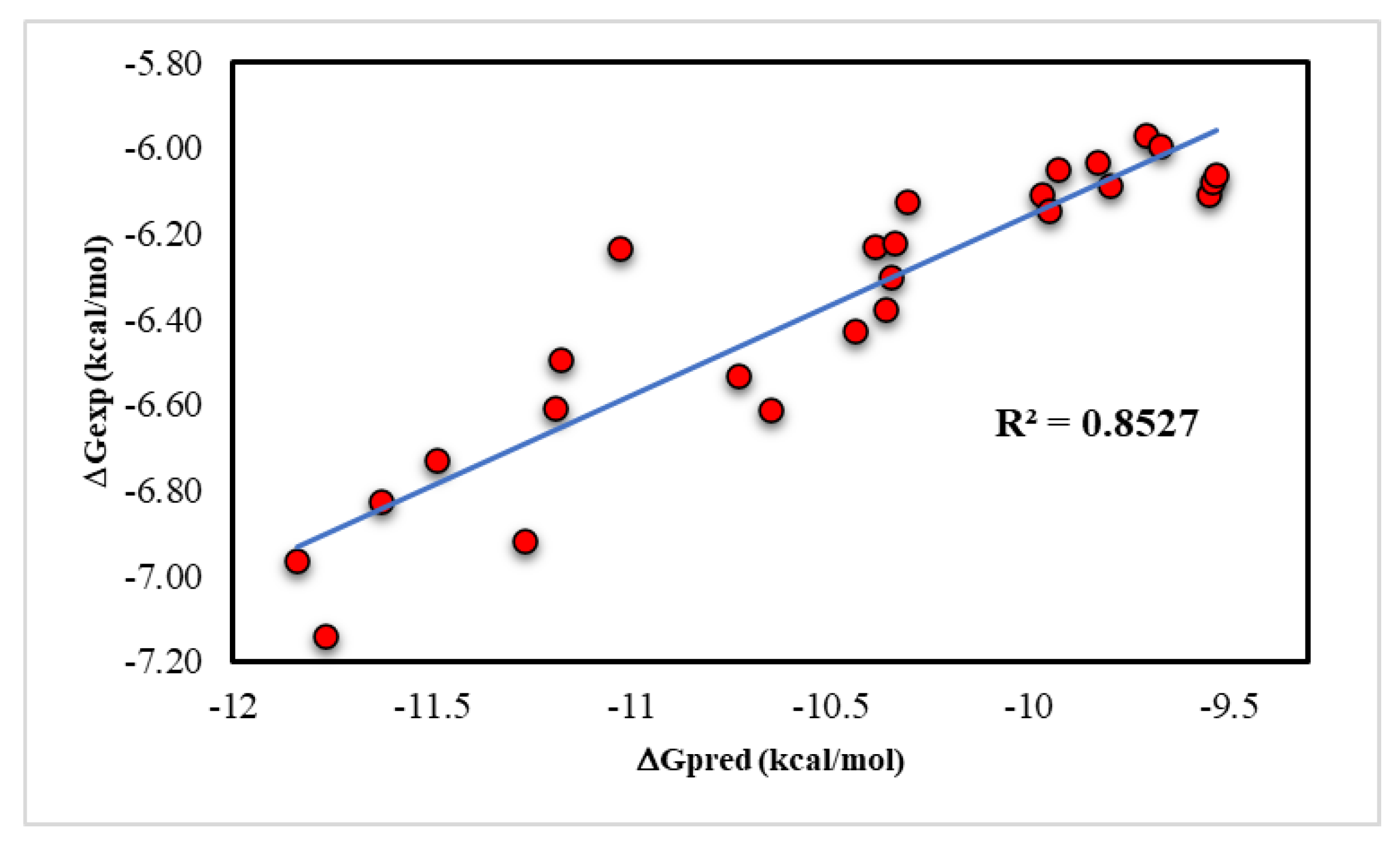
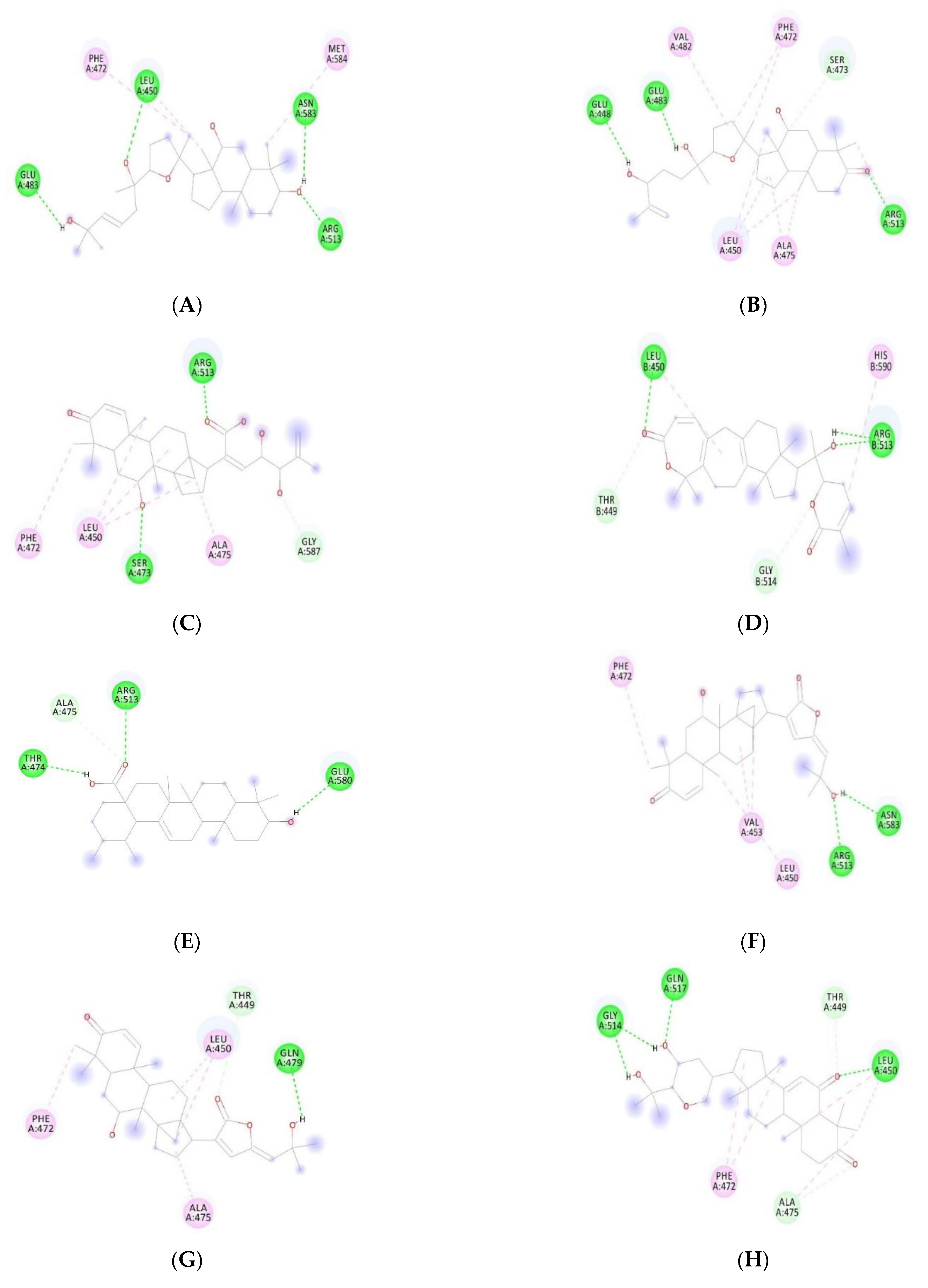
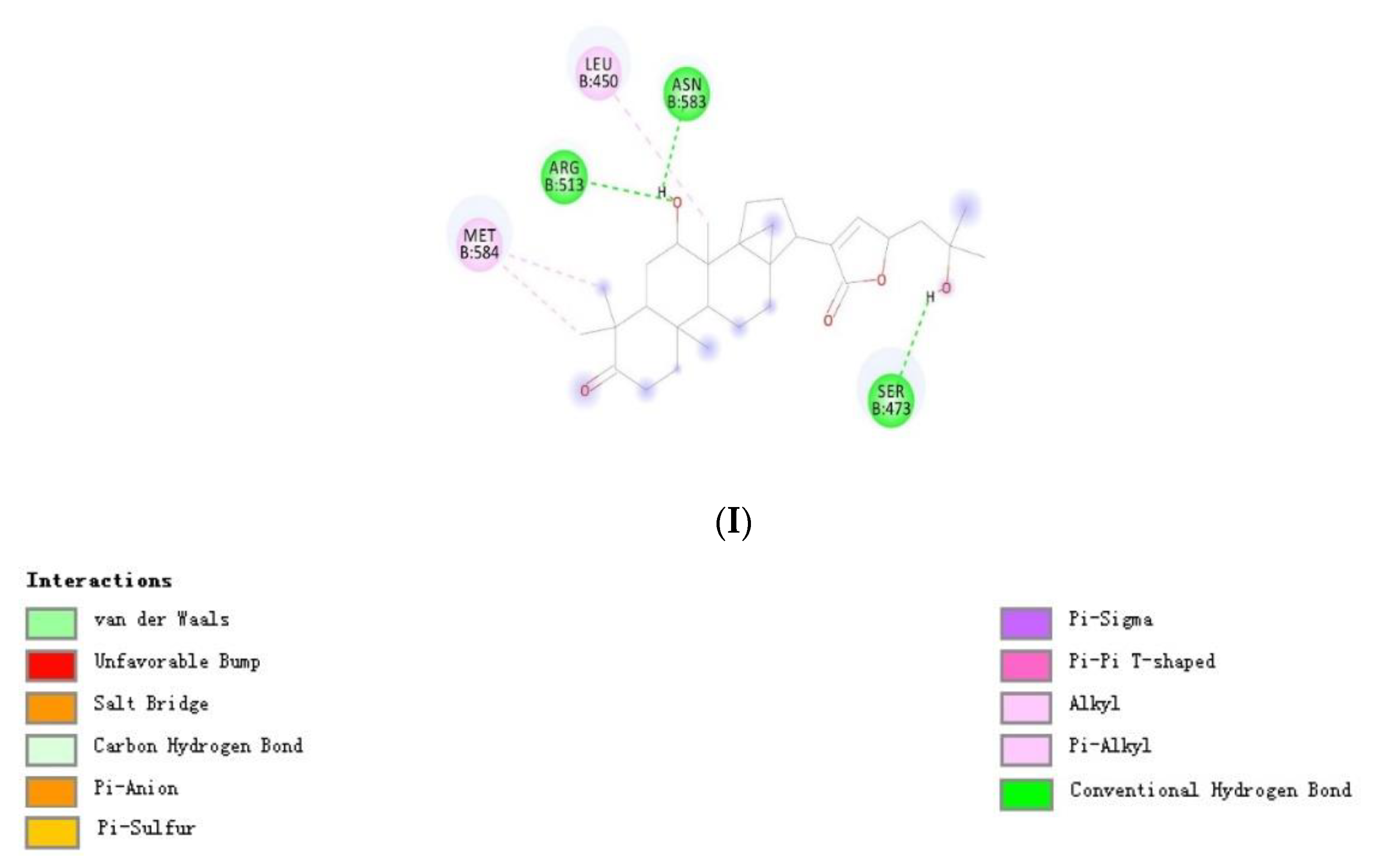
3.3. Docking Studies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Chudzik, M.; Korzonek-Szlacheta, I.; Król, W. Triterpenes as Potentially Cytotoxic Compounds. Molecules 2015, 20, 1610–1625. [Google Scholar] [CrossRef]
- Thimmappa, R.; Geisler, K.; Louveau, T.; O’Maille, P.; Osbourn, A. Triterpene Biosynthesis in Plants. Annu. Rev. Plant Biol. 2014, 65, 225–257. [Google Scholar] [CrossRef] [PubMed]
- Duy, L.X.; Toan, T.Q.; Nghi, D.H.; Thanh, L.T.; Hoang, V.D.; Kim, Y.H.; Cuong, N.M. Triterpenes from Docynia indica fruits and their cytotoxic activity. Vietnam J. Sci. Technol. 2018, 56, 199. [Google Scholar] [CrossRef]
- Liu, X.-Y.; Wang, X.-L.; Shen, T.; Ren, D.-M.; Lou, H.-X.; Wang, X.-N. Two new triterpenoids from the fungus Diplodia cupressi. Nat. Prod. Res. 2019, 34, 2179–2185. [Google Scholar] [CrossRef] [PubMed]
- Qiu, F.; Liu, H.; Duan, H.; Chen, P.; Lu, S.-J.; Yang, G.-Z.; Lei, X.-X. Isolation, Structural Elucidation of Three New Triterpenoids from the Stems and Leaves of Schisandra chinensis (Turcz) Baill. Molecules 2018, 23, 1624. [Google Scholar] [CrossRef]
- Liu, Y.; Gu, W. The complexity of p53-mediated metabolic regulation in tumor suppression. Semin. Cancer Biol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Boysen, M.; Kityk, R.; Mayer, M.P. Hsp70- and Hsp90-mediated regulation of the conformation of p53 DNA binding domain and p53 cancer variants. Mol. Cell 2019, 74, 831–843. [Google Scholar] [CrossRef] [PubMed]
- Yendo, A.C.A.; de Costa, F.; Gosmann, G.; Fett-Neto, A.G. Production of Plant Bioactive Triterpenoid Saponins: Elicitation Strategies and Target Genes to Improve Yields. Mol. Biotechnol. 2010, 46, 94–104. [Google Scholar] [CrossRef] [PubMed]
- Schrodinger, LLC. The PyMOL Molecular Graphics System, Version 1.3, 2010.
- Allouche, A.-R. Gabedit—A graphical user interface for computational chemistry softwares. J. Comput. Chem 2011, 32, 174–182. [Google Scholar] [CrossRef] [PubMed]
- Molinspiration Cheminformatics Free Web Services, Slovensky Grob, Slovakia. Available online: https://www.molinspiration.com (accessed on 20 April 2021).
- Banerjee, P.; Eckert, A.O.; Schrey, A.K.; Preissner, R. ProTox-II: A webserver for the prediction of toxicity of chemicals. Nucleic Acids Res. 2018, 46, W257–W263. [Google Scholar] [CrossRef]
- Amick, J.; Schlanger, S.E.; Wachnowsky, C.; Moseng, M.A.; Emerson, C.C.; Dare, M.; Luo, W.-I.; Ithychanda, S.S.; Nix, J.C.; Cowan, J.A.; et al. Crystal structure of the nucleotide-binding domain of mortalin, the mitochondrial Hsp70 chaperone. Protein Sci. 2014, 23, 833–842. [Google Scholar] [CrossRef] [PubMed]
- Moseng, M.A.; Nix, J.C.; Page, R.C. 2- and N6-functionalized adenosine-5′-diphosphate analogs for the inhibition of mortalin. FEBS Lett. 2019, 593, 2030–2039. [Google Scholar] [CrossRef] [PubMed]
- Grover, A.; Priyandoko, D.; Gao, R.; Shandilya, A.; Widodo, N.; Bisaria, V.S.; Kaul, S.C.; Wadhwa, R.; Sundar, D. Withanone binds to mortalin and abrogates mortalin–p53 complex: Computational and experimental evidence. Int. J. Biochem. Cell Biol. 2012, 44, 496–504. [Google Scholar] [CrossRef]
- Kaul, S.C.; Reddel, R.R.; Mitsui, Y.; Wadhwa, R. An N-terminal Region of Mot-2 Binds to p53 In Vitro. Neoplasia 2001, 3, 110–114. [Google Scholar] [CrossRef]
- Kaul, S.C.; Yaguchi, T.; Taira, K.; Reddel, R.R.; Wadhwa, R. Overexpressed mortalin (mot-2)/mthsp70/GRP75 and hTERT cooperate to extend the in vitro lifespan of human fibroblasts. Exp. Cell Res. 2003, 286, 96–101. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed]
- Vaishnavi, K.; Saxena, N.; Shah, N.; Singh, R.; Manjunath, K.; Uthayakumar, M.; Kanaujia, S.P.; Kaul, S.C.; Sekar, K.; Wadhwa, R. Differential Activities of the Two Closely Related Withanolides, Withaferin A and Withanone: Bioinformatics and Experimental Evidences. PLoS ONE 2012, 7, e44419. [Google Scholar] [CrossRef]
- Pham, M.Q.; Tran, T.H.V.; Pham, Q.L.; Gairin, J.E. In silico analysis of the binding properties of solasonine to mortalin and p53, and in vitro pharmacological studies of its apoptotic and cytotoxic effects on human HepG2 and Hep3b hepatocellular carcinoma cells. Fund Clin. Pharmacol. 2019, 33, 385–396. [Google Scholar] [CrossRef] [PubMed]
- Utomo, D.H.; Widodo, N.; Rifa’i, M. Identifications small molecules inhibitor of p53-mortalin complex for cancer drug using virtual screening. Bioinformation 2012, 8, 426–429. [Google Scholar] [CrossRef]
- Dassault Systèmes BIOVIA. Discovery Studio Visualizer, v17.2.0.16349; Dassault Systèmes: San Diego, CA, USA, 2019. [Google Scholar]
- Laskowski, R.A.; Swindells, M.B. LigPlot+: Multiple Ligand–Protein Interaction Diagrams for Drug Discovery. J. Chem. Inf. Model. 2011, 51, 2778–2786. [Google Scholar] [CrossRef] [PubMed]
- Maestro, S.R. Schrödinger Release 2019-4: Maestro; Schrödinger, LLC: New York, NY, USA, 2019. [Google Scholar]
- Huang, C.; Vaishnavi, K.; Kalra, R.S.; Zhang, Z.; Sekar, K.; Kaul, S.C.; Wadha, R. 3β-Methoxy derivation of Withaferin-A attenuates its anticancer potency: Bioinformatics and molecular evidences. J. Medicinal Aromat. Plants 2015, 4, 1000219. [Google Scholar]
- Lipinski, C.; Hopkins, A. Navigating chemical space for biology and medicine. Nature 2004, 432, 855–861. [Google Scholar] [CrossRef]
- Garcia-Sosa, A.T.; Maran, U.; Hetenyi, C. Molecular property filters describing pharmacokinetics and drug binding. Curr. Med. Chem. 2012, 19, 1646–1662. [Google Scholar] [CrossRef] [PubMed]
- Ganesan, A. The impact of natural products upon modern drug discovery. Curr. Opin. Chem. Biol. 2008, 12, 306–317. [Google Scholar] [CrossRef] [PubMed]
- Khan, T.; Dixit, S.; Ahmad, R.; Raza, S.; Azad, I.; Joshi, S.; Khan, A.R. Molecular docking, PASS analysis, bioactivity score prediction, synthesis, characterization and biological activity evaluation of a functionalized 2-butanone thiosemicarbazone ligand and its complexes. J. Chem. Biol. 2017, 10, 91–104. [Google Scholar] [CrossRef]
- Alam, M.S.; Nam, Y.J.; Lee, D.U. Synthesis and evaluation of (Z)-2,3-diphenylacrylonitrile analogs as anti-cancer and anti-microbial agents. Eur. J. Med. Chem. 2013, 69, 790–797. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.C. The power issue: Determination of KB or Ki from IC50: A closer look at the Cheng–Prusoff equation, the Schild plot and related power equations. J. Pharmacol. Toxicol. Methods. 2001, 46, 61–71. [Google Scholar] [CrossRef]
- Soica, C.; Voicu, M.; Ghiulai, R.; Dehelean, C.; Racoviceanu, R.; Trandafirescu, C.; Rosca, O.J.; Nistor, G.; Mioc, M.; Mioc, A. Natural compounds in sex hormone-dependent cancers: The role of triterpenes as therapeutic agents. Front. Endocrinol. 2021, 11, 612396. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Ma, H.; Shi, W.; Duan, J.; Wang, Y.; Zhang, C.; Li, C.; Lin, J.; Li, S.; Jiagao, L.V.; et al. Inhibition of STAT3 signaling pathway by ursolic acid suppresses growth of hepatocellular carcinoma. Int. J. Oncol. 2017, 51, 555–562. [Google Scholar] [CrossRef] [PubMed]
- Kaul, S.C.; Deocaris, C.C.; Wadhwa, R. Three faces of mortalin: A housekeeper, guardian and killer. Exp. Gerontol. 2007, 42, 263–274. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound Name | Dock Score (kcal/mol) | H-Bond Interacting Residues | ||
|---|---|---|---|---|
| 3N8E | 4KBO | 3N8E | Ref. [19] | |
| Withanone | −12.08 | −10.40 | Leu450, Gln479, Asn583 | Leu450, Arg513, Asn583 |
| Withaferin A | −10.76 | −8.54 | Ser473, Arg513, Asn583 | Glu483, Arg513, Asn583, Gly587 |
| ID | Compound Name | MW | HBD | HBA | LogP | MR (cm3/mol) |
|---|---|---|---|---|---|---|
| 1 | 3-oxo-threo-23,24,25-trihydroxytirucall-7-ene | 474 | 3 | 4 | 5.97 | 137.60 |
| 2 * | 23,24,25,26,27-pentanorlanost-7,9(11)-dien-3b,22-diol | 358 | 2 | 2 | 4.72 | 109.02 |
| 3 * | 23,24,25,26,27-pentanorlanost-8-en-3b,22-diol | 374 | 2 | 2 | 5.51 | 112.06 |
| 4 | 23-hydroxyursolic acid | 472 | 3 | 4 | 6.27 | 134.39 |
| 5 | 28-formyloxy-3ß-hydroxy-urs-12-ene | 470 | 1 | 3 | 7.32 | 138.32 |
| 6 | Ailanaltiolide A | 630 | 3 | 9 | 3.87 | 172.39 |
| 7 | Ailanaltiolide B | 630 | 3 | 9 | 3.87 | 172.39 |
| 8 | Ailanaltiolide C | 612 | 3 | 9 | 3.44 | 167.28 |
| 9 | Ailanaltiolide D | 626 | 2 | 9 | 3.8 | 172.04 |
| 10 | Ailanaltiolide E | 594 | 2 | 8 | 4.28 | 165.57 |
| 11 | Ailanaltiolide F | 586 | 1 | 9 | 3.19 | 154.90 |
| 12 | Ailanaltiolide G | 598 | 3 | 8 | 3.58 | 165.93 |
| 13 | Ailanaltiolide H | 522 | 4 | 6 | 4.54 | 146.84 |
| 14 * | Ailanaltiolide I | 486 | 2 | 5 | 4.27 | 136.62 |
| 15 * | Ailanaltiolide J | 430 | 1 | 4 | 5.3 | 119.37 |
| 16 * | Ailanthusin B | 480 | 2 | 5 | 3.75 | 136.66 |
| 17 * | Ailanthusin A | 480 | 2 | 5 | 3.75 | 136.66 |
| 18 * | Ailanthusin C | 482 | 2 | 5 | 4.43 | 134.73 |
| 19 * | Ailanthusin D | 498 | 4 | 6 | 3.81 | 137.71 |
| 20 * | Ailanthusin E | 362 | 3 | 4 | 4.11 | 97.33 |
| 21 * | Ailanthusin F | 492 | 4 | 5 | 4.19 | 140.53 |
| 22 * | Ailanthusin G | 490 | 3 | 5 | 4.75 | 137.98 |
| 23 | Arjunic acid | 502 | 4 | 5 | 5.92 | 140.16 |
| 24 * | Belamchinenin A | 500 | 2 | 5 | 3.91 | 147.25 |
| 25 | Bourjutinolone A | 472 | 2 | 4 | 5.47 | 135.68 |
| 26 * | Cabralealactone | 414 | 0 | 3 | 6.46 | 117.91 |
| 27 | Corosolic acid | 472 | 3 | 4 | 6.38 | 134.48 |
| 28 | Euscaphic acid | 488 | 4 | 5 | 5.18 | 135.70 |
| 29 | Kadsuphilactone B | 482 | 1 | 5 | 5.04 | 135.03 |
| 30 | Maslinic acid | 472 | 3 | 4 | 6.52 | 134.10 |
| 31 | Oleanolic acid | 470 | 2 | 3 | 7.89 | 137.25 |
| 32 | Pomolic acid | 472 | 3 | 4 | 6.13 | 134.24 |
| 33 * | Schinalactone D | 514 | 1 | 5 | 6.19 | 144.80 |
| 34 * | Schinchinenlactone A | 494 | 0 | 6 | 2.91 | 136.25 |
| 35 | Schisanlactone C | 480 | 1 | 5 | 4.33 | 138.80 |
| 36 | Schisanlactone I | 486 | 1 | 5 | 5.22 | 139.30 |
| 37 * | Schisanlactone J | 478 | 0 | 5 | 4.08 | 138.26 |
| 38 * | Ursolic acid | 456 | 2 | 3 | 7.33 | 123.02 |
| 39 | Tormentic acid | 488 | 4 | 5 | 5.18 | 135.70 |
| 40 | Lanost-8-en-3b,22S,23S-triol | 458 | 3 | 3 | 6.19 | 137.60 |
| 41 | Schisphendilactone B | 494 | 0 | 6 | 3.34 | 136.74 |
| Withanone | 470 | 2 | 6 | 3.49 | 124.51 | |
| Withaferin A | 470 | 2 | 6 | 3.35 | 124.46 |
| Compound ID | Dock Score (kcal/mol) | Compound ID | Dock Score (kcal/mol) |
|---|---|---|---|
| Withanone | −12.25 | 14 | −9.71 |
| 21 | −11.84 | 30 | −9.67 |
| 22 | −11.77 | 40 | −9.55 |
| 19 | −11.63 | 18 | −9.54 |
| 9 | −11.49 | 3 | −9.53 |
| 1 | −11.27 | Withaferin A | −9.51 |
| 34 | −11.19 | 8 | −9.48 |
| 37 | −11.18 | 5 | −9.42 |
| 12 | −11.03 | 35 | −9.34 |
| 38 | −10.73 | 36 | −9.31 |
| 16 | −10.65 | 24 | −9.29 |
| 29 | −10.44 | 2 | −8.85 |
| 7 | −10.39 | 25 | −8.72 |
| 10 | −10.36 | 27 | −8.65 |
| 33 | −10.35 | 23 | −8.54 |
| 15 | −10.34 | 20 | −8.51 |
| 11 | −10.31 | 31 | −8.47 |
| 13 | −9.97 | 32 | −8.33 |
| 41 | −9.95 | 4 | −8.18 |
| 6 | −9.93 | 39 | 7.70 |
| 26 | −9.83 | 28 | −7.35 |
| 17 | −9.80 |
| Compound ID | IC50 (μM) | Compound ID | IC50 (μM) |
|---|---|---|---|
| Withanone | 5.12 ± 0.32 | 14 | 44.74 ± 0.74 |
| 21 | 8.43 ± 0.25 | 30 | 42.83 ± 1.78 |
| 22 | 6.28 ± 1.21 | 40 | 35.54 ± 1.43 |
| 19 | 10.67 ± 1.16 | 18 | 37.43 ± 0.68 |
| 9 | 12.54 ± 0.54 | 3 | 38.26 ± 1.82 |
| 1 | 9.12 ± 0.36 | Withaferin A | 33.50 ± 0.76 |
| 34 | 15.35 ± 0.62 | 8 | 49.23 ± 0.77 |
| 37 | 18.52 ± 0.87 | 5 | 48.75 ± 0.35 |
| 12 | 28.81 ± 0.75 | 35 | >50 |
| 38 | 17.42 ± 0.91 | 36 | >50 |
| 16 | 15.25 ± 1.15 | 24 | 47.33 ± 0.52 |
| 29 | 20.75 ± 1.64 | 2 | >50 |
| 7 | 28.87 ± 0.35 | 25 | >50 |
| 10 | 22.65 ± 0.63 | 27 | >50 |
| 33 | 25.63 ± 0.46 | 23 | >50 |
| 15 | 29.43 ± 0.68 | 20 | >50 |
| 11 | 34.54 ± 0.94 | 31 | >50 |
| 13 | 35.53 ± 0.49 | 32 | >50 |
| 41 | 33.28 ± 0.37 | 4 | >50 |
| 6 | 39.16 ± 0.22 | 39 | >50 |
| 26 | 40.25 ± 0.31 | 28 | >50 |
| 17 | 36.88 ± 1.52 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pham, M.Q.; Le Thi, T.H.; Pham, Q.L.; Le, L.T.; Dao, H.T.; Thi Dang, T.L.; Thuy Nguyen Pham, D.; Pham Thi, H.H. In Silico Assessment and Molecular Docking Studies of Some Phyto-Triterpenoid for Potential Disruption of Mortalin-p53 Interaction. Processes 2021, 9, 1983. https://doi.org/10.3390/pr9111983
Pham MQ, Le Thi TH, Pham QL, Le LT, Dao HT, Thi Dang TL, Thuy Nguyen Pham D, Pham Thi HH. In Silico Assessment and Molecular Docking Studies of Some Phyto-Triterpenoid for Potential Disruption of Mortalin-p53 Interaction. Processes. 2021; 9(11):1983. https://doi.org/10.3390/pr9111983
Chicago/Turabian StylePham, Minh Quan, Thuy Huong Le Thi, Quoc Long Pham, Le Thi Le, Huy Toan Dao, Thanh Le Thi Dang, Dung Thuy Nguyen Pham, and Hai Ha Pham Thi. 2021. "In Silico Assessment and Molecular Docking Studies of Some Phyto-Triterpenoid for Potential Disruption of Mortalin-p53 Interaction" Processes 9, no. 11: 1983. https://doi.org/10.3390/pr9111983
APA StylePham, M. Q., Le Thi, T. H., Pham, Q. L., Le, L. T., Dao, H. T., Thi Dang, T. L., Thuy Nguyen Pham, D., & Pham Thi, H. H. (2021). In Silico Assessment and Molecular Docking Studies of Some Phyto-Triterpenoid for Potential Disruption of Mortalin-p53 Interaction. Processes, 9(11), 1983. https://doi.org/10.3390/pr9111983