
Identifying the Most Potent Dual-Targeting Compound(s) against 3CLprotease and NSP15exonuclease of SARS-CoV-2 from Nigella sativa: Virtual Screening via Physicochemical Properties, Docking and Dynamic Simulation Analysis

 ,
, 
 , ,
, ,  ,
,  , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Compounds and Protein Structure Retrieval
2.2. Calculation of Physicochemical Properties and Prediction of Toxicity Potential
2.3. Molecular Docking
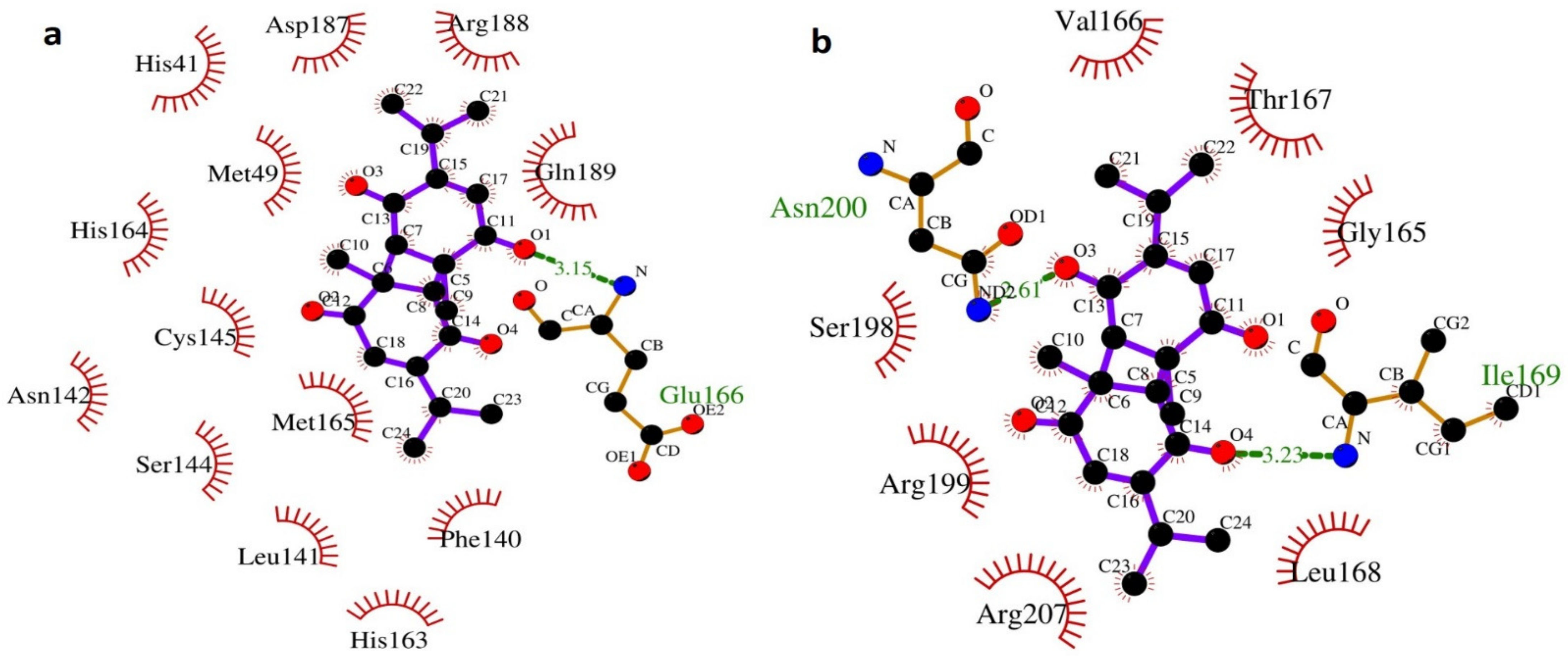
2.4. LIGPLOT+ Analysis
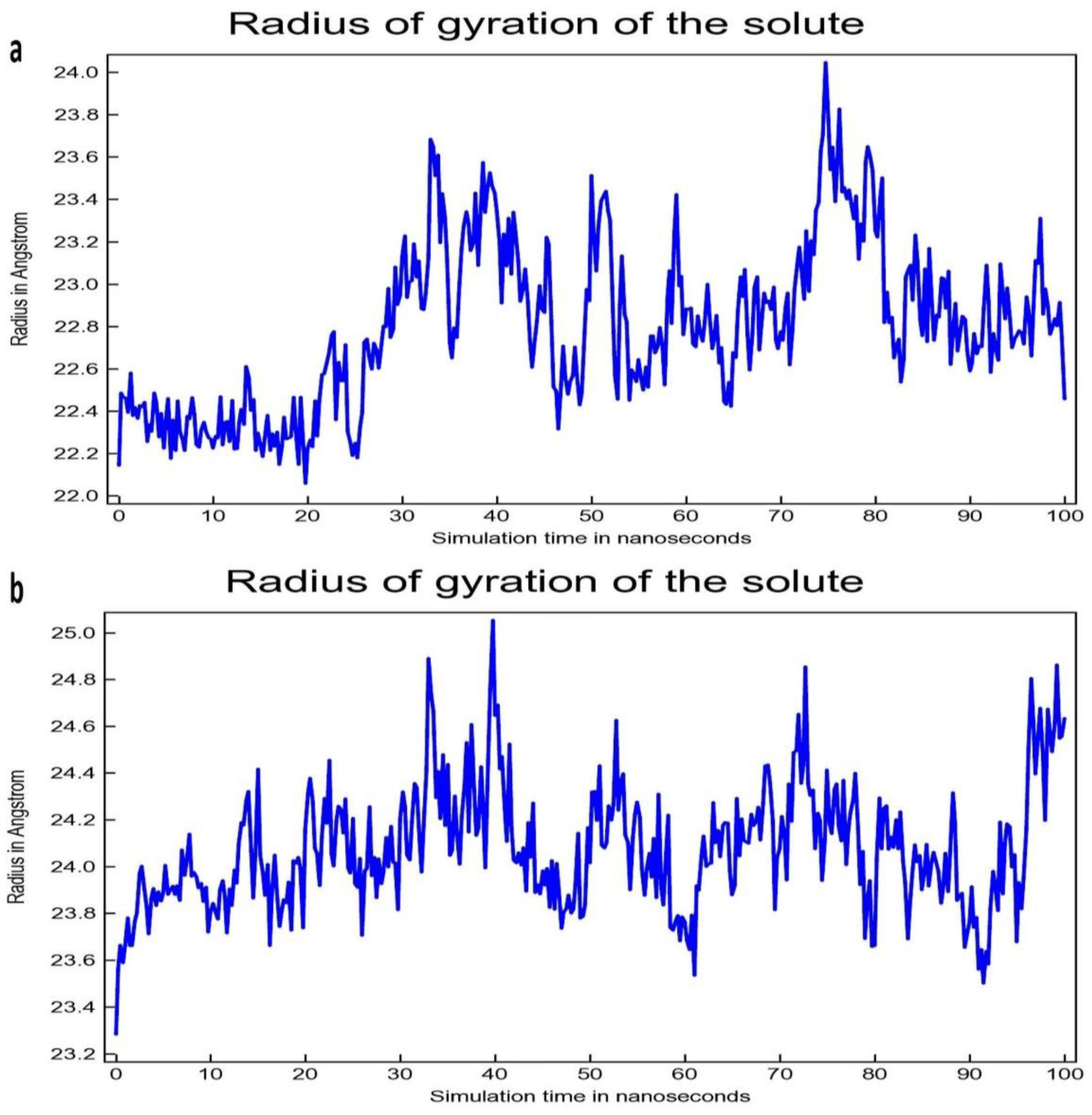
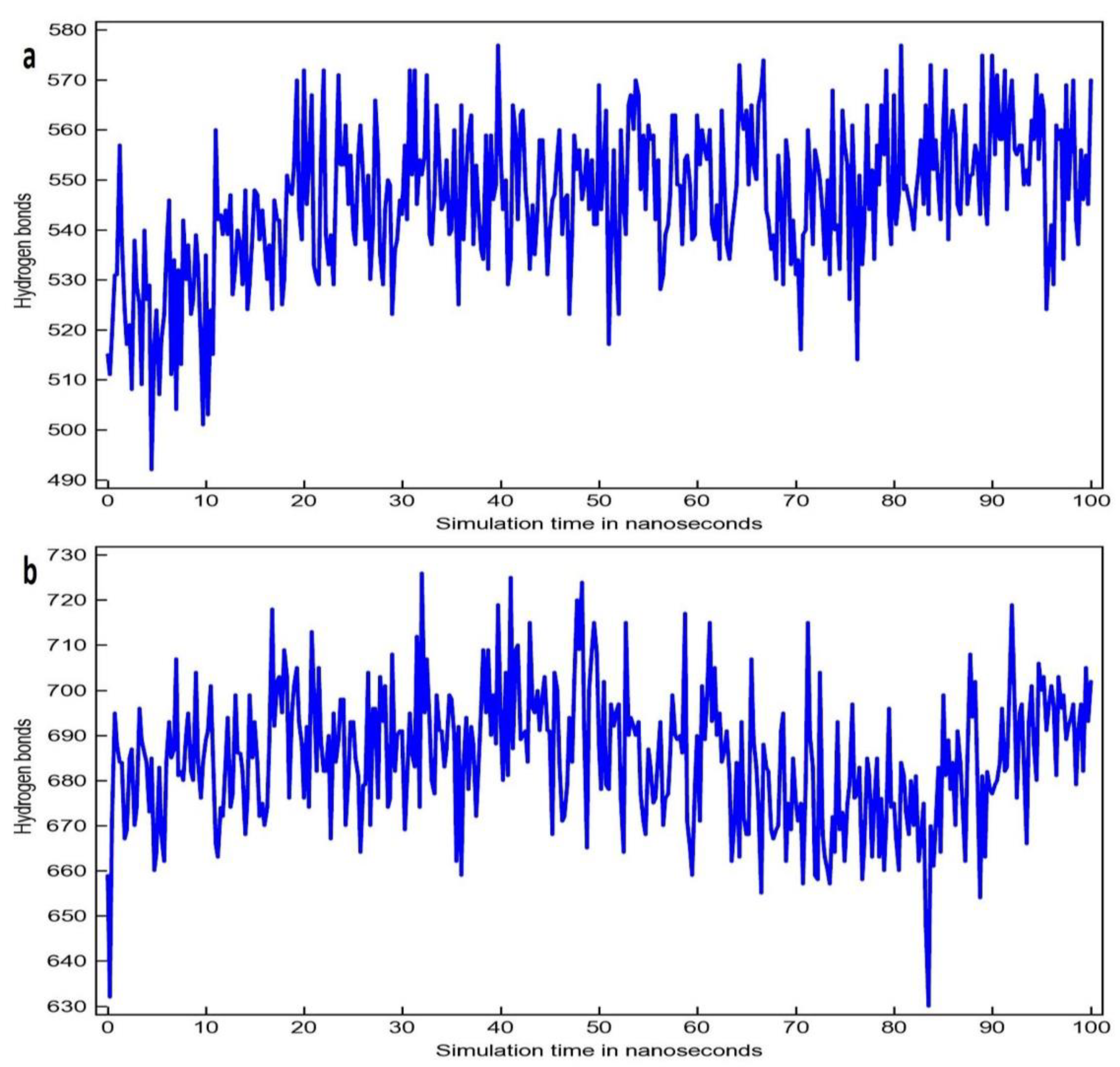
2.5. Molecular Dynamics Simulation Study
3. Results
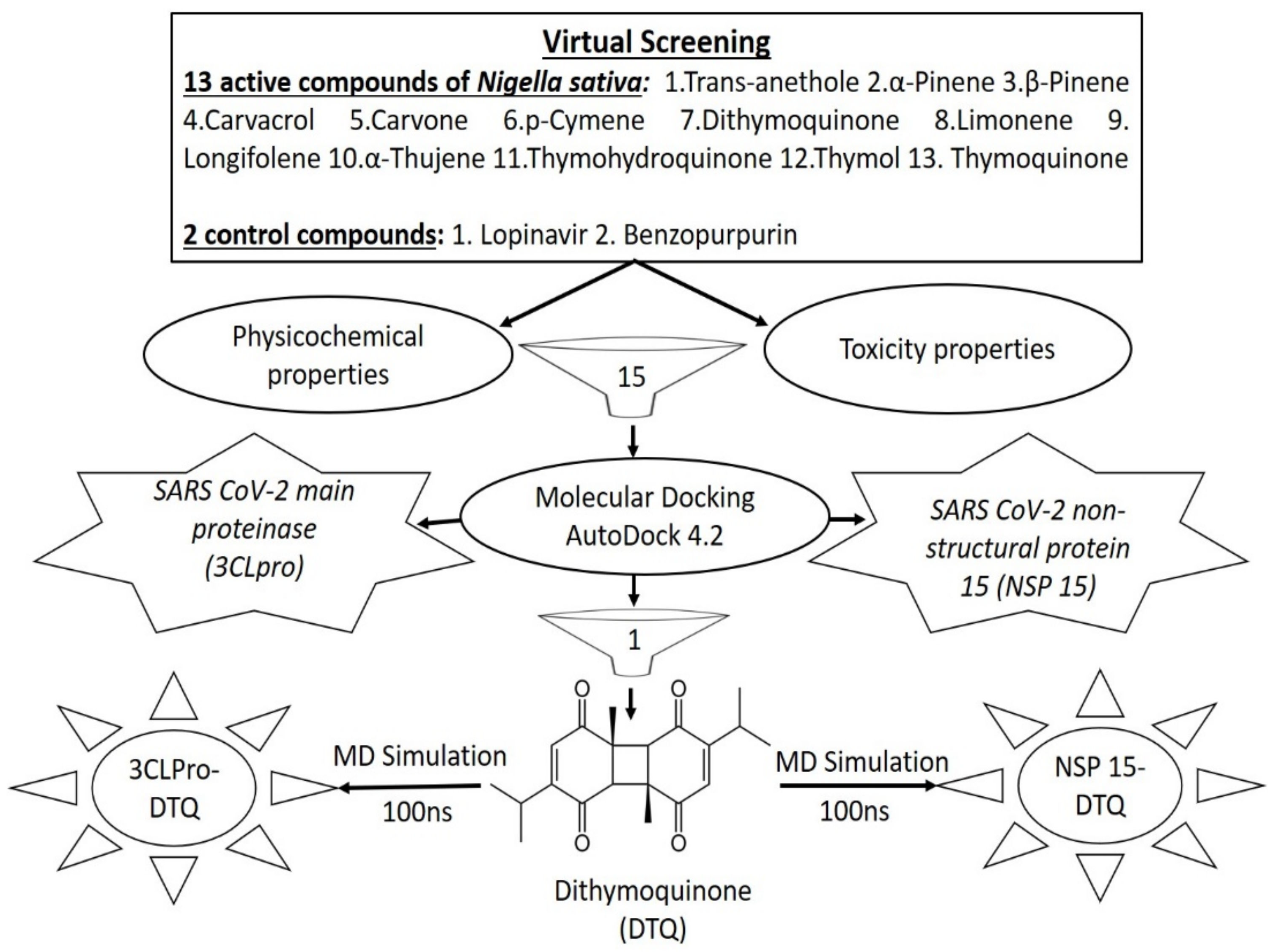
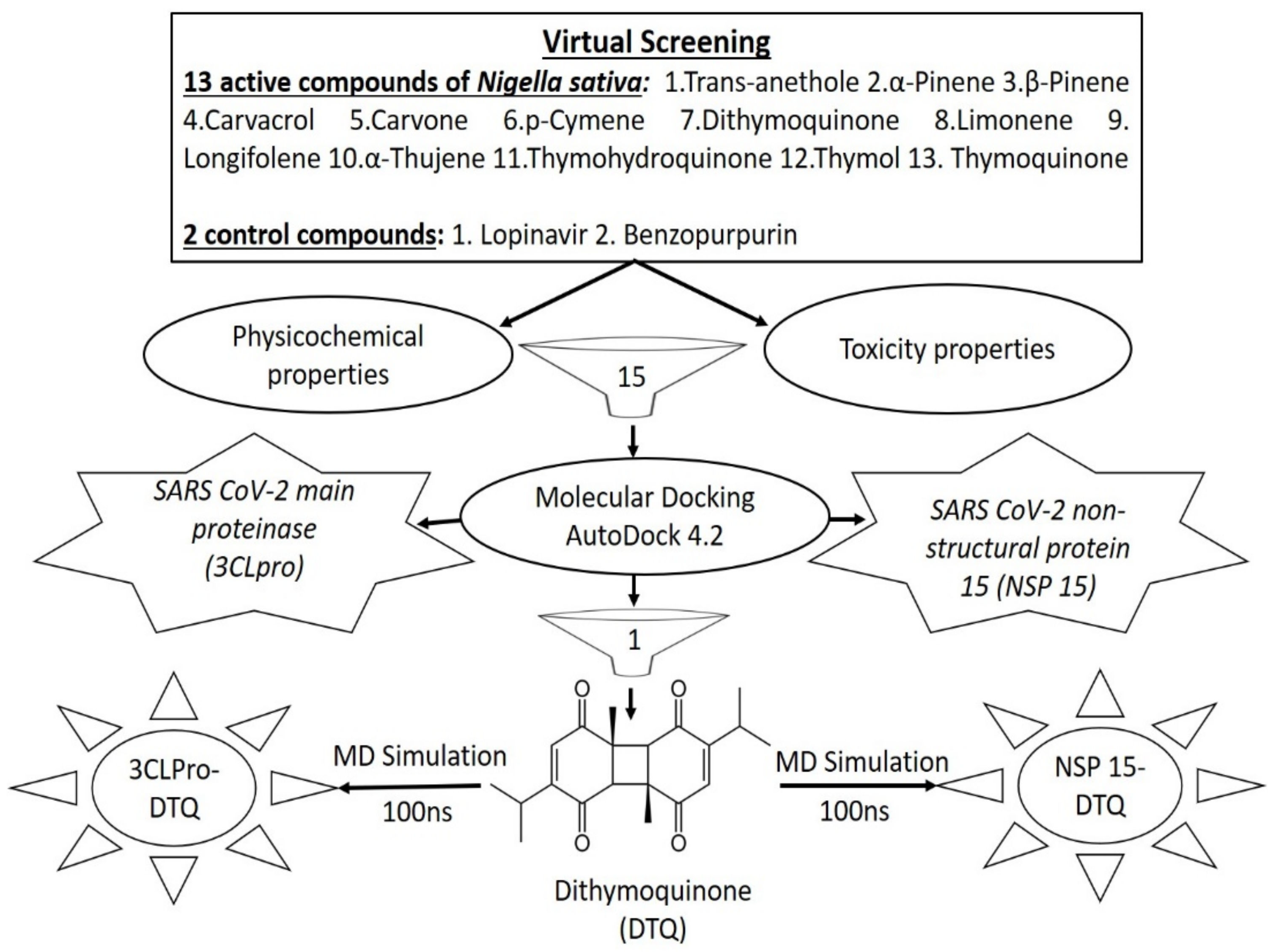
3.1. Virtual Screening
3.1.1. Virtual Screening via Molecular Docking Analysis
3.1.2. Virtual Screening via Physicochemical Properties Analysis
3.1.3. Virtual Screening via Toxicity Assessment
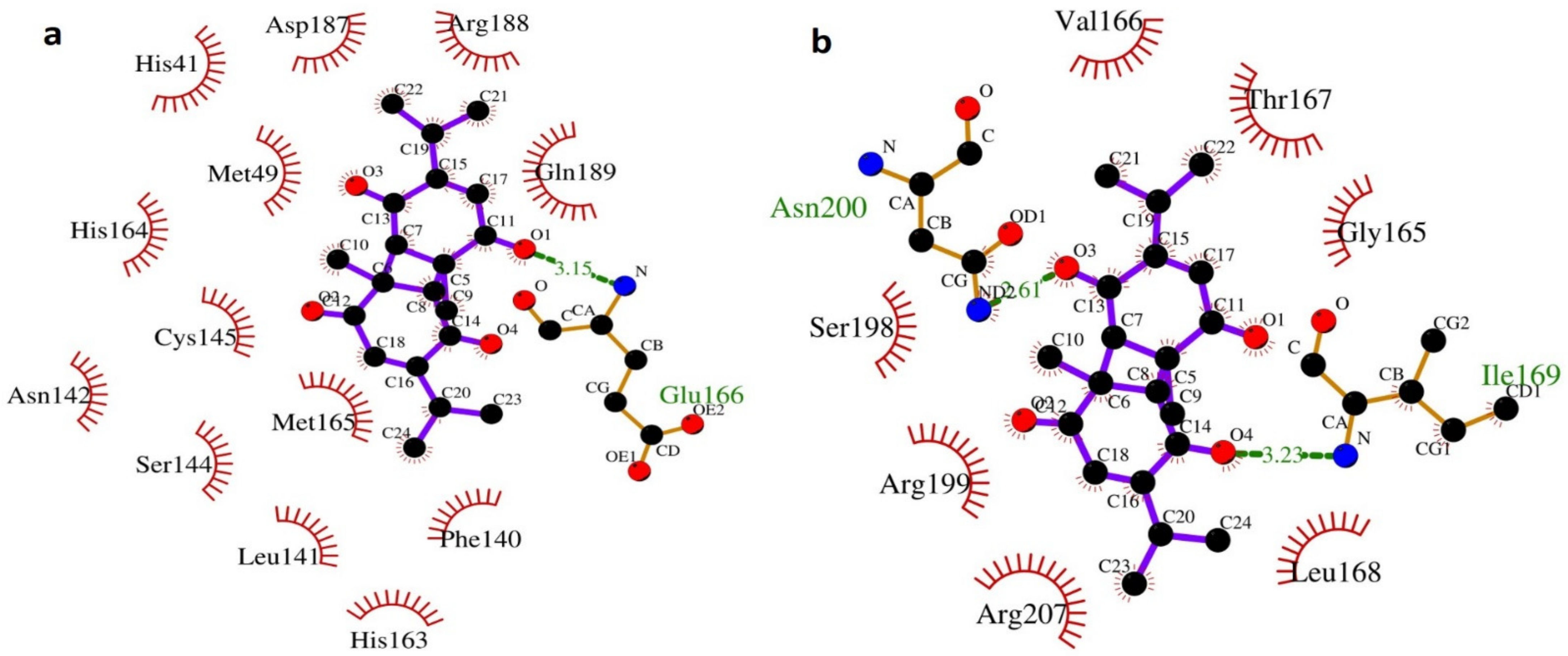
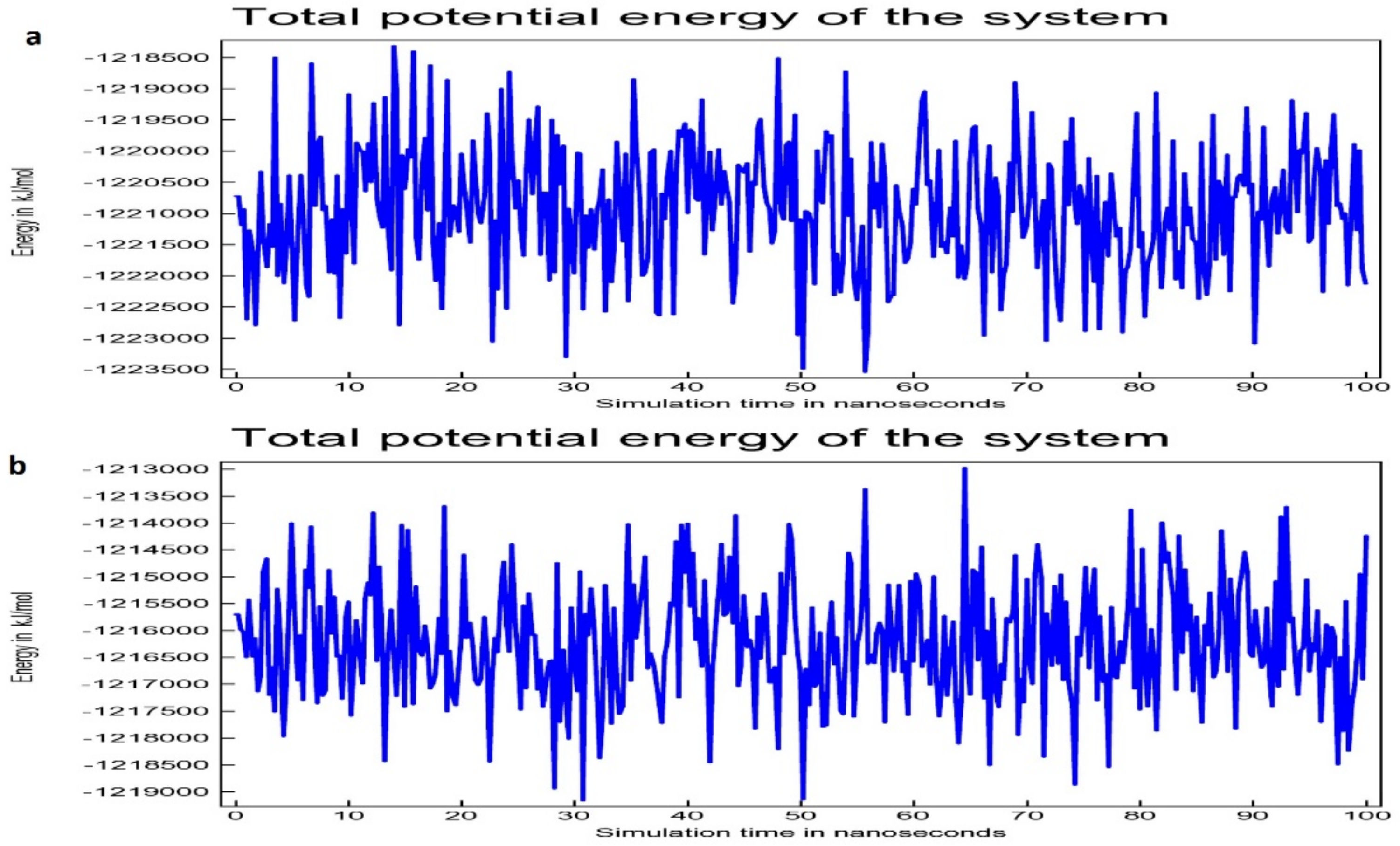
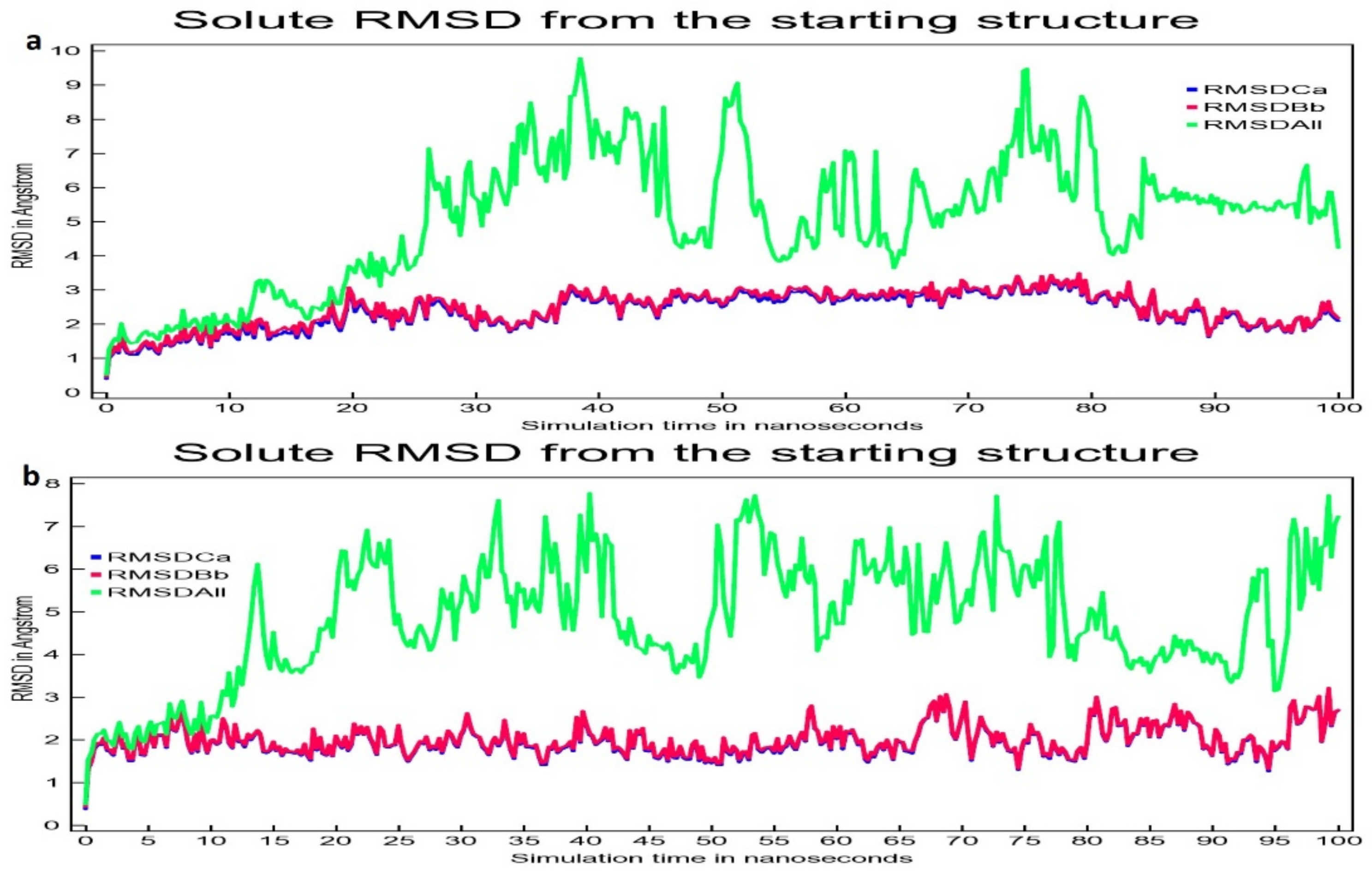
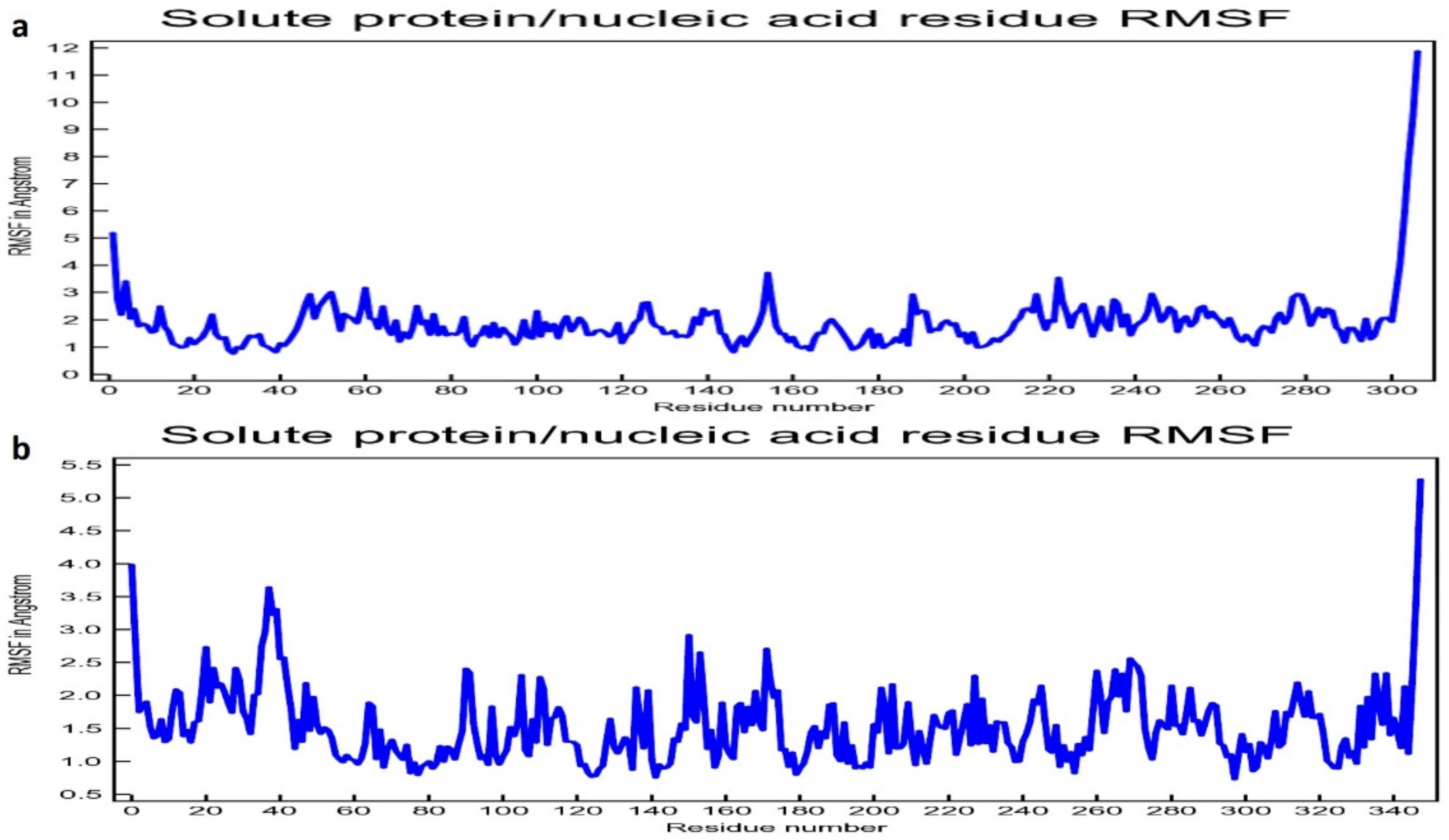
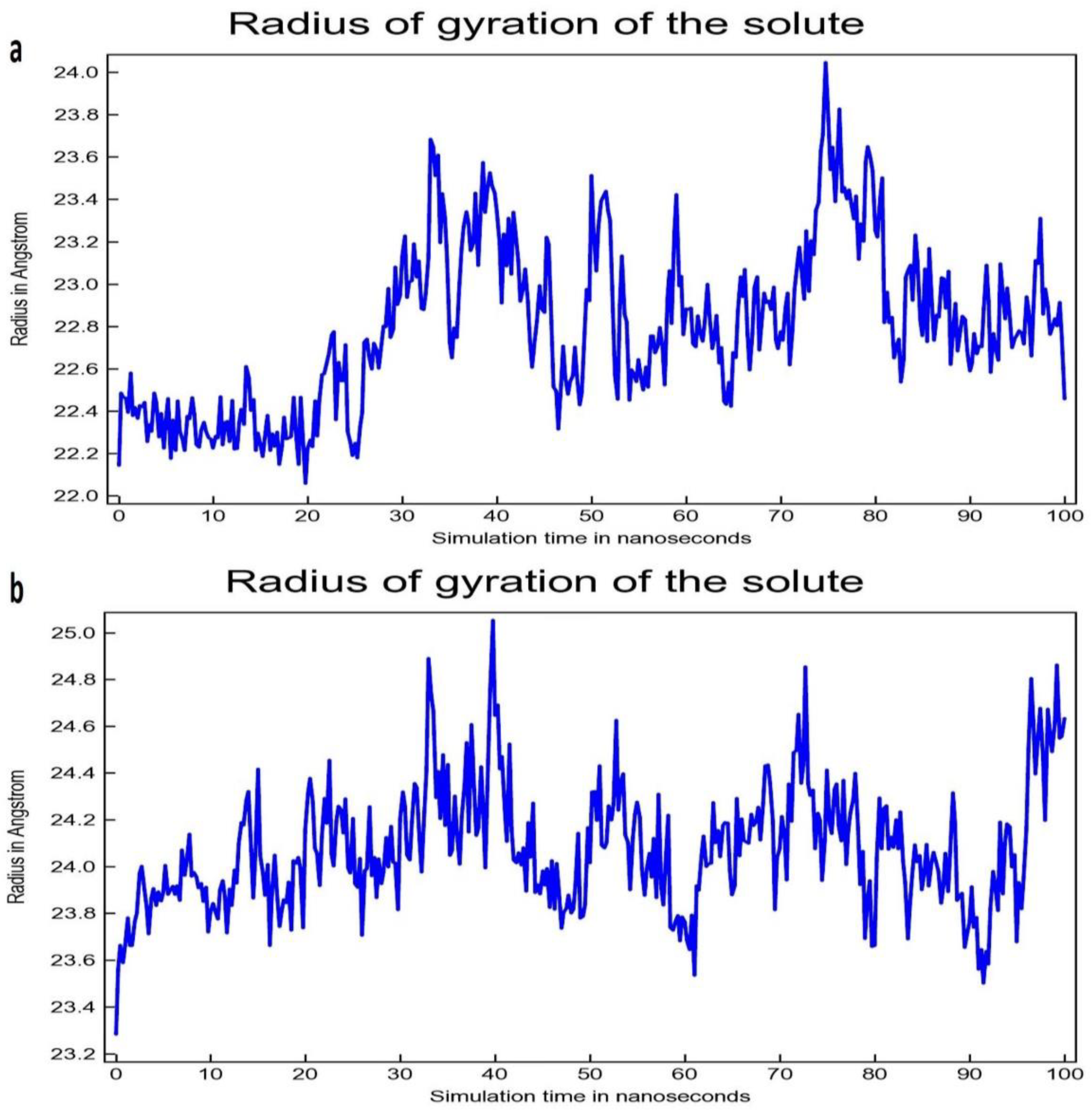
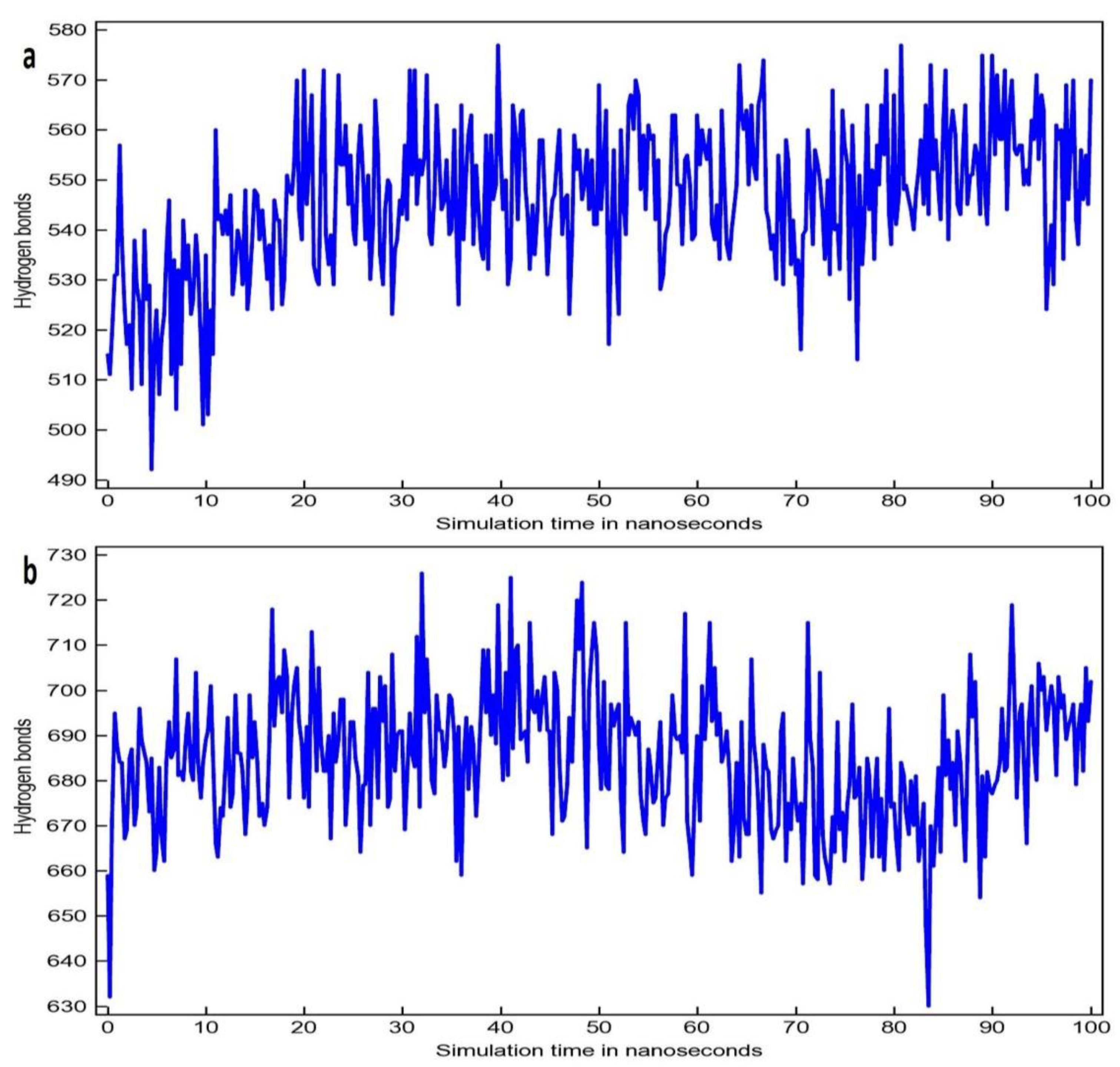
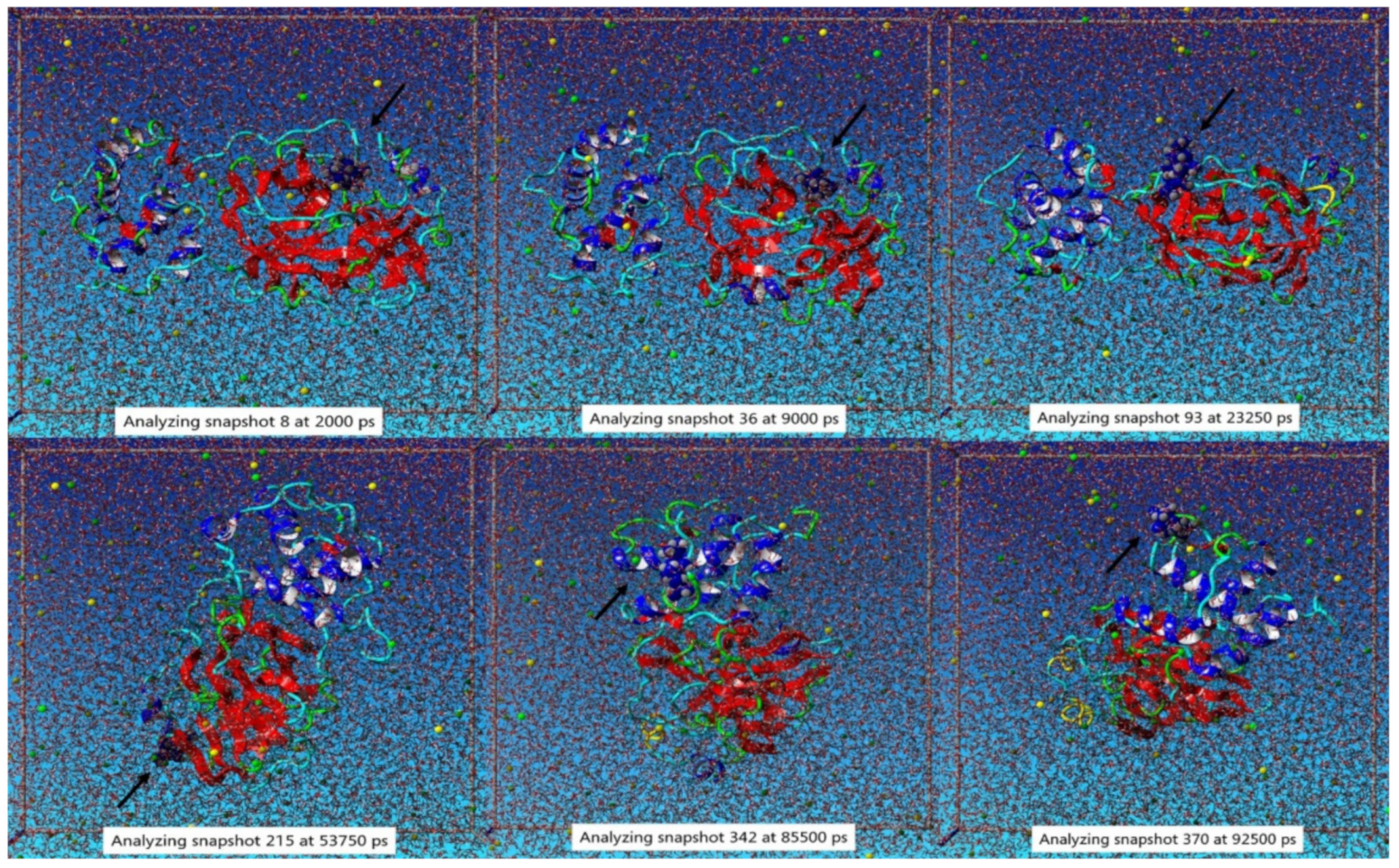
3.2. Molecular Dynamic Simulation Study
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Jakhmola, M.R.; Sehgal, N.; Dogra, N.; Saxena, S.; Pande, K.D. Deciphering underlying mechanism of Sars-CoV-2 infection in humans and revealing the therapeutic potential of bioactive constituents from Nigella sativa to combat COVID19: In-Silico study. J. Biomol. Struct. Dyn. 2020, 1–13, online ahead of print. [Google Scholar] [CrossRef] [PubMed]
- WHO COVID-19, WHO 2020. Available online: https://www.who.int/health-topics/coronavirus#tab=tab_1 (accessed on 13 December 2020).
- McGonagle, D.; Sharif, K.; O’Regan, A.; Bridgewood, C. The Role of Cytokines including Interleukin-6 in COVID-19 induced Pneumonia and Macrophage Activation Syndrome-Like Disease. Autoimmun. Rev. 2020, 19, 102537. [Google Scholar] [CrossRef]
- Wu, C.; Chen, X.; Cai, Y.; Xia, J.; Zhou, X.; Xu, S.; Huang, H.; Zhang, L.; Zhou, X.; Du, C.; et al. Risk Factors Associated with Acute Respiratory Distress Syndrome and Death in Patients with Coronavirus Disease 2019 Pneumonia in Wuhan, China. JAMA Intern. Med. 2020, 180, 934–943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cascella, M.; Rajnik, M.; Cuomo, A.; Dulebohn, S.C.; Di Napoli, R. Features, Evaluation, and Treatment of Coronavirus (COVID-19). In StatPearls [Internet]; StatPearls Publisher: Treasure Island, FL, USA, 2021. [Google Scholar]
- Song, Z.; Xu, Y.; Bao, L.; Zhang, L.; Yu, P.; Qu, Y.; Zhu, H.; Zhao, W.; Han, Y.; Qin, C. From SARS to MERS, Thrusting Coronaviruses into the Spotlight. Viruses 2019, 11, 59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhama, K.; Khan, S.; Tiwari, R.; Sircar, S.; Bhat, S.; Malik, Y.S.; Singh, K.P.; Chaicumpa, W.; Bonilla-Aldana, D.K.; Rodriguez-Morales, A.J. Coronavirus Disease 2019-COVID-19. Clin. Microbiol. Rev. 2020, 33, e00028-20. [Google Scholar] [CrossRef]
- Guo, Y.R.; Cao, Q.D.; Hong, Z.S.; Tan, Y.Y.; Chen, S.D.; Jin, H.J.; Tan, K.S.; Wang, D.Y.; Yan, Y. The origin, transmission and clinical therapies on coronavirus disease 2019 (COVID-19) outbreak—An update on the status. Mil. Med. Res. 2020, 7, 11. [Google Scholar] [CrossRef] [Green Version]
- Boopathi, S.; Poma, A.B.; Kolandaivel, P. Novel 2019 coronavirus structure, mechanism of action, antiviral drug promises and rule out against its treatment. J. Biomol. Struct. Dyn. 2020, 1–10, online ahead of print. [Google Scholar] [CrossRef] [Green Version]
- Mirza, M.U.; Froeyen, M. Structural elucidation of SARS-CoV-2 vital proteins: Computational methods reveal potential drug candidates against main protease, Nsp12 polymerase and Nsp13 helicase. J. Pharm. Anal. 2020, 10, 320–328. [Google Scholar] [CrossRef]
- Angeletti, S.; Benvenuto, D.; Bianchi, M.; Giovanetti, M.; Pascarella, S.; Ciccozzi, M. COVID-2019: The role of the nsp2 and nsp3 in its pathogenesis. J. Med. Virol. 2020, 92, 584–588. [Google Scholar] [CrossRef] [Green Version]
- Chan, J.F.; Kok, K.H.; Zhu, Z.; Chu, H.; To, K.K.; Yuan, S.; Yuen, K.Y. Genomic characterization of the 2019 novel human-pathogenic coronavirus isolated from a patient with atypical pneumonia after visiting Wuhan. Emerg. Microbes Infect. 2020, 9, 221–236. [Google Scholar] [CrossRef] [Green Version]
- Lin, L.T.; Hsu, W.C.; Lin, C.C. Antiviral natural products and herbal medicines. J. Tradit. Complementary Med. 2014, 4, 24–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koshak, D.A.E.; Koshak, P.E.A. Nigella sativa L. as a potential phytotherapy for coronavirus disease 2019: A mini review of in silico studies. Curr. Ther. Res. Clin. Exp. 2020, 93, 100602. [Google Scholar] [CrossRef]
- Ahmad, A.; Husain, A.; Mujeeb, M.; Khan, S.A.; Najmi, A.K.; Siddique, N.A.; Damanhouri, Z.A.; Anwar, F. A review on therapeutic potential of Nigella sativa: A miracle herb. Asian Pac. J. Trop. Biomed. 2013, 3, 337–352. [Google Scholar] [CrossRef] [Green Version]
- Molla, S.; Azad, M.A.K.; Al Hasib, M.A.A.; Hossain, M.M.; Ahammed, S.; Rana, S.; Islam, M.T. A review on antiviral effects of Nigella sativa L. Pharmacol. Online 2019, 2, 47–53. [Google Scholar]
- Barakat, A.B.; Shoman, S.A.; Dina, N.; Alfarouk, O.R. Antiviral activity and mode of action of Dianthus caryophyllus L. and Lupinus termes L. seed extracts against in vitro herpes simplex and hepatitis A viruses infection. J. Microbiol. Antimicrob. 2010, 2, 23–29. [Google Scholar]
- Oyero, O.G.; Toyama, M.; Mitsuhiro, N.; Onifade, A.A.; Hidaka, A.; Okamoto, M.; Baba, M. Selective inhibition of hepatitis c virus replication by alpha-zam, a Nigella sativa seed formulation. Afr. J. Tradit. Complementary Altern. Med. 2016, 13, 144–148. [Google Scholar] [CrossRef]
- Dorra, N.H.; El-Barrawy, M.A.; Sallam, S.M.; Mahmoud, R.S. Evaluation of Antiviral and Antioxidant Activity of Selected Herbal Extracts. J. High Inst. Public Health 2019, 49, 36–40. [Google Scholar] [CrossRef]
- Toma, C.C.; Simu, G.M.; Hanganu, D.A.; Olah, N.; Vata, F.M.; Hammami, C.; Hammami, M. Chemical composition of the Tunisian Nigella sativa. Note I. Profile on essential oil. Farmacia 2010, 58, 458–464. [Google Scholar]
- Sahak, M.K.; Kabir, N.; Abbas, G.; Draman, S.; Hashim, N.H.; Hasan Adli, D.S. The role of Nigella sativa and its active constituents in learning and memory. Evid.-Based Complementary Altern. Med. 2016, 2016, 6075679. [Google Scholar] [CrossRef] [Green Version]
- Ghahramanloo, K.H.; Kamalidehghan, B.; Javar, H.A.; Widodo, R.T.; Majidzadeh, K.; Noordin, M.I. Comparative analysis of essential oil composition of Iranian and Indian Nigella sativa L. extracted using supercritical fluid extraction and solvent extraction. Drug Des. Dev. Ther. 2017, 11, 2221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haseena, S.; Aithal, M.; Das, K.K.; Saheb, S.H. Phytochemical analysis of Nigella sativa and its effect on reproductive system. J. Pharm. Sci. Res. 2015, 7, 514. [Google Scholar]
- Astani, A.; Reichling, J.; Schnitzler, P. Screening for antiviral activities of isolated compounds from essential oils. Evid.-Based Complementary Altern. Med. 2011, 2011, 253643. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.; Wu, N.; Zu, Y.; Fu, Y. Comparative anti-infectious bronchitis virus (IBV) activity of (−)-pinene: Effect on nucleocapsid (N) protein. Molecules 2011, 16, 1044–1054. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wang, D.; Wu, X.; Xu, R.; Li, Y. Antiviral mechanism of carvacrol on HSV-2 infectivity through inhibition of RIP3-mediated programmed cell necrosis pathway and ubiquitin-proteasome system in BSC-1 cells. BMC Infect. Dis. 2020, 20, 832. [Google Scholar] [CrossRef]
- Lasarte-Cia, A.; Lozano, T.; Pérez-González, M.; Gorraiz, M.; Iribarren, K.; Hervás-Stubbs, S.; Sarobe, P.; Rabal, O.; Cuadrado-Tejedor, M.; García-Osta, A.; et al. Immunomodulatory properties of carvone inhalation and its effects on contextual fear memory in mice. Front. Immunol. 2018, 9, 68. [Google Scholar] [CrossRef] [Green Version]
- Sharifi-Rad, J.; Salehi, B.; Schnitzler, P.; Ayatollahi, S.A.; Kobarfard, F.; Fathi, M.; Eisazadeh, M.; Sharifi-Rad, M. Susceptibility of herpes simplex virus type 1 to monoterpenes thymol, carvacrol, p-cymene and essential oils of Sinapis arvensis L., Lallemantia royleana Benth. and Pulicaria vulgaris Gaertn. Cell. Mol. Biol. 2017, 63, 42–47. [Google Scholar] [CrossRef]
- Ryabchenko, B.; Tulupova, E.; Schmidt, E.; Wlcek, K.; Buchbauer, G.; Jirovetz, L. Investigation of anticancer and antiviral properties of selected aroma samples. Nat. Prod. Commun. 2008, 3, 1085–1087. [Google Scholar] [CrossRef] [Green Version]
- Gavanji, S.; Sayedipour, S.S.; Larki, B.; Bakhtari, A. Antiviral activity of some plant oils against herpes simplex virus type 1 in Vero cell culture. J. Acute Med. 2015, 5, 62–68. [Google Scholar] [CrossRef] [Green Version]
- Haddad, J.G.; Picard, M.; Bénard, S.; Desvignes, C.; Desprès, P.; Diotel, N.; El Kalamouni, C. Ayapana triplinervis essential oil and its main component thymohydroquinone dimethyl ether inhibit Zika virus at doses devoid of toxicity in zebrafish. Molecules 2019, 24, 3447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, W.L.; Chuang, H.S.; Lee, M.H.; Wei, C.L.; Lin, C.F.; Tsai, Y.C. Inhibition of herpes simplex virus type 1 by thymol-related monoterpenoids. Planta Med. 2012, 78, 1636–1638. [Google Scholar] [CrossRef] [Green Version]
- Badary, O.A.; Hamza, M.S.; Tikamdas, R. Thymoquinone: A Promising Natural Compound with Potential Benefits for COVID-19 Prevention and Cure. Drug Des. Dev. Ther. 2021, 15, 1819. [Google Scholar] [CrossRef]
- Chakravarty, N. Inhibition of histamine release from mast cells by nigellone. Ann Allergy Asthma Immunol. 1993, 70, 237–242. [Google Scholar]
- El-Dakhakhny, M.; Madi, N.J.; Lembert, N.; Ammon, H.P. Nigella sativa oil, nigellone and derived thymoquinone inhibit synthesis of 5-lipoxygenase products in polymorphonuclear leukocytes from rats. J. Ethnopharmacol. 2002, 81, 161–164. [Google Scholar] [CrossRef]
- Faiza, M.; Abdullah, T.; Wang, P. Dithymoquinone as a novel inhibitor for 3-carboxy-4-methyl-5-propyl-2-furanpropanoic acid (CMPF) to prevent renal failure. arXiv 2017, arXiv:1709.03813. [Google Scholar]
- Jin, Z.; Du, X.; Xu, Y.; Deng, Y.; Liu, M.; Zhao, Y.; Zhang, B.; Li, X.; Zhang, L.; Peng, C.; et al. Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature 2020, 582, 289–293. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.; Jedrzejczak, R.; Maltseva, N.I.; Wilamowski, M.; Endres, M.; Godzik, A.; Michalska, K.; Joachimiak, A. Crystal structure of Nsp15 endoribonuclease NendoU from SARS-CoV-2. Protein Sci. 2020, 29, 1596–1605. [Google Scholar] [CrossRef] [PubMed]
- www.openmolecules.org. Available online: http://www.openmolecules.org/datawarrior/download.html (accessed on 7 December 2020).
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Rizvi, S.M.D.; Shakil, S.; Haneef, M. A simple click by click protocol to perform docking: AutoDock 4.2 made easy for non-bioinformaticians. EXCLI J. 2013, 12, 831–857. [Google Scholar] [PubMed]
- Laskowski, R.A.; Swindells, M.B. LigPlot+: Multiple ligand-protein interaction diagrams for drug discovery. J. Chem. Inf. Model. 2011, 51, 2778–2786. [Google Scholar] [CrossRef]
- Krieger, E.; Vriend, G. YASARA View—Molecular graphics for all devices—From smartphones to workstations. Bioinformatics 2014, 30, 2981–2982. [Google Scholar] [CrossRef] [Green Version]
- Krieger, E.; Dunbrack, R.L., Jr.; Hooft, R.W.; Krieger, B. Assignment of protonation states in proteins and ligands: Combining pKa prediction with hydrogen bonding network optimization. Methods Mol. Biol. 2012, 819, 405–421. [Google Scholar]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jakalian, A.; Jack, D.B.; Bayly, C.I. Fast, efficient generation of high-quality atomic charges. AM1-BCC model: II. Parameterization and validation. J. Comput. Chem. 2002, 23, 1623–1641. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef] [PubMed]
- Krieger, E.; Vriend, G. New ways to boost molecular dynamics simulations. J. Comput. Chem. 2015, 36, 996–1007. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef] [Green Version]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Keretsu, S.; Bhujbal, S.P.; Cho, S.J. Rational approach toward COVID-19 main protease inhibitors via molecular docking, molecular dynamics simulation and free energy calculation. Sci. Rep. 2020, 10, 17716. [Google Scholar] [CrossRef]
- Islam, R.; Parves, M.R.; Paul, A.S.; Uddin, N.; Rahman, S.; Mamun, A.A.; Hossain, N.; Ali, A.; Halim, M.A. A molecular modeling approach to identify effective antiviral phytochemicals against the main protease of SARS-CoV-2. J. Biomol. Struct. Dyn. 2020, 1–12, online ahead of print. [Google Scholar] [CrossRef]
- Lobo-Galo, N.; Terrazas-López, M.; Martínez-Martínez, A.; Díaz-Sánchez, Á.G. FDA-approved thiol-reacting drugs that potentially bind into the SARS-CoV-2 main protease, essential for viral replication. J. Biomol. Struct. Dyn. 2020, 1–9, online ahead of print. [Google Scholar] [CrossRef]
- Deng, X.; Hackbart, M.; Mettelman, R.C.; O’Brien, A.; Mielech, A.M.; Yi, G.; Kao, C.C.; Baker, S.C. Coronavirus nonstructural protein 15 mediates evasion of dsRNA sensors and limits apoptosis in macrophages. Proc. Natl. Acad. Sci. USA 2017, 114, E4251–E4260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sinha, S.K.; Shakya, A.; Prasad, S.K.; Singh, S.; Gurav, N.S.; Prasad, R.S.; Gurav, S.S. An in-silico evaluation of different Saikosaponins for their potency against SARS-CoV-2 using NSP15 and fusion spike glycoprotein as targets. J. Biomol. Struct. Dyn. 2020, 1–12, online ahead of print. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | 3Clpro | Nsp15 | ||
|---|---|---|---|---|
| Binding Energy (ΔG) kcal/mol | Inhibition Constant (Ki) | Binding Energy (ΔG) kcal/mol | Inhibition Constant (Ki) | |
| Trans-anethole | −4.92 | 246.75 μM | −5.16 | 166.25 μM |
| α-Pinene | −5.76 | 60.23 μM | −5.36 | 118.03 μM |
| β-Pinene | −5.80 | 55.60 μM | −5.45 | 100.81 μM |
| Carvacrol | −5.20 | 153.32 μM | −5.50 | 92.57 μM |
| Carvone | −5.42 | 107.14 μM | −5.55 | 85.02 μM |
| p-Cymene | −4.80 | 300.76 μM | −5.19 | 157.67 μM |
| Dithymoquinone | −8.56 | 531.10 nM | −8.31 | 803.86 nM |
| Limonene | −5.19 | 157.44 μM | −5.28 | 134.92 μM |
| Longifoline | −6.48 | 17.89 μM | −6.17 | 29.92 μM |
| α-Thujene | −5.22 | 150.37 μM | −4.97 | 226.02 μM |
| Thymohydroquinone | −5.35 | 120.74 μM | −5.90 | 47.45 μM |
| Thymol | −5.19 | 157.16 μM | −5.27 | 136.00 μM |
| Thymoquinone | −5.25 | 142.75 μM | −5.66 | 71.50 μM |
| Lopinavir * | −7.95 | 1.48 μM | - | - |
| Benzopurpurin B * | - | - | −5.87 | 50.15 μM |
| Compounds | Physiochemical Parameters | |||||
|---|---|---|---|---|---|---|
| Molecular Weight (g/mol) | cLogP ** | Hydrogen Bond Donors | Hydrogen Bond Acceptors | Number of Rotatable Bonds | Lipinski’s Violation | |
| Rule | <500 | ≤5 | <5 | <10 | ≤10 | ≤1 |
| Trans-anethole | 148.20 | 2.68 | 0 | 1 | 2 | 0 |
| α-Pinene | 136.23 | 2.72 | 0 | 0 | 0 | 0 |
| β-Pinene | 136.23 | 2.79 | 0 | 0 | 0 | 0 |
| Carvacrol | 150.22 | 2.84 | 1 | 1 | 1 | 0 |
| Carvone | 150.22 | 2.65 | 0 | 1 | 1 | 0 |
| p-Cymene | 134.22 | 3.19 | 0 | 0 | 1 | 0 |
| Dithymoquinone | 328.40 | 2.73 | 0 | 4 | 2 | 0 |
| Limonene | 136.23 | 3.36 | 0 | 0 | 1 | 0 |
| Longifoline | 204.35 | 4.06 | 0 | 0 | 0 | 0 |
| α-Thujene | 136.23 | 2.78 | 0 | 0 | 1 | 0 |
| Thymohydroquinone | 166.21 | 2.49 | 2 | 2 | 1 | 0 |
| Thymol | 150.22 | 2.84 | 1 | 1 | 1 | 0 |
| Thymoquinone | 164.20 | 1.63 | 0 | 2 | 1 | 0 |
| Lopinavir * | 628.81 | 4.84 | 4 | 9 | 15 | 2 |
| Benzopurpurin B * | 680.76 | 4.98 | 4 | 12 | 7 | 2 |
| Compounds | Toxicity Risks | |||
|---|---|---|---|---|
| Mutagenic | Tumorigenic | Reproductive Effect | Irritant | |
| Trans-anethole | High | High | High | None |
| α-Pinene | None | None | None | High |
| β-Pinene | None | None | None | None |
| Carvacrol | None | None | None | High |
| Carvone | None | None | None | Low |
| p-Cymene | None | Low | None | High |
| Dithymoquinone | None | None | None | None |
| Limonene | None | None | None | Low |
| Longifoline | None | None | None | None |
| α-Thujene | None | None | None | Low |
| Thymohydroquinone | High | Low | None | None |
| Thymol | High | None | High | None |
| Thymoquinone | High | None | None | None |
| Lopinavir * | None | None | None | High |
| Benzopurpurin B * | High | High | High | High |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rizvi, S.M.D.; Hussain, T.; Moin, A.; Dixit, S.R.; Mandal, S.P.; Adnan, M.; Jamal, Q.M.S.; Sharma, D.C.; Alanazi, A.S.; Unissa, R. Identifying the Most Potent Dual-Targeting Compound(s) against 3CLprotease and NSP15exonuclease of SARS-CoV-2 from Nigella sativa: Virtual Screening via Physicochemical Properties, Docking and Dynamic Simulation Analysis. Processes 2021, 9, 1814. https://doi.org/10.3390/pr9101814
Rizvi SMD, Hussain T, Moin A, Dixit SR, Mandal SP, Adnan M, Jamal QMS, Sharma DC, Alanazi AS, Unissa R. Identifying the Most Potent Dual-Targeting Compound(s) against 3CLprotease and NSP15exonuclease of SARS-CoV-2 from Nigella sativa: Virtual Screening via Physicochemical Properties, Docking and Dynamic Simulation Analysis. Processes. 2021; 9(10):1814. https://doi.org/10.3390/pr9101814
Chicago/Turabian StyleRizvi, Syed Mohd Danish, Talib Hussain, Afrasim Moin, Sheshagiri R. Dixit, Subhankar P. Mandal, Mohd Adnan, Qazi Mohammad Sajid Jamal, Dinesh C. Sharma, Abulrahman Sattam Alanazi, and Rahamat Unissa. 2021. "Identifying the Most Potent Dual-Targeting Compound(s) against 3CLprotease and NSP15exonuclease of SARS-CoV-2 from Nigella sativa: Virtual Screening via Physicochemical Properties, Docking and Dynamic Simulation Analysis" Processes 9, no. 10: 1814. https://doi.org/10.3390/pr9101814
APA StyleRizvi, S. M. D., Hussain, T., Moin, A., Dixit, S. R., Mandal, S. P., Adnan, M., Jamal, Q. M. S., Sharma, D. C., Alanazi, A. S., & Unissa, R. (2021). Identifying the Most Potent Dual-Targeting Compound(s) against 3CLprotease and NSP15exonuclease of SARS-CoV-2 from Nigella sativa: Virtual Screening via Physicochemical Properties, Docking and Dynamic Simulation Analysis. Processes, 9(10), 1814. https://doi.org/10.3390/pr9101814