Development of Carbonization and a Relatively High-Temperature Halogenation Process for the Removal of Radionuclides from Spent Ion Exchange Resins

 ,
,
Abstract
1. Introduction
2. Principle of Carbonization/Halogenation Treatment
2.1. Characteristics of a Target Waste Stream
2.2. Principle of the Proposed Process
3. Experimental and Numerical Optimization Study for Key Processes
3.1. Procedure of Experimental and Numerical Modeling Study
3.2. Establishment of the Carbonization Reactor Condition
3.2.1. Analysis of Pyrolysis and Carbonization Steps
3.2.2. Establishment of the Carbonization Reactor Condition
3.3. Determination of Operating Condition of the UHC Combustor
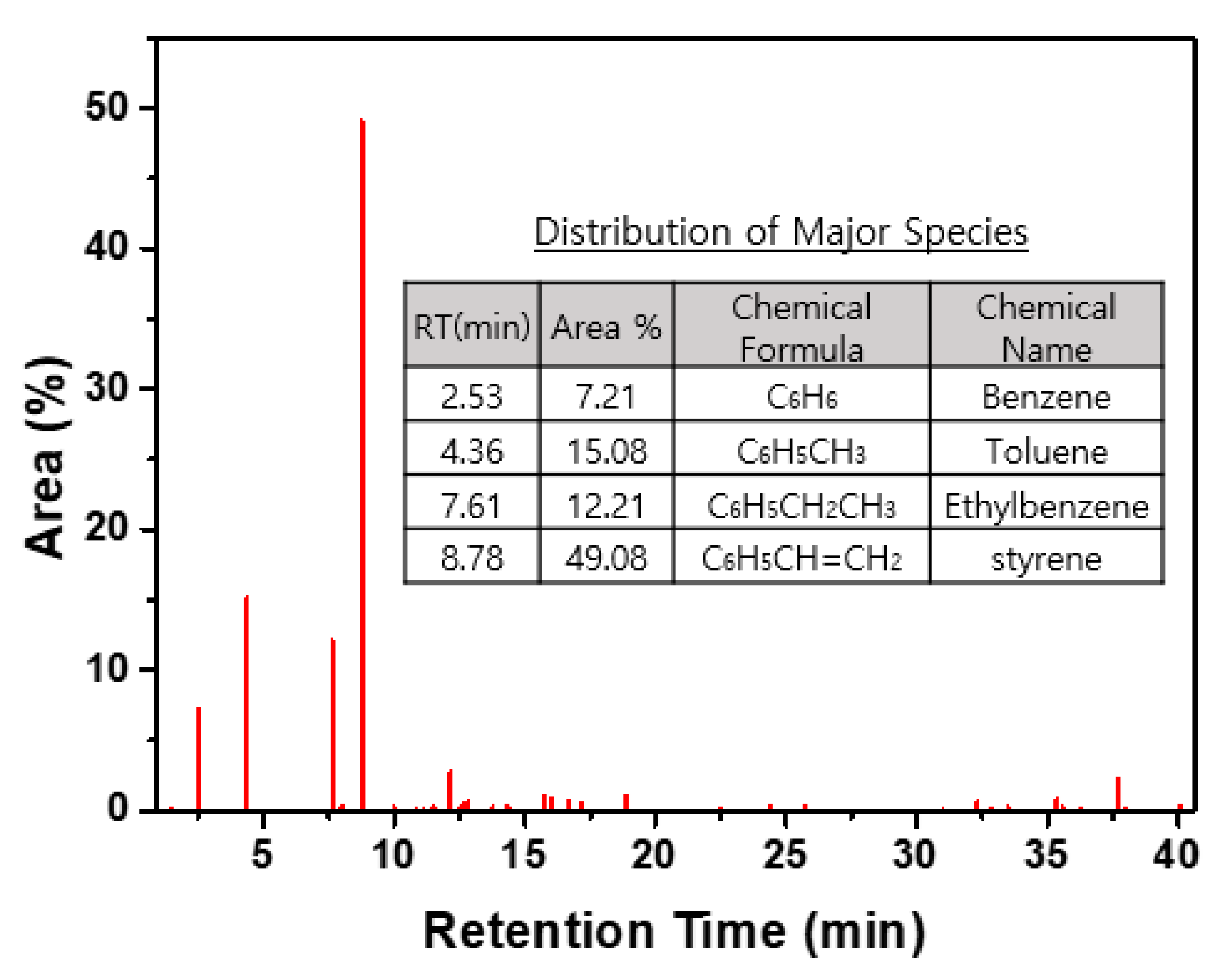
3.3.1. UHCs Generated by the Carbonization of CER
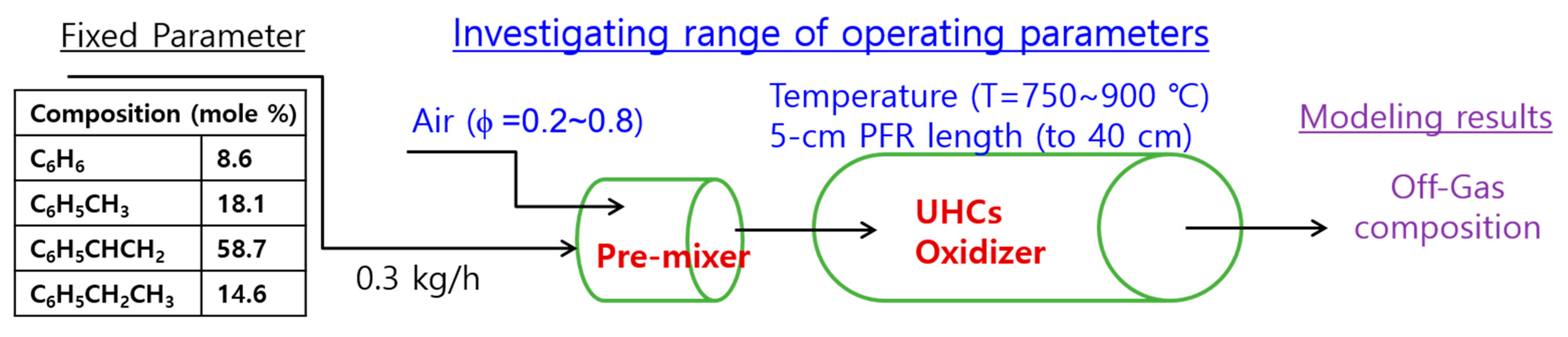
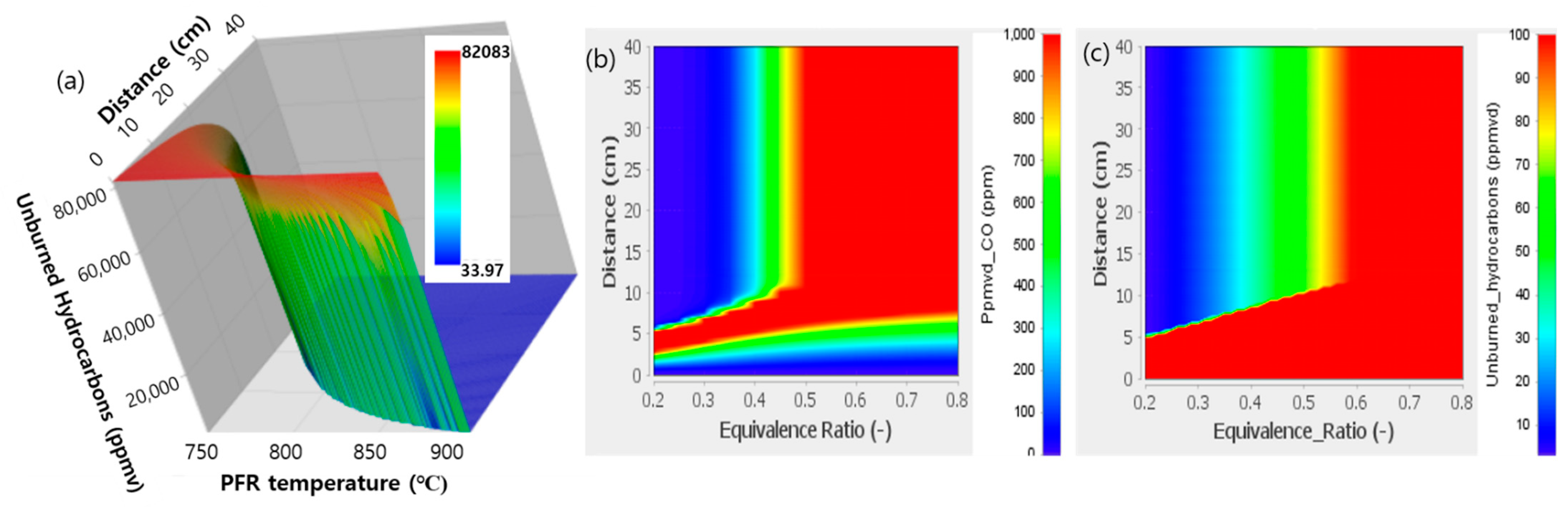
3.3.2. Numerical Determination of the UHC Combustor Operating Condition
3.4. Determination of the Appropriate Halogenation Condition
4. Installation of a Bench-Scale Process and Test Operation
4.1. Process Description
4.2. Detailed Design of the Multifunctional Reactor and Test Results
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Zhang, J.; Qian, H. Thermal behavior of weak basic ion exchange resin. J. Therm. Anal. Calorim. 2014, 115, 875–880. [Google Scholar] [CrossRef]
- Mohammad, A.; Inamuddin, A.A.; Nuashad, M.; Eldesoky, G.E. Forward ion-exchange kinetics of heavy metal ions on the surface of carboxymethyl cellulose Sn(IV) phosphate composite nano-rod-like exchanger. J. Therm. Anal. Calorim. 2012, 110, 715–723. [Google Scholar] [CrossRef]
- Wang, J.; Wan, Z. Treatment and disposal of spent radioactive ion-exchange resins produced in the nuclear industry. Prog. Nucl. Energy 2015, 78, 47–55. [Google Scholar] [CrossRef]
- Chun, U.K.; Choi, K.S.; Yang, K.H.; Park, J.K.; Song, M.J. Waste minimization pretreatment via pyrolysis and oxidative pyrolysis of organic ion exchange resin. Waste Manag. 1998, 18, 183–196. [Google Scholar] [CrossRef]
- Antonetti, P.; Claire, Y.; Massit, H.; Lessart, P.; Van Cang, C.P.; Perichaud, A. Pyrolysis of cobalt and caesium doped cationic ion-exchange resin. J. Anal. Appl. Pyrolysis 2000, 55, 81–92. [Google Scholar] [CrossRef]
- Barton, R.G.; Clark, W.G.; Seeker, W.R. Fate of metals in waste combustion systems. Combust. Sci. Technol. 1990, 74, 328–336. [Google Scholar] [CrossRef]
- Georg, B.; Rainer, S. Pyrolysis of spent ion exchange resins. In Proceedings of the Nuclear Plant Chemistry Conference 2012 (NPC 2010), Quebec City, QC, Canada, 3–7 October 2012; pp. 23–27. [Google Scholar]
- Masami, M.; Kiyomi, F.; Makoto, K. Decomposition of Ion Exchange Resins by Pyrolysis. Nucl. Technol. 1986, 75, 187–982. [Google Scholar]
- Peterson, S.; Kemmler, G. Experience of Resin Pyrolysis; Waste Management: Tuscon, AZ, USA, 1984. [Google Scholar]
- Matsuda, M.; Funabashi, K.; Yusa, H. Influence of functional sulfonic acid group on pyrolysis characteristics for cation exchange resin. J. Nucl. Sci. Technol. 1987, 24, 124–128. [Google Scholar] [CrossRef]
- Michlink, D.L.; Marchall, R.W.; Smith, K.R.; Turner, V.C.; Vander Wall, E.M. The Feasibility of Spent Resins Incineration at Nuclear Power Plants; Waste Management: Tuscon, AZ, USA, 1983; pp. 439–441. [Google Scholar]
- Chiara, S.; Alessio, F.; Alberto, C.; Tiziano, F.; Eliseo, R. A wide range kinetic modeling study of pyrolysis and oxidation of benzene. Combust. Flame 2013, 160, 1168–1190. [Google Scholar]
- Ho, T.C.; Kuo, T.H.; Hopper, J.R. Thermodynamic study of the behavior of uranium and plutonium during thermal treatment under reducing and oxidizing modes. Waste Manag. 2000, 50, 355–361. [Google Scholar] [CrossRef]
- Barbin, N.M.; Terentiev, D.I.; Alexeev, S.G.; Barbina, T.M. Thermodynamic analysis of radionuclides behaviour in products of vapour phase hydrothermal oxidation of radioactive graphite. J. Radioanal. Nucl. Chem. 2016, 307, 1459–1470. [Google Scholar] [CrossRef]
- Yang, H.C.; Lee, S.Y.; Choi, Y.C.; Yang, I.H.; Chung, D.C. Thermokinetic analysis of spent ion-exchange resins for the optimization of carbonization reactor condition. J. Therm. Anal. Calorim. 2017, 127, 587–595. [Google Scholar] [CrossRef]
- Vyazovkin, S.; Burnham, A.K.; Criado, J.M.; Pérez-Marqueda, L.A.; Popescu, C.; Sbirrazzuoli, N. ICTAC Kinetics Committee recommendations for performing kinetic computations on thermal analysis data. Thermochim. Acta 2011, 520, 1–19. [Google Scholar] [CrossRef]
- Friedman, H.L. Kinetics of thermal degradation of char-forming plastics from thermogravimetry—Application to a phenolic plastic. J. Polym. Sci. 1964, 6, 183–195. [Google Scholar] [CrossRef]
- Roduit, B.; Borgeat, C.; Berger, B.; Folly, P.; Alonso, B.; Aebischer, J.N.; Stoessel, F. Advanced kinetic tools for the evaluation of decomposition reactions. J. Therm. Anal. Calorim. 2005, 80, 229–236. [Google Scholar] [CrossRef]
- Yang, H.C.; Lee, M.W.; Eun, H.C.; Kim, J.H.; Lee, K.; Seo, B.K. Thermal decontamination of spent activated carbon contaminated with radiocarbon and tritium. Processes 2020, 8, 1359. [Google Scholar] [CrossRef]
- ANSYS. Chemkin-Pro, Chemistry Solution for More Efficient Designs; ANSYS, Inc.: Canonsburg, PA, USA, 2016; Available online: https://www.ansys.co17m/-/media/ansys/corporate/resourcelibrary/brochure/chemkin-pro-brochure.pdf (accessed on 11 November 2020).
- Westbrook, C.K.; Pitz, W.J.; Herbinet, O.H.J.; Curran, H.J.; Silke, E.J. A detailed chemical kinetic reaction mechanism for n-alkane hydrocarbons from n-Octane to n-Hexadecane. Combust. Flame 2009, 156, 181–199. [Google Scholar] [CrossRef]
- Yang, H.C.; Eun, H.C.; Lee, D.G.; Oh, W.Z.; Lee, K.W. Behavior of radioactive elements during thermal treatment of nuclear graphite–thermodynamic model analysis. J. Nucl. Sci. Technol. 2005, 42, 869–876. [Google Scholar] [CrossRef]
- Durlak, S.K.; Biswas, P.; Shi, J. Equilibrium analysis of the affect of moisture and sodium content on heavy metal emissions from municipal solid waste incinerators. J. Hazard. Mater. 1996, 56, 1–20. [Google Scholar] [CrossRef]
- Outokumpu Research. HSC Chemistry 7.1—Chemical Reaction and Equilibrium Software with Extensive Thermodynamic Database; Outotec Research Center: Pori, Finland, 2015. [Google Scholar]
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Carbonized CER (Mole %) | Chlorination Gas (mole %) | ||||
|---|---|---|---|---|---|
| Non-Radioactive Compounds (wt.%) | Radioactive Compounds (×10−6) | ||||
| C | 0.80 | 134/137Cs2SO4 | 0.007 | Cl2 | 0.1 |
| Na2SO4 | 0.025 | 58/60CoSO4 | 0.012 | N2 | 0.8998 |
| CaSO4 | 0.0006 | 90SrSO4 | 0.008 | O2 | 0.0001 |
| Fe(+59Fe) SO4 | 0.0023 | 54MnSO4 | 0.01 | H2O | 0.00005 |
| Cr2(SO4)3 | 0.0011 | 65ZnSO4 | 0.006 | CO2 | 0.00002 |
| MgSO4 | 0.00046 | 95Zr (SO4)2 | 0.004 | ||
| NiSO4 | 0.000041 | ||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, H.-C.; Park, H.-O.; Park, K.-T.; Kim, S.-J.; Kim, H.-J.; Eun, H.-C.; Lee, K. Development of Carbonization and a Relatively High-Temperature Halogenation Process for the Removal of Radionuclides from Spent Ion Exchange Resins. Processes 2021, 9, 96. https://doi.org/10.3390/pr9010096
Yang H-C, Park H-O, Park K-T, Kim S-J, Kim H-J, Eun H-C, Lee K. Development of Carbonization and a Relatively High-Temperature Halogenation Process for the Removal of Radionuclides from Spent Ion Exchange Resins. Processes. 2021; 9(1):96. https://doi.org/10.3390/pr9010096
Chicago/Turabian StyleYang, Hee-Chul, Hyeon-Oh Park, Kyu-Tae Park, Sung-Jun Kim, Hyung-Ju Kim, Hee-Chul Eun, and Keunyoung Lee. 2021. "Development of Carbonization and a Relatively High-Temperature Halogenation Process for the Removal of Radionuclides from Spent Ion Exchange Resins" Processes 9, no. 1: 96. https://doi.org/10.3390/pr9010096
APA StyleYang, H.-C., Park, H.-O., Park, K.-T., Kim, S.-J., Kim, H.-J., Eun, H.-C., & Lee, K. (2021). Development of Carbonization and a Relatively High-Temperature Halogenation Process for the Removal of Radionuclides from Spent Ion Exchange Resins. Processes, 9(1), 96. https://doi.org/10.3390/pr9010096