Machine Learning for Ionic Liquid Toxicity Prediction



Abstract
1. Introduction
2. Experimental Data
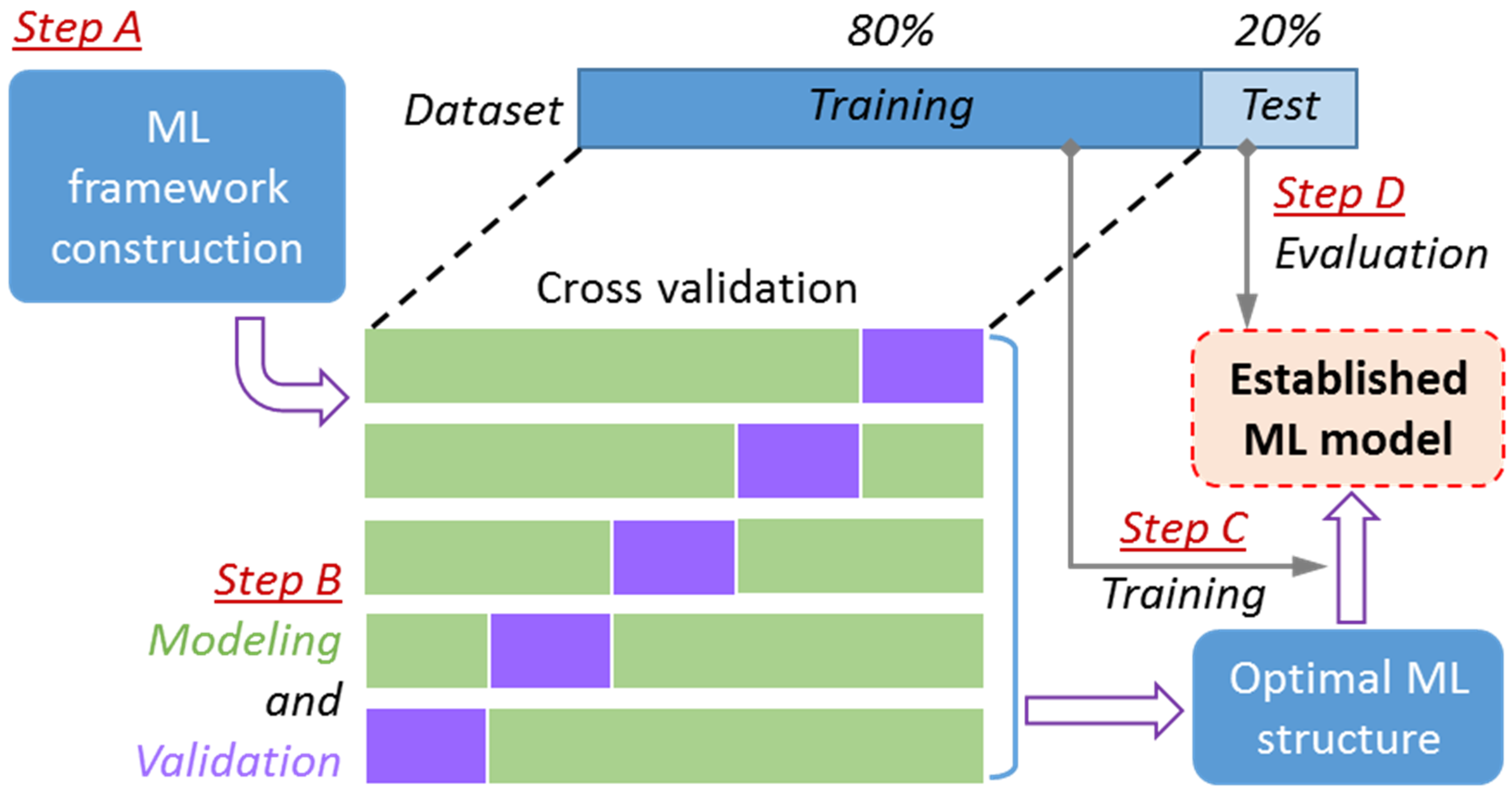
3. ML Modeling
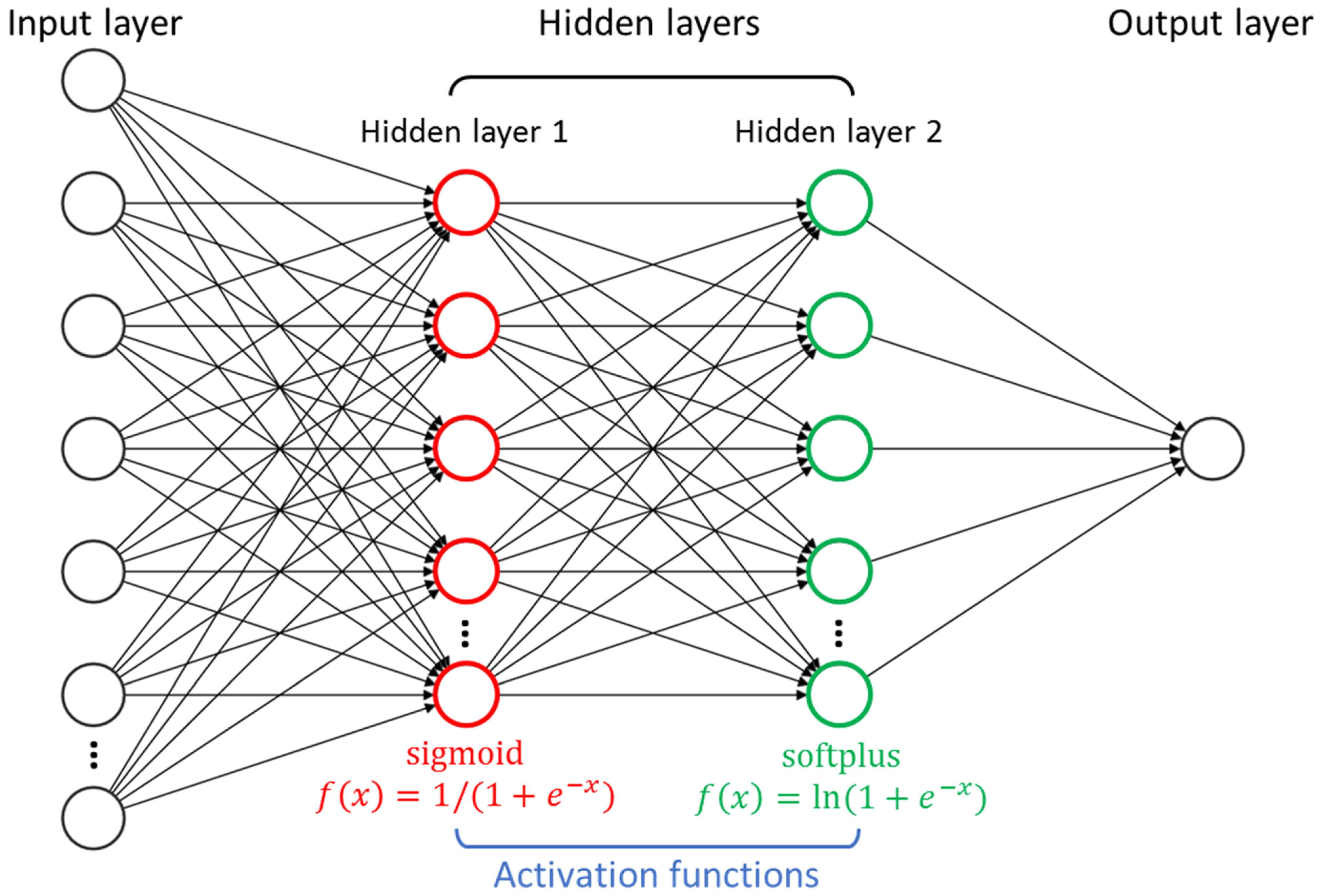
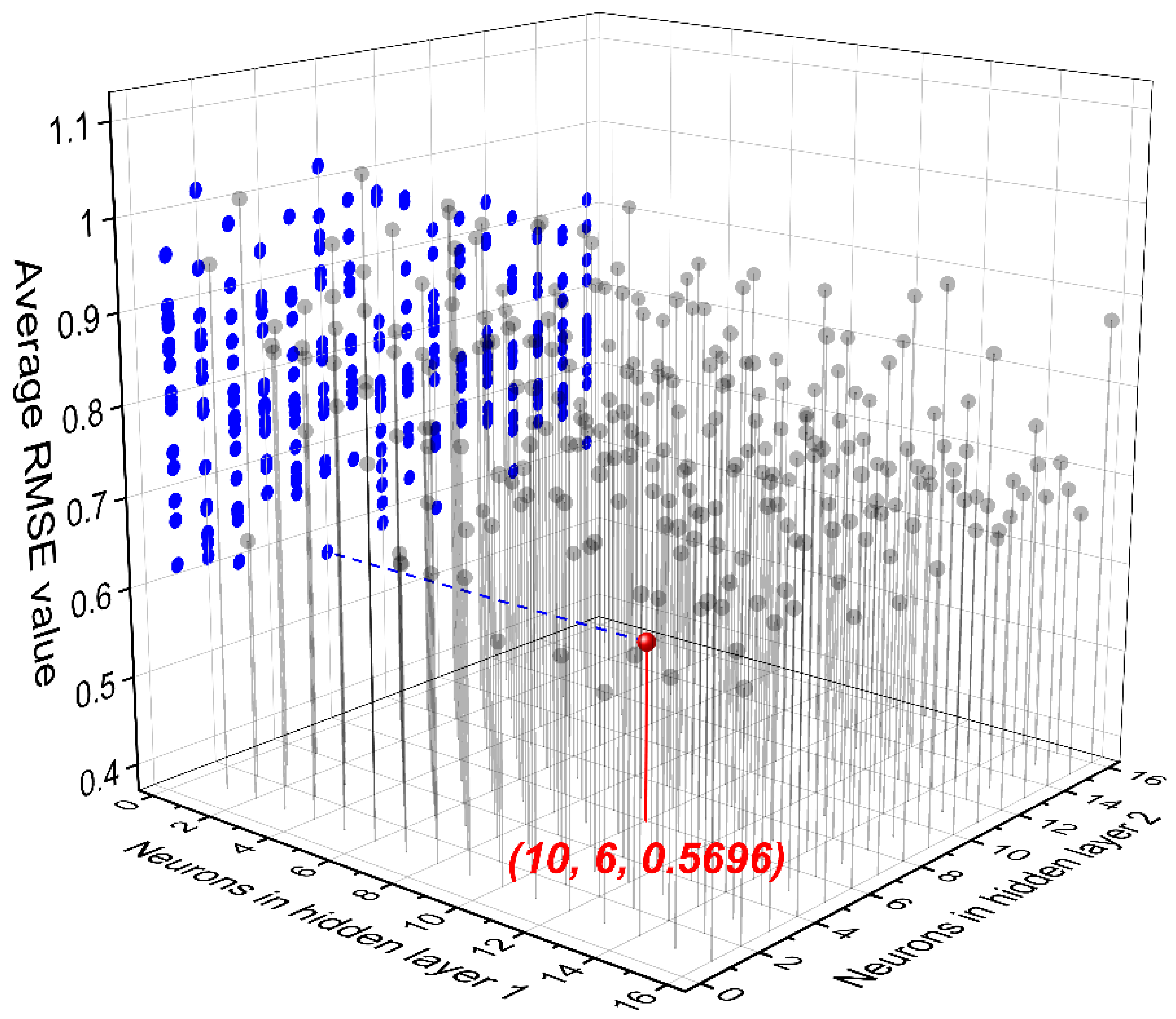
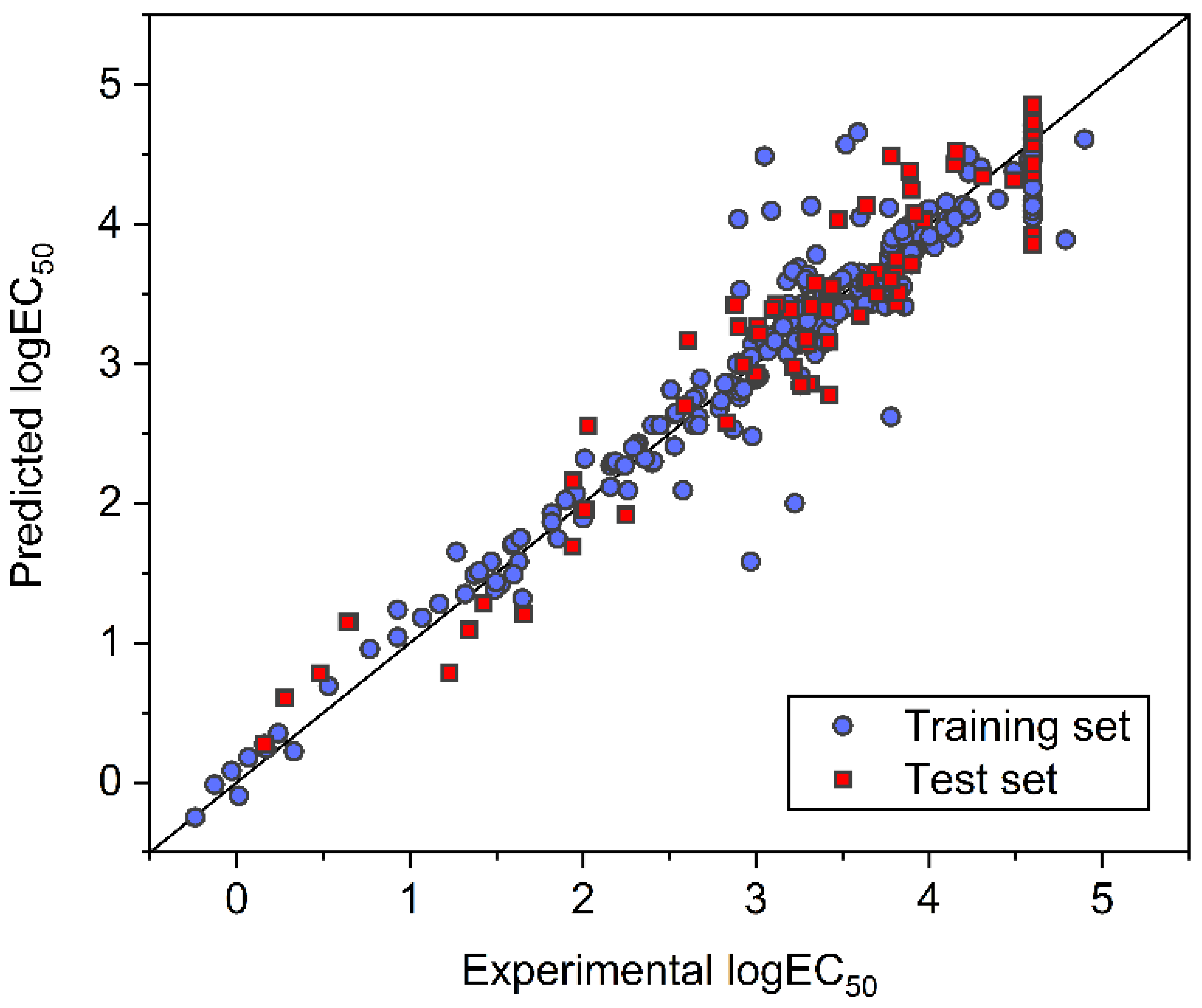
3.1. FNN Modeling
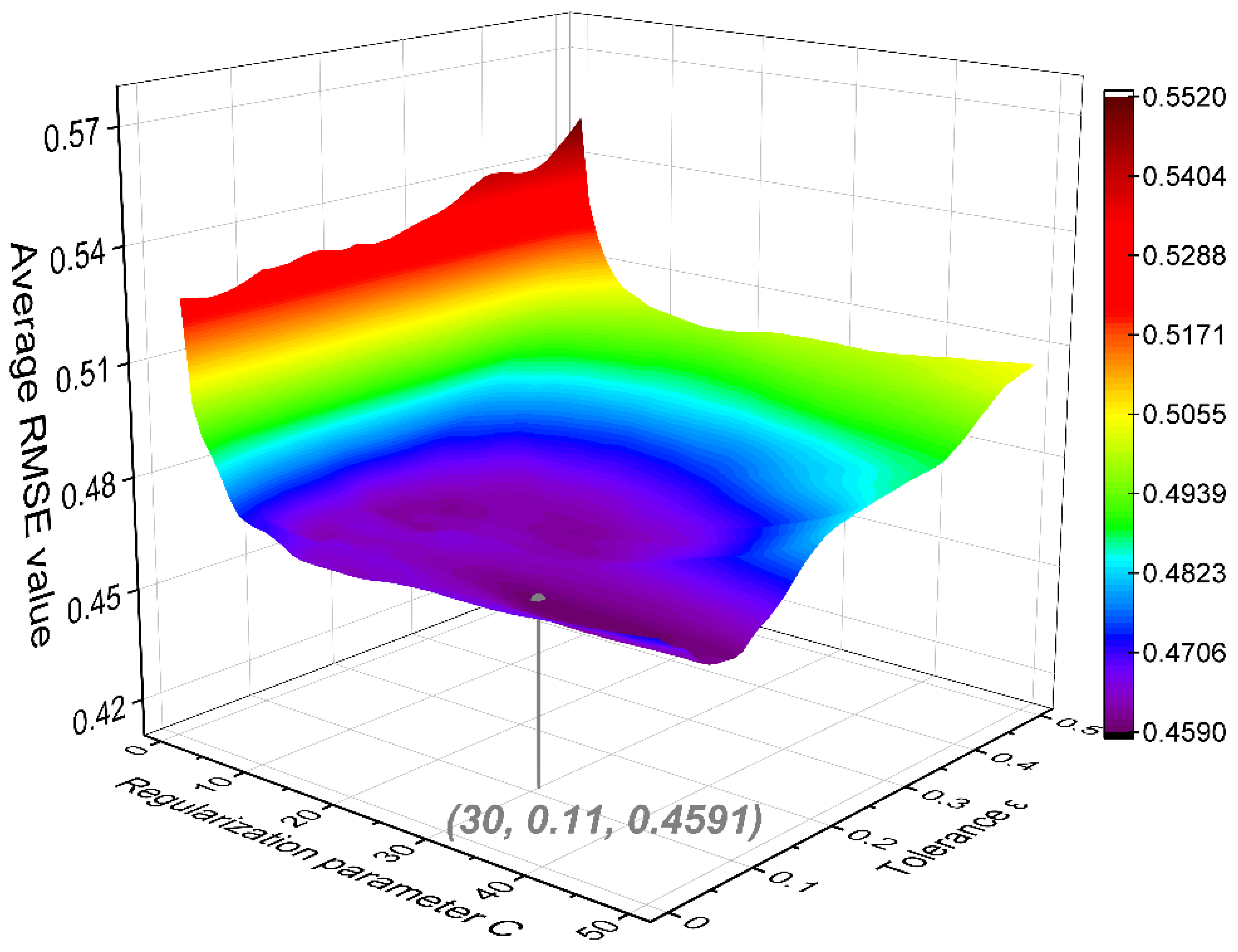
3.2. SVM Modeling
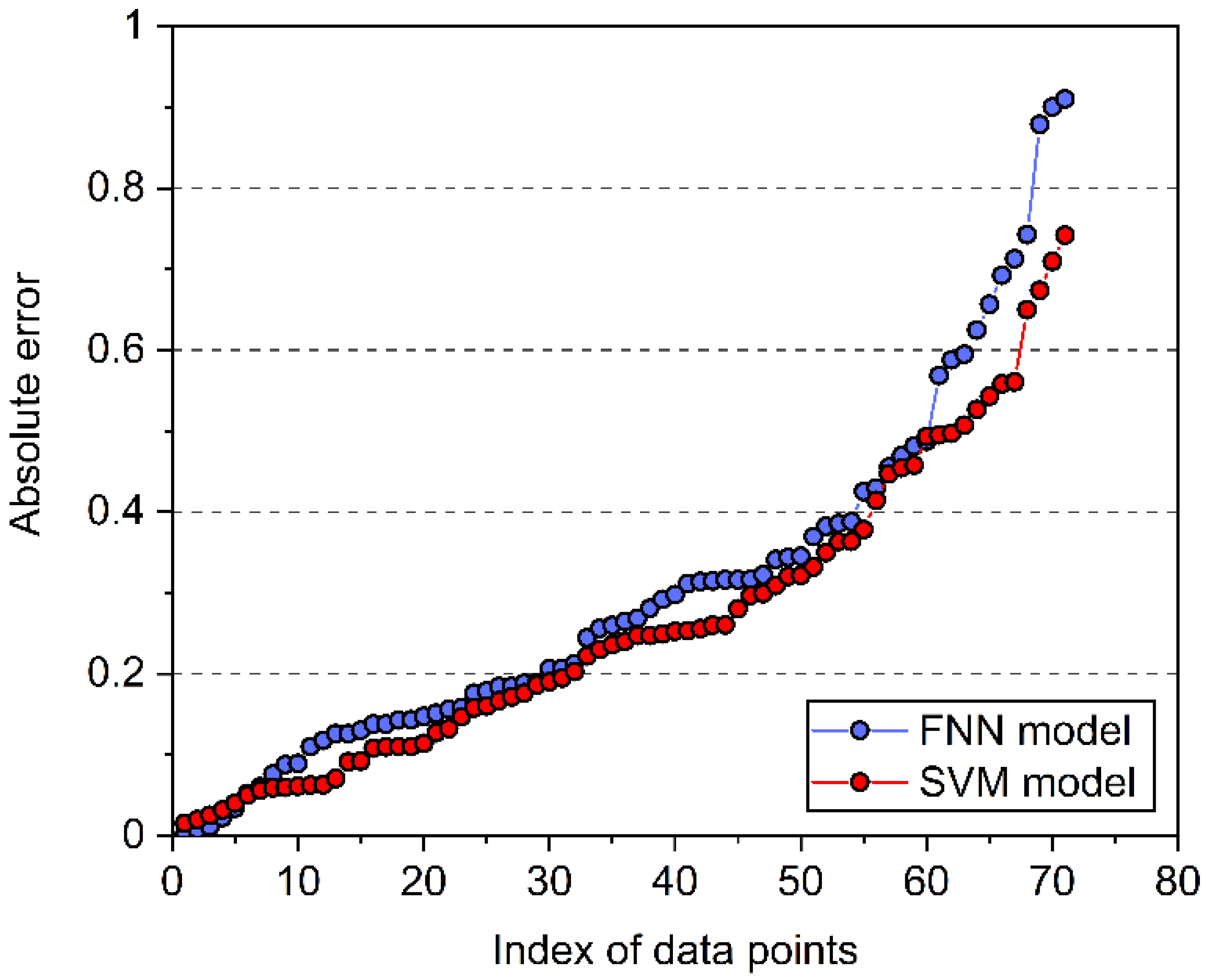
4. Model Comparison
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Watanabe, M.; Thomas, M.L.; Zhang, S.; Ueno, K.; Yasuda, T.; Dokko, K. Application of ionic liquids to energy storage and conversion materials and devices. Chem. Rev. 2017, 117, 7190–7239. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhang, X.; Dong, H.; Zhao, Z.; Zhang, S.; Huang, Y. Carbon capture with ionic liquids: Overview and progress. Energy Environ. Sci. 2012, 5, 6668–6681. [Google Scholar] [CrossRef]
- Song, Z.; Hu, X.; Zhou, Y.; Zhou, T.; Qi, Z.; Sundmacher, K. Rational design of double salt ionic liquids as extraction solvents: Separation of thiophene/n-octane as example. AIChE J. 2019, 65, e16625. [Google Scholar] [CrossRef]
- Song, Z.; Zhou, T.; Qi, Z.; Sundmacher, K. Systematic method for screening ionic liquids as extraction solvents exemplified by an extractive desulfurization process. ACS Sustain. Chem. Eng. 2017, 5, 3382–3389. [Google Scholar] [CrossRef]
- Gharagheizi, F.; Sattari, M.; Ilani-Kashkouli, P.; Mohammadi, A.H.; Ramjugernath, D.; Richon, D. A “non-linear” quantitative structure–property relationship for the prediction of electrical conductivity of ionic liquids. Chem. Eng. Sci. 2013, 101, 478–485. [Google Scholar] [CrossRef]
- Gharagheizi, F.; Sattari, M.; Ilani-Kashkouli, P.; Mohammadi, A.H.; Ramjugernath, D.; Richon, D. Quantitative structure—property relationship for thermal decomposition temperature of ionic liquids. Chem. Eng. Sci. 2012, 84, 557–563. [Google Scholar] [CrossRef]
- Huang, Y.; Dong, H.; Zhang, X.; Li, C.; Zhang, S. A new fragment contribution-corresponding states method for physicochemical properties prediction of ionic liquids. AIChE J. 2013, 59, 1348–1359. [Google Scholar] [CrossRef]
- Zhou, T.; Chen, L.; Ye, Y.; Chen, L.; Qi, Z.; Freund, H.; Sundmacher, K. An overview of mutual solubility of ionic liquids and water predicted by COSMO-RS. Ind. Eng. Chem. Res. 2012, 51, 6256–6264. [Google Scholar] [CrossRef]
- Song, Z.; Zhang, C.; Qi, Z.; Zhou, T.; Sundmacher, K. Computer-aided design of ionic liquids as solvents for extractive desulfurization. AIChE J. 2018, 64, 1013–1025. [Google Scholar] [CrossRef]
- Chong, F.K.; Eljack, F.T.; Atilhan, M.; Foo, D.C.; Chemmangattuvalappil, N.G. A systematic visual methodology to design ionic liquids and ionic liquid mixtures: Green solvent alternative for carbon capture. Comput. Chem. Eng. 2016, 91, 219–232. [Google Scholar] [CrossRef]
- Chong, F.K.; Foo, D.C.Y.; Eljack, F.T.; Atilhan, M.; Chemmangattuvalappil, N.G. A systematic approach to design task-specific ionic liquids and their optimal operating conditions. Mol. Syst. Des. Eng. 2016, 1, 109–121. [Google Scholar] [CrossRef]
- Zhou, T.; Shi, H.; Ding, X.; Zhou, Y. Thermodynamic modeling and rational design of ionic liquids for pre-combustion carbon capture. Chem. Eng. Sci. 2021, 229, 116076. [Google Scholar] [CrossRef]
- Zhou, T.; McBride, K.; Linke, S.; Song, Z.; Sundmacher, K. Computer-aided solvent selection and design for efficient chemical processes. Curr. Opin. Chem. Eng. 2020, 27, 35–44. [Google Scholar] [CrossRef]
- Shi, H.; Zhang, X.; Zhou, T.; Sundmacher, K. Model-based optimal design of phase change ionic liquids for efficient thermal energy storage. Green Energy Environ. 2021. [Google Scholar] [CrossRef]
- Ranke, J.; Stolte, S.; Störmann, R.; Arning, J.; Jastorff, B. Design of sustainable chemical products the example of ionic liquids. Chem. Rev. 2007, 107, 2183–2206. [Google Scholar] [CrossRef]
- Stolte, S.; Matzke, M.; Arning, J.; Böschen, A.; Pitner, W.R.; Welz-Biermann, U.; Jastorff, B.; Ranke, J. Effects of different head groups and functionalised side chains on the aquatic toxicity of ionic liquids. Green Chem. 2007, 9, 1170–1179. [Google Scholar] [CrossRef]
- Yan, F.; Xia, S.; Wang, Q.; Ma, P. Predicting the toxicity of ionic liquids in leukemia rat cell line by the quantitative structure–activity relationship method using topological indexes. Ind. Eng. Chem. Res. 2012, 51, 13897–13901. [Google Scholar] [CrossRef]
- Sosnowska, A.; Grzonkowska, M.; Puzyn, T. Global versus local QSAR models for predicting ionic liquids toxicity against IPC-81 leukemia rat cell line: The predictive ability. J. Mol. Liq. 2017, 231, 333–340. [Google Scholar] [CrossRef]
- Wu, T.; Li, W.; Chen, M.; Zhou, Y.; Zhang, Q. Estimation of Ionic Liquids Toxicity against Leukemia Rat Cell Line IPC-81 based on the Empirical-like Models using Intuitive and Explainable Fingerprint Descriptors. Mol. Inf. 2020, 39, 2000102. [Google Scholar] [CrossRef]
- Varnek, A.; Baskin, I. Machine learning methods for property prediction in chemoinformatics: Quo vadis? J. Chem. Inf. Model. 2012, 52, 1413–1437. [Google Scholar] [CrossRef]
- Liu, Q.; Zhang, L.; Tang, K.; Liu, L.; Du, J.; Meng, Q.; Gani, R. A machine learning-based atom contribution method for the prediction of charge density profiles and solvent design. AIChE J. 2020, e17110. [Google Scholar] [CrossRef]
- Zhang, L.; Mao, H.; Liu, L.; Du, J.; Gani, R. A machine learning based computer-aided molecular design/screening methodology for fragrance molecules. Comput. Chem. Eng. 2018, 115, 295–308. [Google Scholar] [CrossRef]
- Su, Y.; Wang, Z.; Jin, S.; Shen, W.; Ren, J.; Eden, M.R. An architecture of deep learning in QSPR modeling for the prediction of critical properties using molecular signatures. AIChE J. 2019, 65, e16678. [Google Scholar] [CrossRef]
- Wang, Z.; Su, Y.; Shen, W.; Jin, S.; Clark, J.H.; Ren, J.; Zhang, X. Predictive deep learning models for environmental properties: The direct calculation of octanol-water partition coefficients from molecular graphs. Green Chem. 2019, 21, 4555–4565. [Google Scholar] [CrossRef]
- Zhou, T.; Jhamb, S.; Liang, X.; Sundmacher, K.; Gani, R. Prediction of acid dissociation constants of organic compounds using group contribution methods. Chem. Eng. Sci. 2018, 183, 95–105. [Google Scholar] [CrossRef]
- Venkatraman, V.; Evjen, S.; Knuutila, H.K.; Fiksdahl, A.; Alsberg, B.K. Predicting ionic liquid melting points using machine learning. J. Mol. Liq. 2018, 264, 318–326. [Google Scholar] [CrossRef]
- Song, Z.; Shi, H.; Zhang, X.; Zhou, T. Prediction of CO2 solubility in ionic liquids using machine learning methods. Chem. Eng. Sci. 2020, 223, 115752. [Google Scholar] [CrossRef]
- Zhao, Y.; Zhang, X.; Deng, L.; Zhang, S. Prediction of viscosity of imidazolium-based ionic liquids using MLR and SVM algorithms. Comput. Chem. Eng. 2016, 92, 37–42. [Google Scholar] [CrossRef]
- Zhao, Y.; Zhao, J.; Huang, Y.; Zhou, Q.; Zhang, X.; Zhang, S. Toxicity of ionic liquids: Database and prediction via quantitative structure-activity relationship method. J. Hazard. Mater. 2014, 278, 320–329. [Google Scholar] [CrossRef]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B.; et al. PubChem 2019 update: Improved access to chemical data. Nucleic Acids Res. 2019, 47, D1102–D1109. [Google Scholar] [CrossRef]
- Wang, Z.; Su, Y.; Jin, S.; Shen, W.; Ren, J.; Zhang, X.; Clark, J.H. A novel unambiguous strategy of molecular feature extraction in machine learning assisted predictive models for environmental properties. Green Chem. 2020, 22, 3867–3876. [Google Scholar] [CrossRef]
- RDKit: Open-Source Cheminformatics Software. Available online: https://www.rdkit.org/ (accessed on 14 October 2020).
- PyTorch. Available online: https://pytorch.org/ (accessed on 14 October 2020).
- Kingma, D.P.; Ba, J. Adam: A method for stochastic optimization. arXiv 2014, arXiv:1412.6980. [Google Scholar]
- Rumelhart, D.E.; Hinton, G.E.; Williams, R.J. Learning representations by back-propagating errors. Nature 1986, 323, 533–536. [Google Scholar] [CrossRef]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V.; et al. Scikit-learn: Machine learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Model | Sample Number | RMSE | MAE | R2 |
|---|---|---|---|---|
| MLR model (Sosnowska et al. [18]) | 304 | 0.51 | - | 0.77 |
| MLR model (Wu et al. [19]) | 304 | 0.43 | 0.34 | - |
| FNN model (this work) | 355 | 0.3089 | 0.2294 | 0.9157 |
| SVM model (this work) | 355 | 0.2875 | 0.1935 | 0.9270 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Z.; Song, Z.; Zhou, T. Machine Learning for Ionic Liquid Toxicity Prediction. Processes 2021, 9, 65. https://doi.org/10.3390/pr9010065
Wang Z, Song Z, Zhou T. Machine Learning for Ionic Liquid Toxicity Prediction. Processes. 2021; 9(1):65. https://doi.org/10.3390/pr9010065
Chicago/Turabian StyleWang, Zihao, Zhen Song, and Teng Zhou. 2021. "Machine Learning for Ionic Liquid Toxicity Prediction" Processes 9, no. 1: 65. https://doi.org/10.3390/pr9010065
APA StyleWang, Z., Song, Z., & Zhou, T. (2021). Machine Learning for Ionic Liquid Toxicity Prediction. Processes, 9(1), 65. https://doi.org/10.3390/pr9010065