Biomimetic Vanadate and Molybdate Systems for Oxidative Upgrading of Iono- and Organosolv Hard- and Softwood Lignins


 , , ,
, , ,  , ,
, ,  and
and 
Abstract
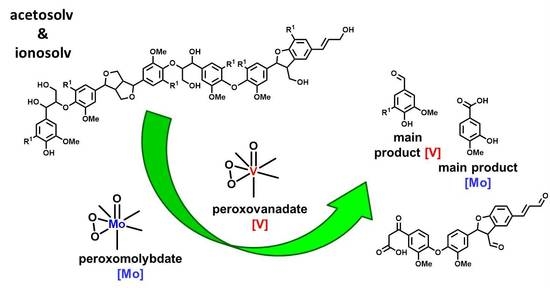
1. Introduction
2. Materials and Methods
3. Results and Discussion
3.1. Catalytic Systems
3.2. Lignin Starting Materials
3.3. Catalytic Degradation of Softwood Lignins
3.4. Catalytic Degradation of Hardwood Lignins
3.5. Mechanistic Considerations
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Aresta, M.; Dibenedetto, A.; Dumeignil, F. Biorefineries, An Introduction; De Gruyter: Berlin, Germany; Boston, MA, USA, 2015; ISBN 978-3-11-033158-5. [Google Scholar]
- Fernando, S.; Adhikari, S.; Chandrapal, C.; Murali, N. Biorefineries: Current Status, Challenges, and Future Direction. Energy Fuels 2006, 20, 1727–1737. [Google Scholar] [CrossRef]
- Takkellapati, S.; Li, T.; Gonzalez, M.A. An overview of biorefinery-derived platform chemicals from a cellulose and hemicellulose biorefinery. Clean Technol. Environ. Policy 2018, 20, 1615–1630. [Google Scholar] [CrossRef] [PubMed]
- Yabushita, M.; Kobayashi, H.; Fukuoka, A. Catalytic transformation of cellulose into platform chemicals. Appl. Catal. B Environ. 2014, 145, 1–9. [Google Scholar] [CrossRef]
- Delidovich, I.; Leonhard, K.; Palkovits, R. Cellulose and hemicellulose valorisation: An integrated challenge of catalysis and reaction engineering. Energy Environ. Sci. 2014, 7, 2803–2830. [Google Scholar] [CrossRef]
- Argyropoulos, D.S.; Crestini, C. A Perspective on Lignin Refining, Functionalization, and Utilization. ACS Sustain. Chem. Eng. 2016, 4, 5089. [Google Scholar] [CrossRef]
- Fiorani, G.; Crestini, C.; Selva, M.; Perosa, A. Advancements and Complexities in the Conversion of Lignocellulose Into Chemicals and Materials. Front. Chem. 2020, 8. [Google Scholar] [CrossRef]
- Mobley, J.K. Conversion of Lignin to Value-added Chemicals via Oxidative Depolymerization. In Chemical Catalysts for Biomass Upgrading; Crocker, M., Santillan-Jimenez, E., Eds.; Wiley: Hoboken, NJ, USA, 2020; pp. 357–393. ISBN 978-3-527-34466-6. [Google Scholar]
- Lange, H.; Decina, S.; Crestini, C. Oxidative upgrade of lignin—Recent routes reviewed. Eur. Polym. J. 2013, 49, 1151–1173. [Google Scholar] [CrossRef]
- Crestini, C.; Crucianelli, M.; Orlandi, M.; Saladino, R. Oxidative strategies in lignin chemistry: A new environmental friendly approach for the functionalisation of lignin and lignocellulosic fibers. Catal. Today 2010, 156, 8–22. [Google Scholar] [CrossRef]
- Zakzeski, J.; Bruijnincx, P.C.A.; Jongerius, A.L.; Weckhuysen, B.M. The Catalytic Valorization of Lignin for the Production of Renewable Chemicals. Chem. Rev. 2010, 110, 3552–3599. [Google Scholar] [CrossRef]
- Argyropoulos, D.S. Oxidative Delignification Chemistry: Fundamentals and Catalysis; ACS Symposium Series; American Chemical Society: Washington, DC, USA, 2001; ISBN 978-0-8412-3738-4. [Google Scholar]
- Crestini, C.; Pro, P.; Neri, V.; Saladino, R. Methyltrioxorhenium: A new catalyst for the activation of hydrogen peroxide to the oxidation of lignin and lignin model compounds. Bioorg. Med. Chem. 2005, 13, 2569–2578. [Google Scholar] [CrossRef]
- Crestini, C.; Caponi, M.C.; Argyropoulos, D.S.; Saladino, R. Immobilized methyltrioxo rhenium (MTO)/H2O2 systems for the oxidation of lignin and lignin model compounds. Bioorg. Med. Chem. 2006, 14, 5292–5302. [Google Scholar] [CrossRef]
- Guadix-Montero, S.; Sankar, M. Review on Catalytic Cleavage of C–C Inter-unit Linkages in Lignin Model Compounds: Towards Lignin Depolymerisation. Top. Catal. 2018, 61, 183–198. [Google Scholar] [CrossRef]
- Chan, J.M.W.; Bauer, S.; Sorek, H.; Sreekumar, S.; Wang, K.; Toste, F.D. Studies on the Vanadium-Catalyzed Nonoxidative Depolymerization of Miscanthus giganteus-Derived Lignin. ACS Catal. 2013, 3, 1369–1377. [Google Scholar] [CrossRef]
- Son, S.; Toste, F.D. Non-Oxidative Vanadium-Catalyzed C-O Bond Cleavage: Application to Degradation of Lignin Model Compounds. Angew. Chem. Int. Ed. 2010, 49, 3791–3794. [Google Scholar] [CrossRef] [PubMed]
- Amadio, E.; Di Lorenzo, R.; Zonta, C.; Licini, G. Vanadium catalyzed aerobic carbon–carbon cleavage. Coord. Chem. Rev. 2015, 301–302, 147–162. [Google Scholar] [CrossRef]
- Bozell, J.J.; Hoberg, J.O.; Dimmel, D.R. Heteropolyacid Catalyzed Oxidation of Lignin and Lignin Models to Benzoquinones. J. Wood Chem. Technol. 2000, 20, 19–41. [Google Scholar] [CrossRef]
- Mobley, J.K.; Jennings, J.A.; Morgan, T.; Kiefer, A.; Crocker, M. Oxidation of Benzylic Alcohols and Lignin Model Compounds with Layered Double Hydroxide Catalysts. Inorganics 2018, 6, 75. [Google Scholar] [CrossRef]
- Voitl, T.; Rudolf von Rohr, P. Oxidation of Lignin Using Aqueous Polyoxometalates in the Presence of Alcohols. ChemSusChem 2008, 1, 763–769. [Google Scholar] [CrossRef]
- Weinstock, I.A.; Atalla, R.H.; Reiner, R.S.; Moen, M.A.; Hammel, K.E.; Houtman, C.J.; Hill, C.L.; Harrup, M.K. A new environmentally benign technology for transforming wood pulp into paper. Engineering polyoxometalates as catalysts for multiple processes. J. Mol. Catal. A Chem. 1997, 116, 59–84. [Google Scholar] [CrossRef]
- Mottweiler, J.; Puche, M.; Räuber, C.; Schmidt, T.; Concepción, P.; Corma, A.; Bolm, C. Copper- and Vanadium-Catalyzed Oxidative Cleavage of Lignin using Dioxygen. ChemSusChem 2015, 8, 2106–2113. [Google Scholar] [CrossRef]
- Hao, K.; Zhang, L.-L.; Song, L.; Guan, H.-Y.; Li, C.-M.; Liu, T.; Yu, Q.; Zeng, J.-M.; Wang, Z.-W. Highly active Mo-V-based bifunctional catalysts for catalytic conversion of lignin dimer model compounds at room temperature. Inorg. Chem. Commun. 2020, 116, 107910. [Google Scholar] [CrossRef]
- Drago, G.A.; Gibson, T.D. Enzyme Stability and Stabilisation: Applications and Case Studies. In Engineering and Manufacturing for Biotechnology; Hofman, M., Thonart, P., Eds.; Focus on Biotechnology; Springer: Dordrecht, The Netherlands, 2002; pp. 361–376. ISBN 978-0-306-46889-6. [Google Scholar]
- Crestini, C.; Pastorini, A.; Tagliatesta, P. Metalloporphyrins immobilized on motmorillonite as biomimetic catalysts in the oxidation of lignin model compounds. J. Mol. Catal. A Chem. 2004, 208, 195–202. [Google Scholar] [CrossRef]
- Crestini, C.; Tagliatesta, P. Metalloporphyrins in the Biomimetic Oxidation of Lignin and Lignin Model Compounds: Development of Alternative Delignification Strategies. Porphyr. Handb. Bioinorg. Bioorg. Chem. 2003, 11, 161. [Google Scholar]
- Crestini, C.; Tagliatesta, P.; Saladino, R. A biomimetic approach to lignin degradation, metalloporphyrins catalysed oxidation of lignin and lignin model compounds. Oxid. Delignification Chem. Fundam. Catal. 2001, 213–225. [Google Scholar]
- Giannì, P.; Lange, H.; Crestini, C. Lipoxygenase: Unprecedented Carbon-Centered Lignin Activation. ACS Sustain. Chem. Eng. 2018, 6, 5085–5096. [Google Scholar] [CrossRef]
- Wong, D.W.S. Structure and Action Mechanism of Ligninolytic Enzymes. Appl. Biochem. Biotechnol. 2009, 157, 174–209. [Google Scholar] [CrossRef]
- Penín, L.; Lange, H.; Santos, V.; Crestini, C.; Parajó, J.C. Characterization of Eucalyptus nitens Lignins Obtained by Biorefinery Methods Based on Ionic Liquids. Molecules 2020, 25, 425. [Google Scholar] [CrossRef]
- Penín, L.; Peleteiro, S.; Santos, V.; Alonso, J.L.; Parajó, J.C. Selective fractionation and enzymatic hydrolysis of Eucalyptus nitens wood. Cellulose 2019, 26, 1125–1139. [Google Scholar] [CrossRef]
- Penín, L.; Santos, V.; del Río, J.C.; Parajó, J.C. Assesment on the chemical fractionation of Eucalyptus nitens wood: Characterization of the products derived from the structural components. Bioresour. Technol. 2019, 281, 269–276. [Google Scholar] [CrossRef]
- Meng, X.; Crestini, C.; Ben, H.; Hao, N.; Pu, Y.; Ragauskas, A.J.; Argyropoulos, D.S. Determination of hydroxyl groups in biorefinery resources via quantitative 31 P NMR spectroscopy. Nat. Protoc. 2019, 14, 2627–2647. [Google Scholar] [CrossRef]
- Granata, A.; Argyropoulos, D.S. 2-Chloro-4,4,5,5-tetramethyl-1,3,2-dioxaphospholane, a Reagent for the Accurate Determination of the Uncondensed and Condensed Phenolic Moieties in Lignins. J. Agric. Food Chem. 1995, 43, 1538–1544. [Google Scholar] [CrossRef]
- Argyropoulos, D.S. 31P NMR in Wood Chemistry: A Review of Recent Progress. Res. Chem. Intermed. 1995, 21, 373–395. [Google Scholar] [CrossRef]
- Amini, M.; Haghdoost, M.M.; Bagherzadeh, M. Oxido-peroxido molybdenum(VI) complexes in catalytic and stoichiometric oxidations. Coord. Chem. Rev. 2013, 257, 1093–1121. [Google Scholar] [CrossRef]
- Conte, V.; Floris, B. Vanadium and molybdenum peroxides: Synthesis and catalytic activity in oxidation reactions. Dalton Trans. 2011, 40, 1419–1436. [Google Scholar] [CrossRef] [PubMed]
- Schwendt, P.; Tatiersky, J.; Krivosudský, L.; Šimuneková, M. Peroxido complexes of vanadium. Coord. Chem. Rev. 2016, 318, 135–157. [Google Scholar] [CrossRef]
- Conte, V.; Floris, B. Vanadium catalyzed oxidation with hydrogen peroxide. Inorg. Chim. Acta 2010, 363, 1935–1946. [Google Scholar] [CrossRef]
- Conte, V.; Coletti, A.; Floris, B.; Licini, G.; Zonta, C. Mechanistic aspects of vanadium catalysed oxidations with peroxides. Coord. Chem. Rev. 2011, 255, 2165–2177. [Google Scholar] [CrossRef]
- Galloni, P.; Mancini, M.; Floris, B.; Conte, V. A sustainable two-phase procedure for V-catalyzed toluene oxidative bromination with H2O2–KBr. Dalton Trans. 2013, 42, 11963–11970. [Google Scholar] [CrossRef]
- Langeslay, R.R.; Kaphan, D.M.; Marshall, C.L.; Stair, P.C.; Sattelberger, A.P.; Delferro, M. Catalytic Applications of Vanadium: A Mechanistic Perspective. Chem. Rev. 2019, 119, 2128–2191. [Google Scholar] [CrossRef]
- Floris, B.; Sabuzi, F.; Coletti, A.; Conte, V. Sustainable vanadium-catalyzed oxidation of organic substrates with H2O2. Catal. Today 2017, 285, 49–56. [Google Scholar] [CrossRef]
- Kirillova, M.V.; Kuznetsov, M.L.; Romakh, V.B.; Shul’pina, L.S.; Fraústo da Silva, J.J.R.; Pombeiro, A.J.L.; Shul’pin, G.B. Mechanism of oxidations with H2O2 catalyzed by vanadate anion or oxovanadium(V) triethanolaminate (vanadatrane) in combination with pyrazine-2-carboxylic acid (PCA): Kinetic and DFT studies. J. Catal. 2009, 267, 140–157. [Google Scholar] [CrossRef]
- Garau, G.; Palma, A.; Lauro, G.P.; Mele, E.; Senette, C.; Manunza, B.; Deiana, S. Detoxification Processes from Vanadate at the Root Apoplasm Activated by Caffeic and Polygalacturonic Acids. PLoS ONE 2015, 10, e0141041. [Google Scholar] [CrossRef] [PubMed]
- Linskens, H.F.; Jackson, J.F. (Eds.) Plant Cell Wall Analysis; Modern Methods of Plant Analysis; Springer: Berlin/Heidelberg, Germany, 1996; Volume 17, ISBN 978-3-642-64644-7. [Google Scholar]
- Crestini, C.; Jurasek, L.; Argyropoulos, D.S. On the Mechanism of the Laccase-Mediator System in the Oxidation of Lignin. Chem. Eur. J. 2003, 9, 5371–5378. [Google Scholar] [CrossRef] [PubMed]
- Dakin, H.D. The oxidation of hydroxy derivatives of benzaldehyde, acetophenone and related substances. Am. Chem. J. 1909, 42, 477–498. [Google Scholar]



{kind=link}
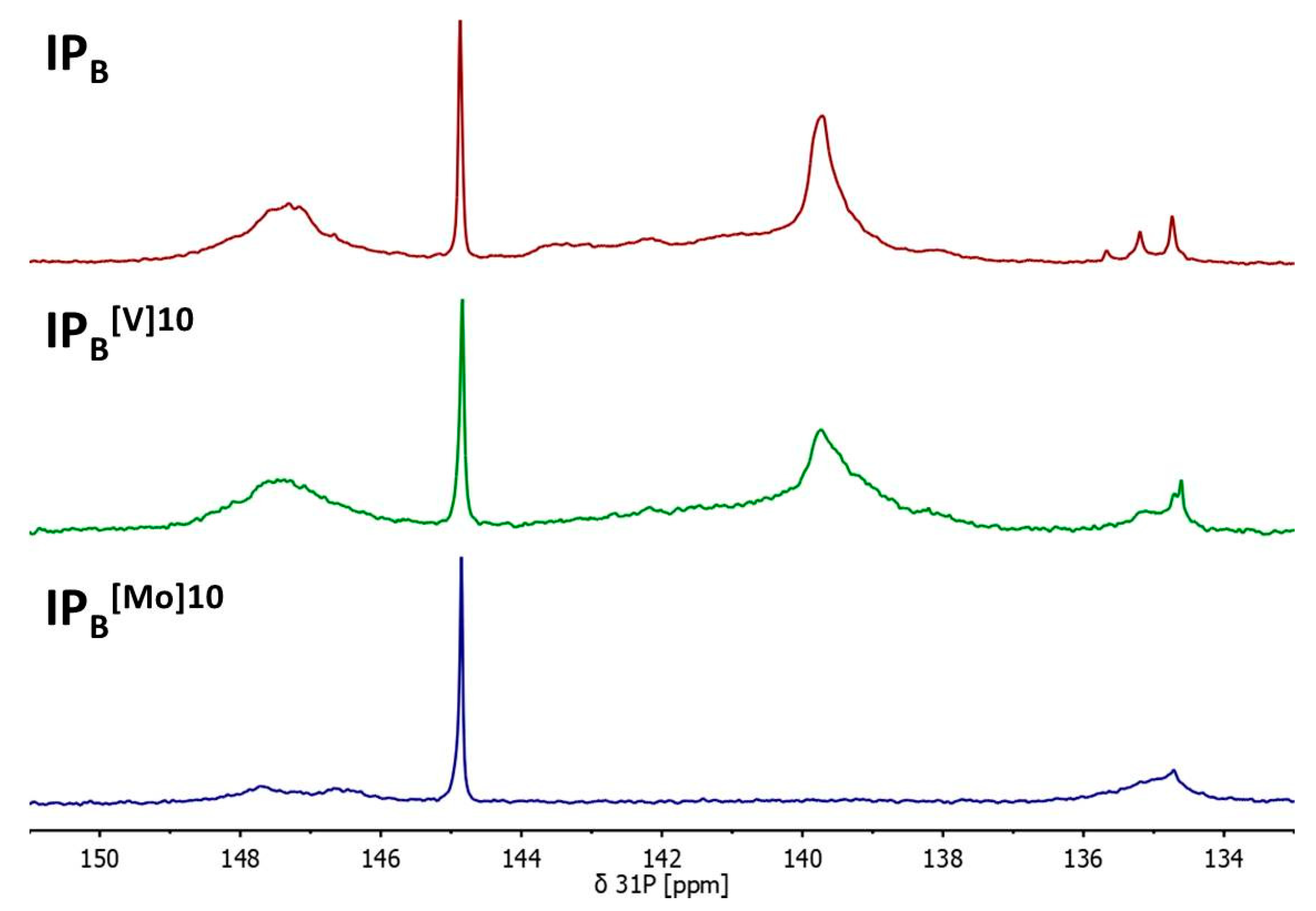{kind=link}
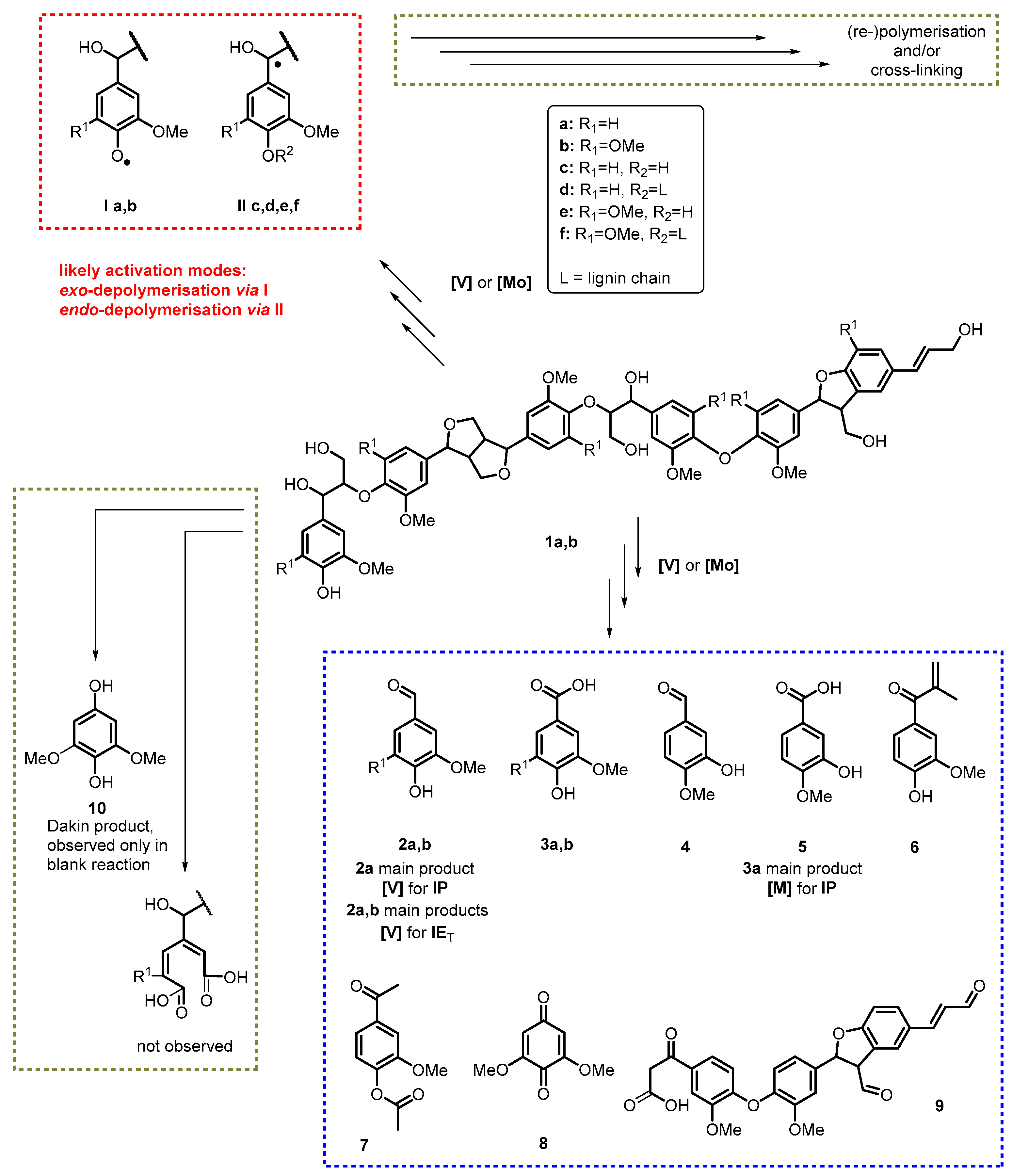{kind=link}
| Lignins a | AP | IPB | AE | IEB | IPB[V]10 | IPB[Mo]10 |
|---|---|---|---|---|---|---|
| OH-group | Abundance (mmol/g) | |||||
| aliphatic OH | 0.23 | 1.36 | 0.34 | 1.28 | 1.31 | 0.80 b |
| Condensed | 0.96 | 0.95 | 2.90 | 3.28 | 0.89 | 0.06 b |
| o-disub. phenols (S units) | --- | --- | 1.25 | 1.23 | --- | --- |
| 4-O-5′ + 5-5′ | 0.96 | 0.95 | 1.65 | 2.05 | 0.89 | 0.06 b |
| o-monosub. phenols (G units) | 1.14 | 1.56 | 0.60 | 0.73 | 1.22 | 0.07 b |
| p-OH phenols | 0.21 | 0.24 | 0.15 | 0.14 | 0.36 | 0.04 b |
| total phenolic OH | 2.32 | 2.75 | 3.65 | 4.16 | 2.47 | 0.17 b |
| carboxylic OH | 0.24 | 0.22 | 0.31 | 0.05 | 0.33 | 0.75 b |
| total phenolic OH/aliphatic OH | 9.9 | 2.0 | 10.8 | 3.2 | 1.9 | 0.2 b |
| total phenolic OH/ condensed phenolic OH | 1.9 | 2.2 | 1.2 | 1.2 | 2.8 | 2.8 b |
| Mn [kDa] (PDI) c | 1.75 (7.3) | 1.40 (5.8) | 1.20 (2.4) | 1.20 (2.5) | n.d. d | n.d. d |
| Lignin a | AP | IPB | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Catalyst System b | [V] | [Mo] | [V] | [Mo] | ||||||||||
| Loading [% (w/w)] | Blank | 5 | 7.5 | 10 | 5 | 7.5 | 10 | Blank | 5 | 7.5 | 10 | 5 | 7.5 | 10 |
| compound c | abundance [%] d | abundance [%] d | ||||||||||||
| vanillin (2a) | --- e | --- e | --- e | 50 | --- e | --- e | --- e | --- e | 67 | 90 | 100 | 7.4 | 10 | 11 |
| vanillic acid (3a) | --- e | 33 | 33 | --- e | 100 | 100 | 100 | --- e | 24 | --- e | --- e | 93 | 90 | --- e |
| isovanillin (4) | --- e | 50 | 17 | 17 | --- e | --- e | --- e | --- e | --- e | --- e | --- e | --- e | --- e | --- e |
| isovanillic acid (5) | --- e | --- e | --- e | --- e | --- e | --- e | --- e | --- e | --- e | --- e | --- e | --- e | --- e | 89 |
| 1-(4-hydroxy-3-methoxy-phenyl)-2-methylprop-2-en-1-one (6) | --- e | --- e | 17 | 33 | --- e | --- e | --- e | --- e | 9 | 10 | --- e | --- e | --- e | --- e |
| 4-acetoxy-3-methoxyacetophenone (7) | --- e | 17 | 33 | --- e | --- e | --- e | --- e | --- e | --- e | --- e | --- e | --- e | --- e | --- e |
| depolymerisation yield [%] f | 6.0 | 23 | 26 | 31 | 26 | 69 | 72 | 9.8 | 31 | 33 | 34 | 58 | 57 | 62 |
| Mn [kDa] (PDI) g | 2.80 (6.0) | n.d. h | n.d. h | 1.95 (2.6) i | n.d. h | n.d. h | 3.20 (4.8) i | 1.80 (8.3) | n.d. h | 1.95 (3.2) i | n.d. h | n.d. h | 2.50 (3.7) i | n.d. h |
| Lignin a | AE | IEB | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Catalyst System b | blank | [V] | [Mo] | blank | [V] | [Mo] | ||||
| Loading [% (w/w)] | --- | 5 | 7.5 | 10 | 7.5 | --- | 5 | 7.5 | 10 | 5 |
| compound c | abundance [%] d | abundance [%] d | ||||||||
| vanillin (2a) | --- e | --- e | --- e | --- e | --- e | --- e | 23 | 24 | 49 | --- e |
| syringaldehyde (2b) | --- e | 61 | 100 | 69 | --- e | --- e | 77 | 65 | 42 | --- e |
| vanillic acid (3a) | --- e | --- e | --- e | --- e | 100 | --- e | --- e | --- e | --- e | 100 |
| syringic acid (3b) | --- e | --- e | --- e | --- e | --- e | --- e | --- e | --- e | --- e | --- e |
| isovanillin (4) | --- e | 12 | --- e | --- e | --- e | --- e | --- e | --- e | --- e | --- e |
| 1-(4-hydroxy-3-methoxy-phenyl)-2-methylprop-2-en-1-one (6) | --- e | 27 | --- e | 31 | --- e | --- e | --- e | 6 | 9 | --- e |
| 2,6-dimethoxy-benzoquinone (8) | --- e | --- e | --- e | --- e | --- e | --- e | --- e | 5 | --- e | --- e |
| depolymerisation yield [%] f | 22 | 46 | 38 | 53 | 84 | 25 | 38 | 37 | 38 | 78 |
| Mn [kDa] (PDI) g | 2.00 (3.8) | n.d. h | n.d. h | 3.35 (9.3) | 5.40 (3.8) | 2.10 (3.5) | n.d. h | n.d. h | 3.00 (14) | 3.60 (9.9) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Penín, L.; Gigli, M.; Sabuzi, F.; Santos, V.; Galloni, P.; Conte, V.; Parajó, J.C.; Lange, H.; Crestini, C. Biomimetic Vanadate and Molybdate Systems for Oxidative Upgrading of Iono- and Organosolv Hard- and Softwood Lignins. Processes 2020, 8, 1161. https://doi.org/10.3390/pr8091161
Penín L, Gigli M, Sabuzi F, Santos V, Galloni P, Conte V, Parajó JC, Lange H, Crestini C. Biomimetic Vanadate and Molybdate Systems for Oxidative Upgrading of Iono- and Organosolv Hard- and Softwood Lignins. Processes. 2020; 8(9):1161. https://doi.org/10.3390/pr8091161
Chicago/Turabian StylePenín, Lucía, Matteo Gigli, Federica Sabuzi, Valentín Santos, Pierluca Galloni, Valeria Conte, Juan Carlos Parajó, Heiko Lange, and Claudia Crestini. 2020. "Biomimetic Vanadate and Molybdate Systems for Oxidative Upgrading of Iono- and Organosolv Hard- and Softwood Lignins" Processes 8, no. 9: 1161. https://doi.org/10.3390/pr8091161
APA StylePenín, L., Gigli, M., Sabuzi, F., Santos, V., Galloni, P., Conte, V., Parajó, J. C., Lange, H., & Crestini, C. (2020). Biomimetic Vanadate and Molybdate Systems for Oxidative Upgrading of Iono- and Organosolv Hard- and Softwood Lignins. Processes, 8(9), 1161. https://doi.org/10.3390/pr8091161