Thermal Biomass Conversion: A Review


Abstract
1. Introduction
2. Theoretical Background
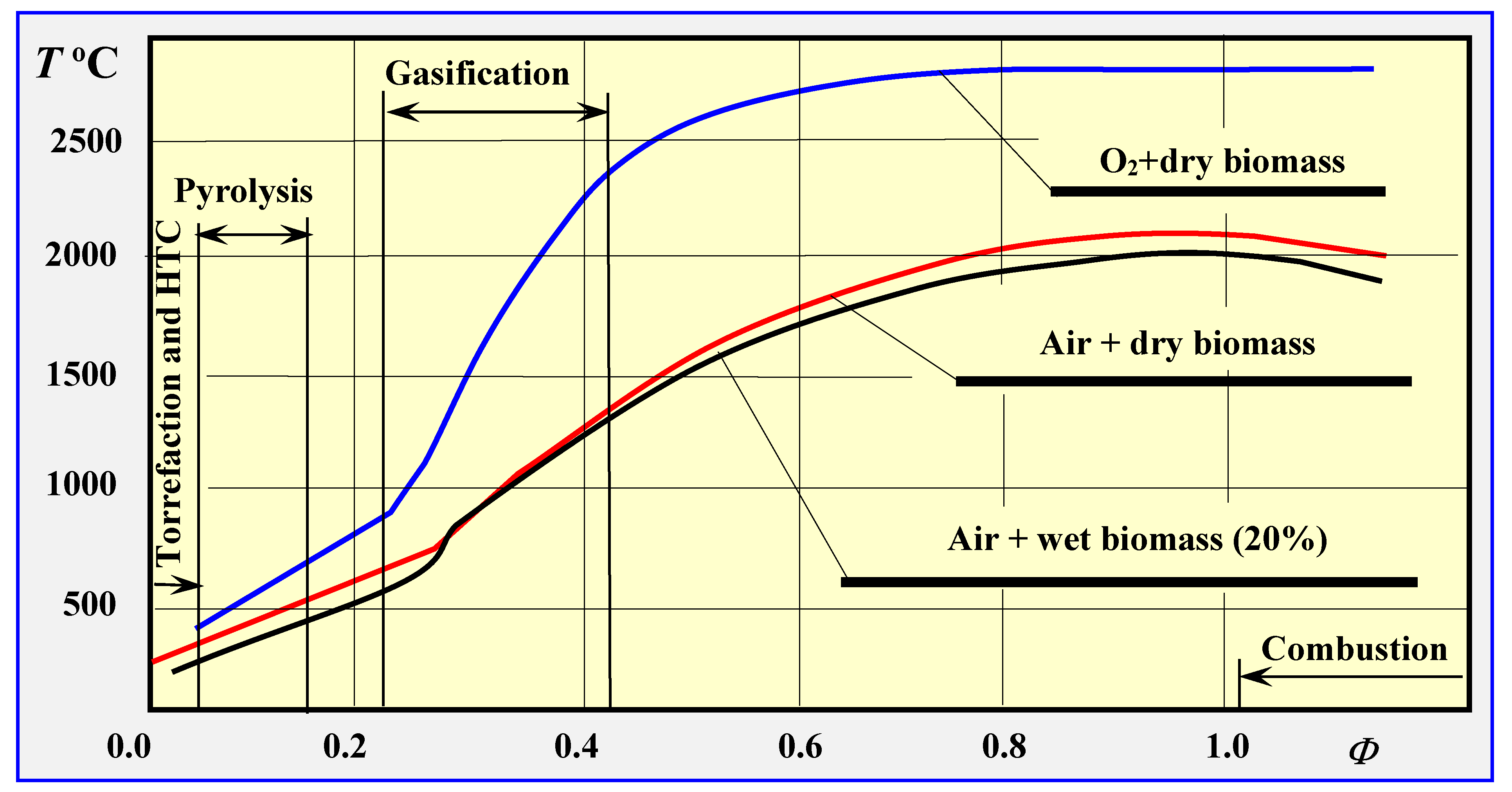
2.1. Methods of Thermal Biomass Conversion
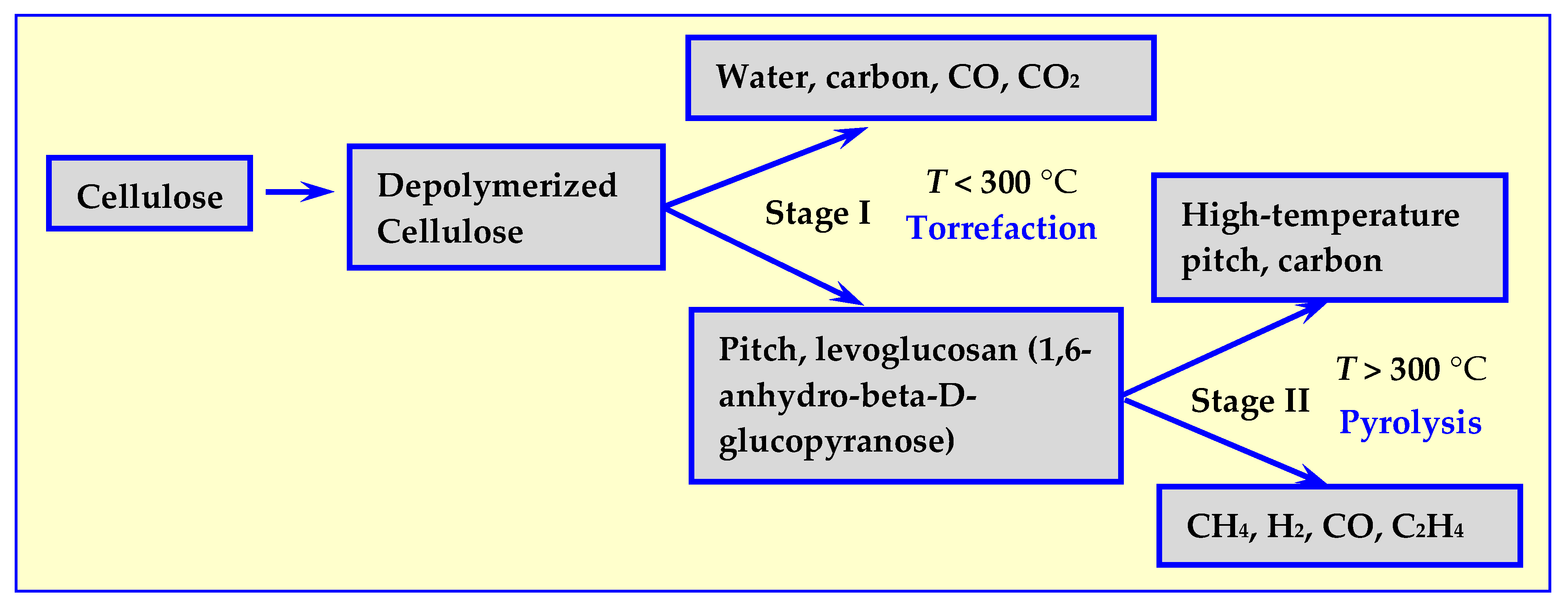
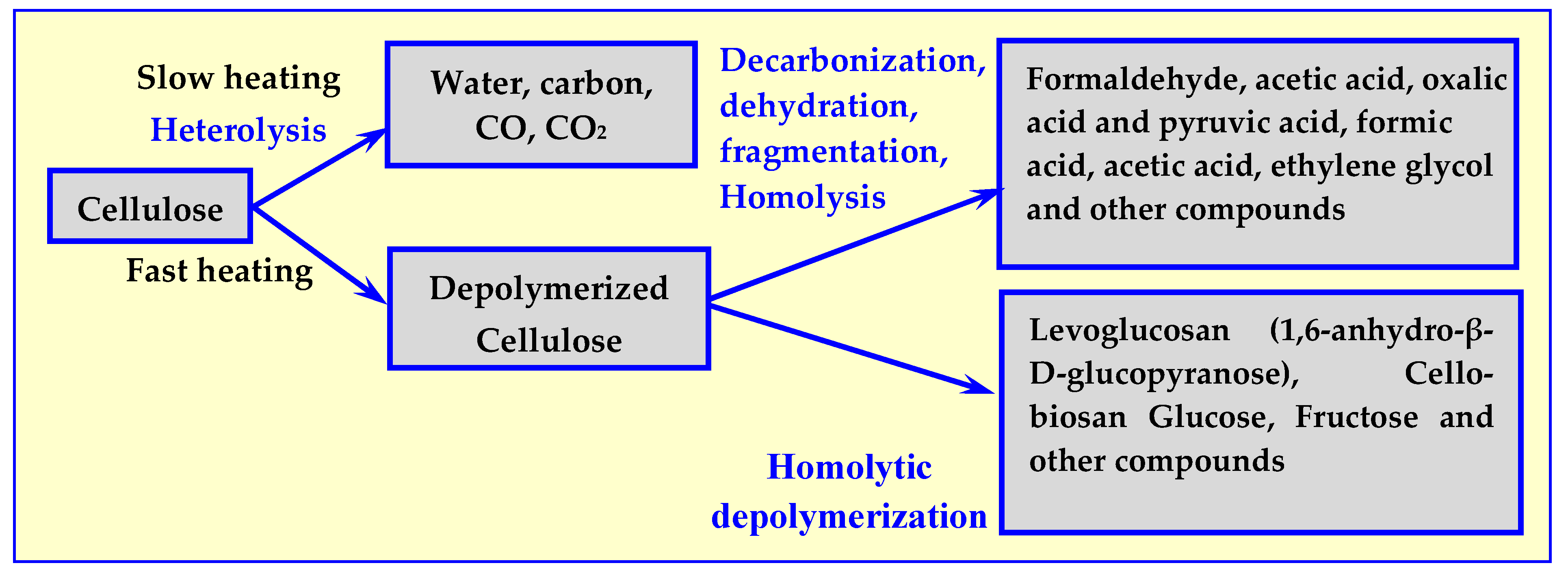
2.2. Mechanisms of Dry Thermal Decomposition of Biomass
3. Torrefaction
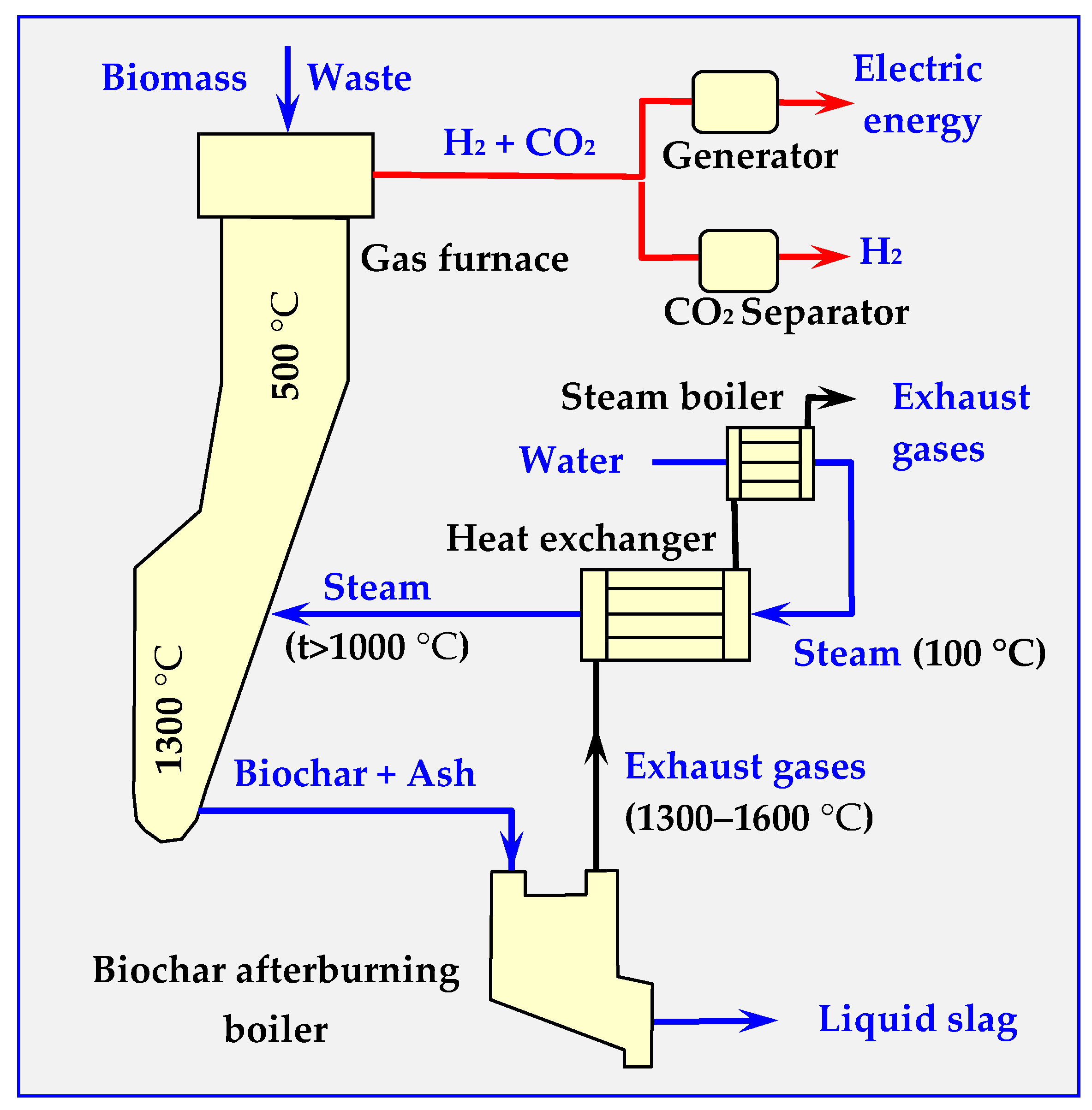
3.1. Biochar
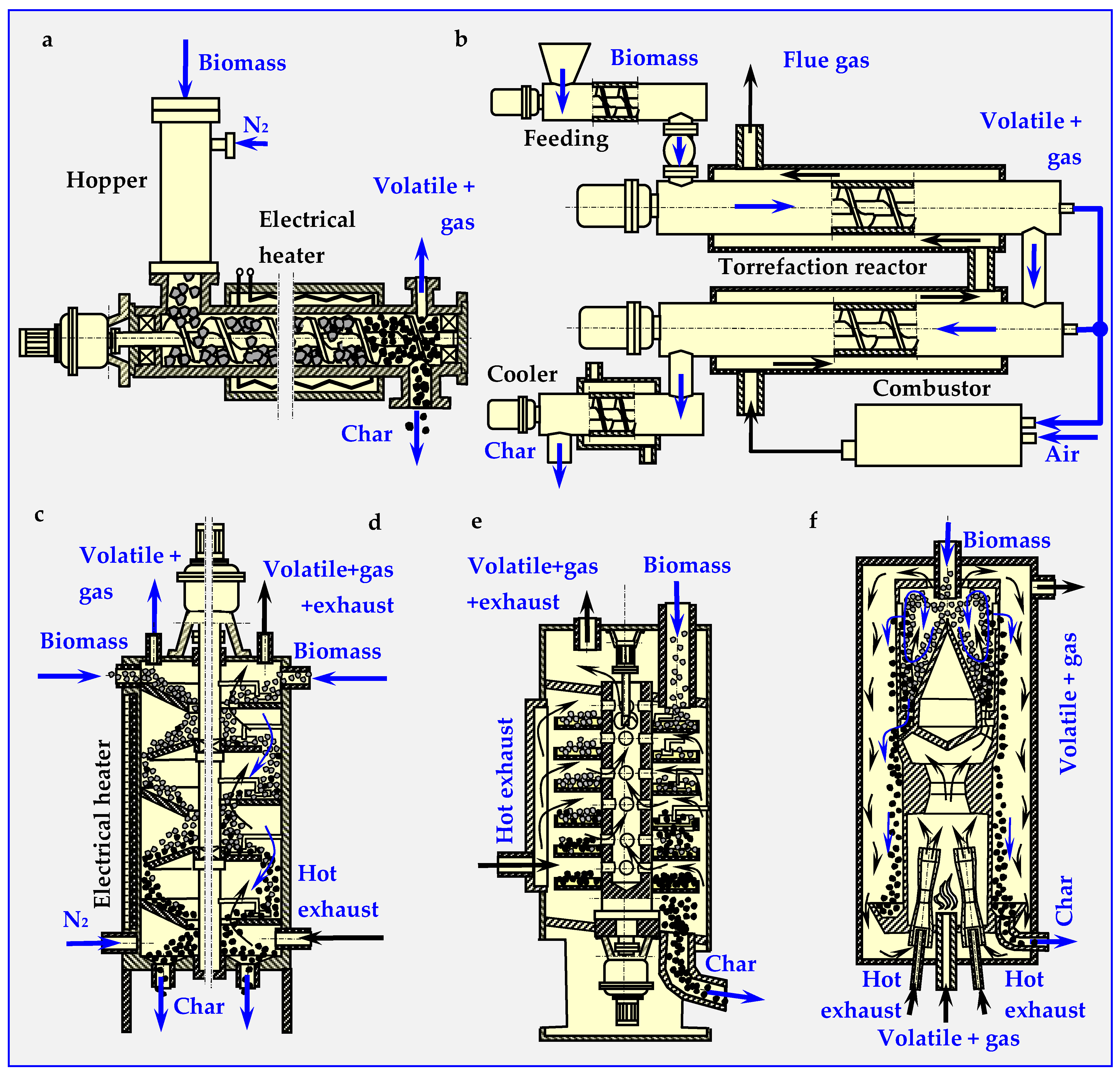
3.2. Torrefaction Reactors and Technologies
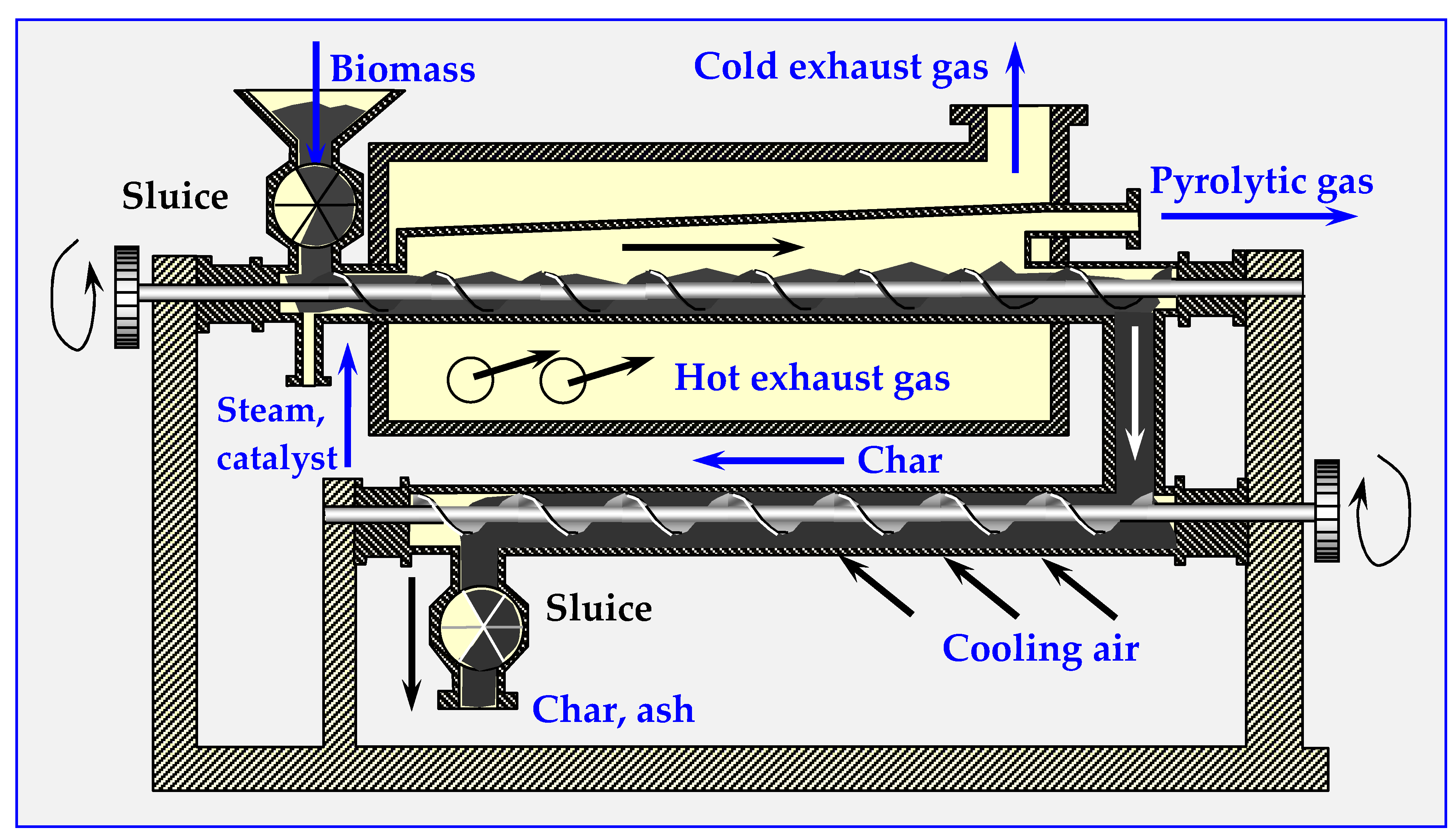
3.2.1. Auger Reactor
3.2.2. Rotary Drum Reactor
3.2.3. Multiple Hearth Furnace (MHF)
3.2.4. Torbed Reactor Technology
3.2.5. Reactor with Moving Bed
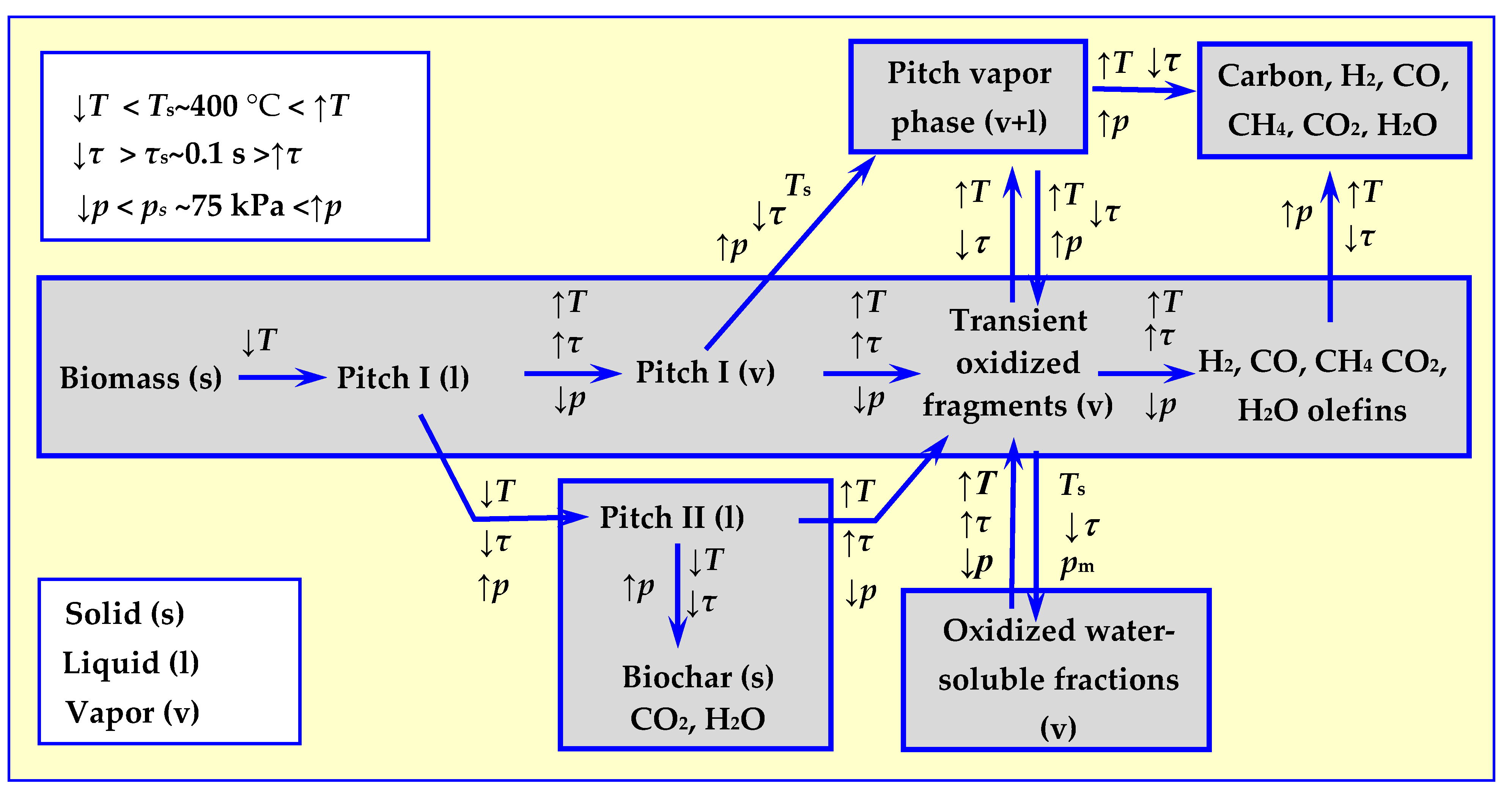
4. Pyrolysis
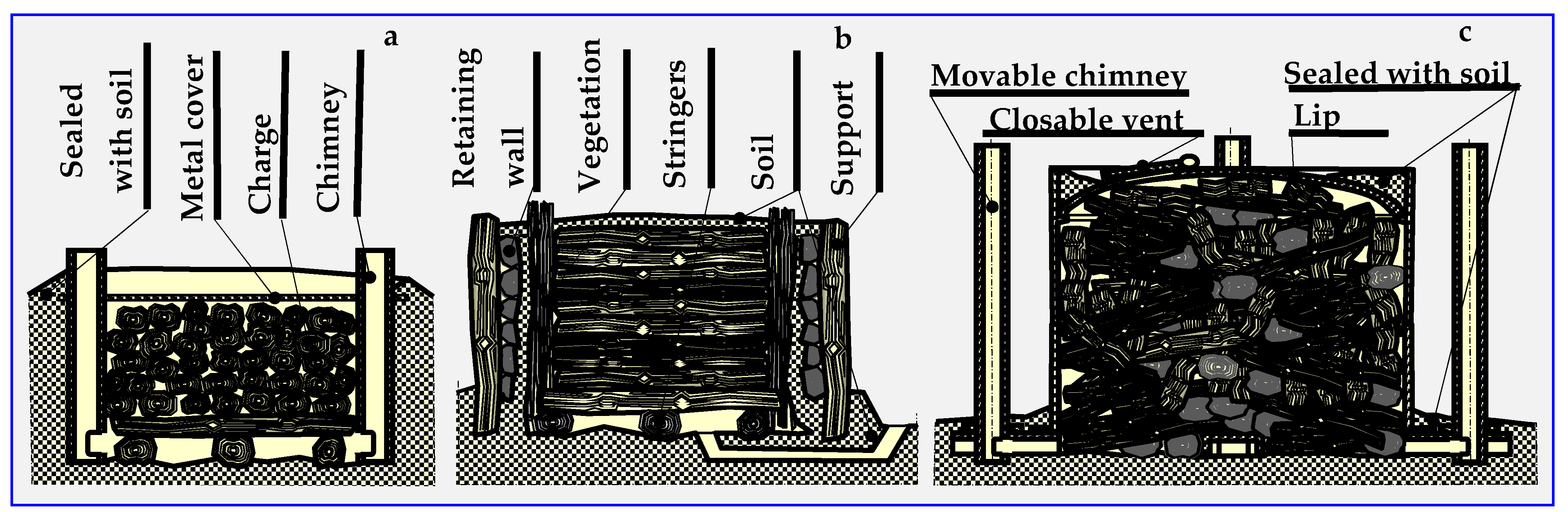
4.1. Slow Pyrolysis—Carbonization
4.2. Historical Outline of Carbonisation and Pyrolysis
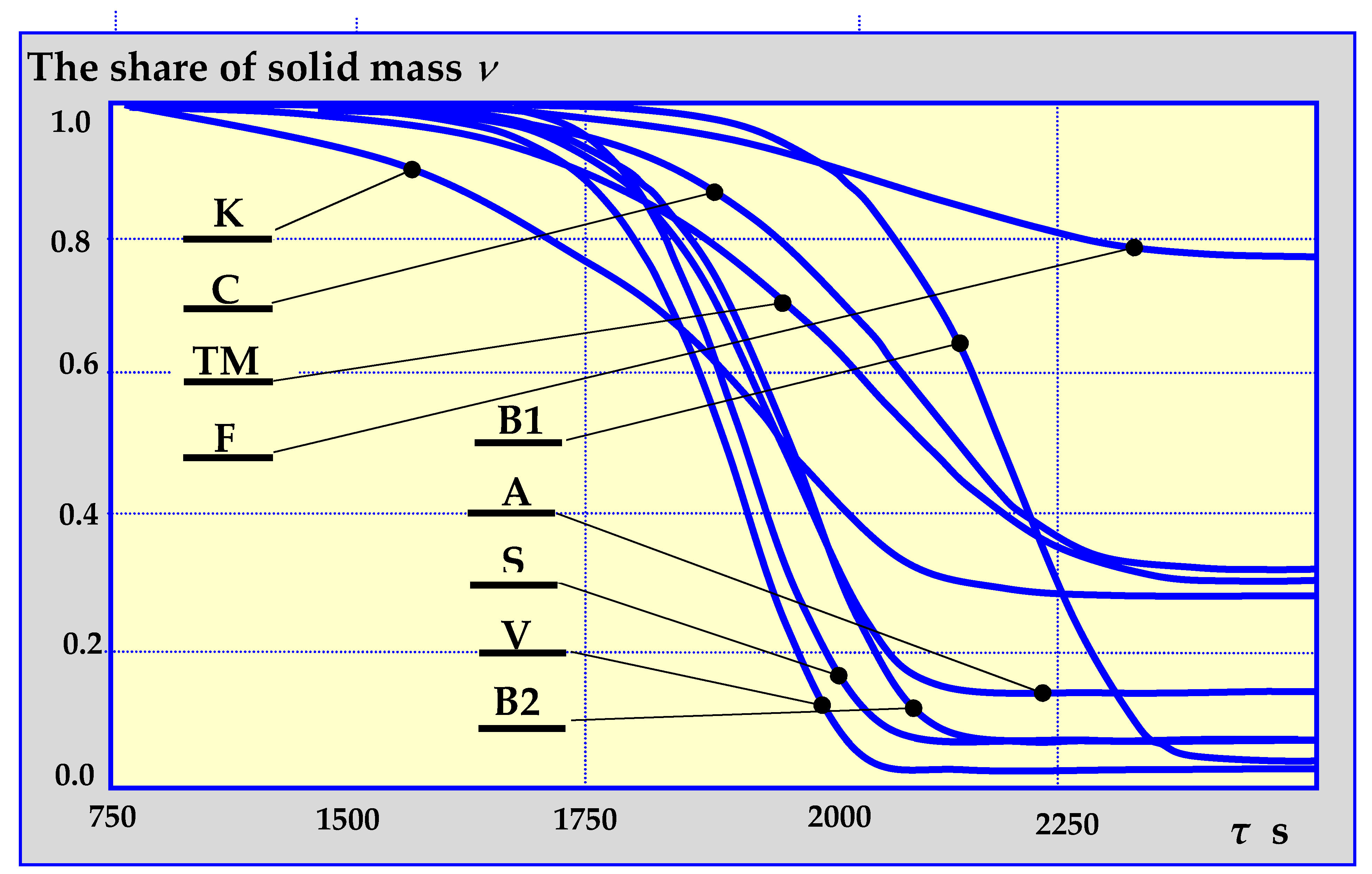
4.3. Biomass Pyrolysis Kinetics
- -
- The first—parallel degassing and drying, which assumes that the total volatile output equals the content of the dominant component determined analytically. The accuracy of this division model depends on parameters that are determined experimentally or are derived from literature data [75].
- -
4.3.1. Pyrolysis Kinetics according to the Broido Mechanism
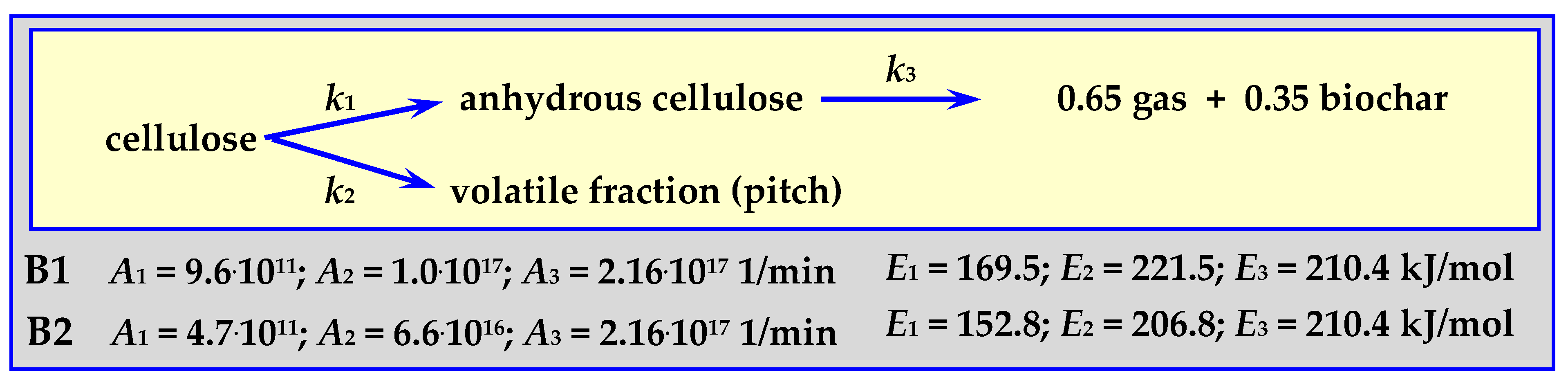
4.3.2. Pyrolysis Kinetics According to the Shafizadeh Mechanism
4.3.3. Pyrolysis Kinetics according to the Broido-Shafizadeh Mechanism
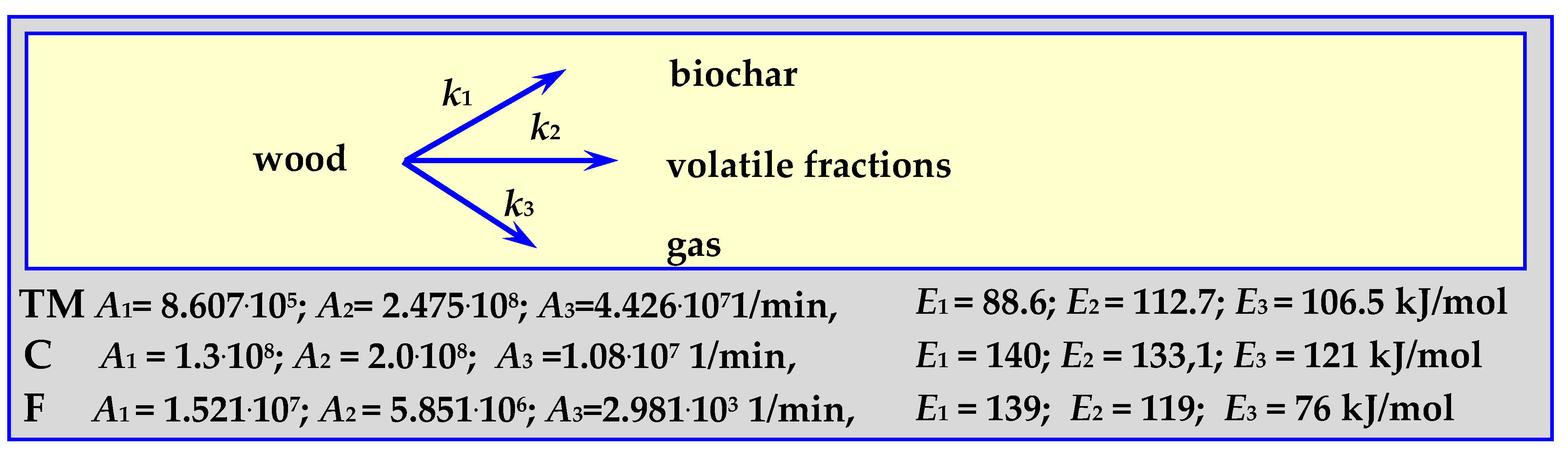
4.3.4. Pyrolysis Kinetics According to the Koufopanos Mechanism
4.3.5. Pyrolysis Kinetics According to the Three-Phase Mechanism
4.3.6. Summary of Pyrolysis Kinetics Mechanisms
4.4. Experimental Research on Biomass Pyrolysis
4.5. Technical Issues of Biomass Pyrolysis
- Solid:
- Biochar—charcoal (in case of wood decomposition) and ash,
- Gas:
- Oil and pitch (tar), which are a mixture of hydrocarbons, phenols and about 200 various aliphatic and aromatic organic compounds, saturated and unsaturated,
- Gas:
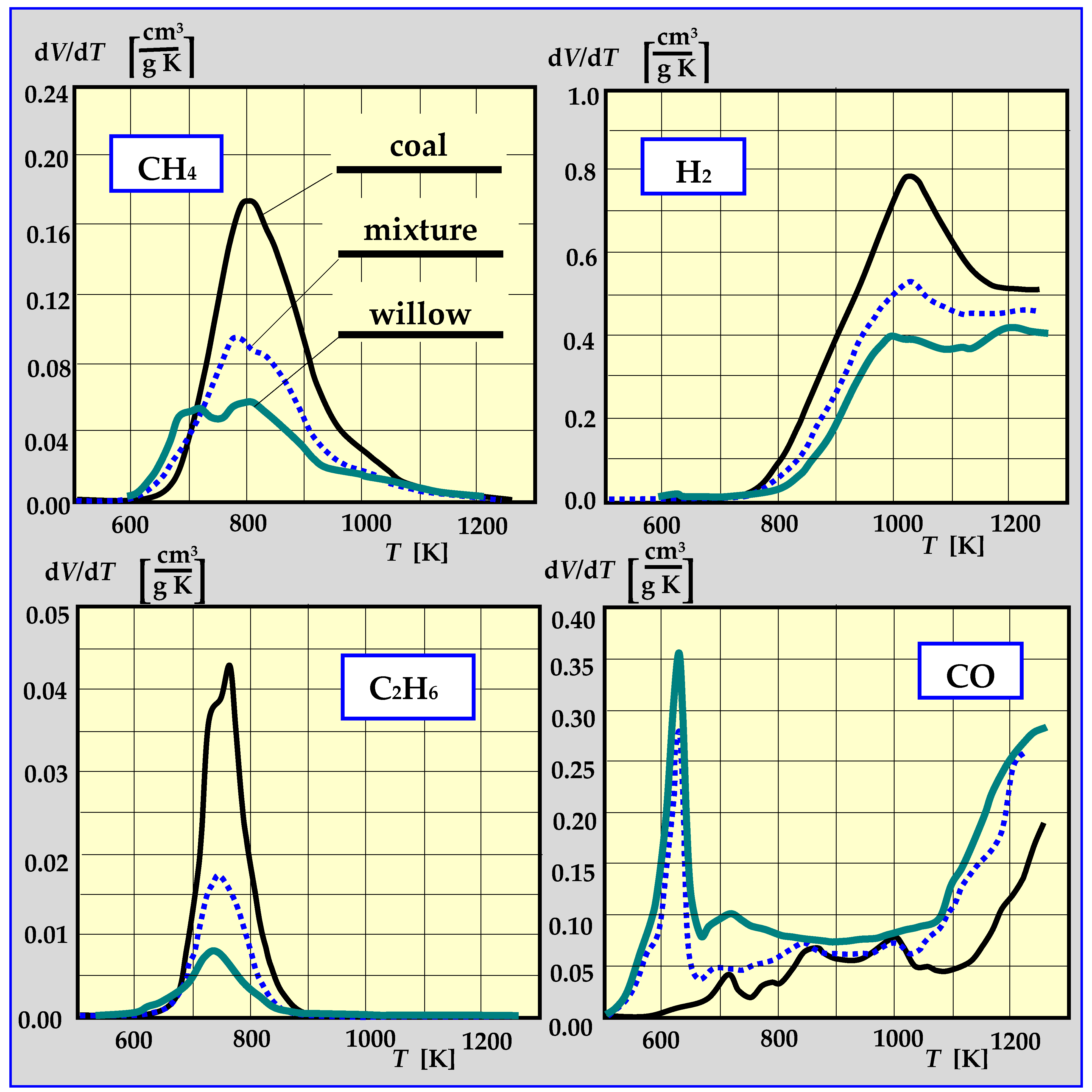
- A mixture of CO, CO2, H2, gas hydrocarbons (CH4, C2H6) and liquid hydrocarbons vapors, e.g., C3H8, C4H10 and steam, with a calorific value of approx. 12 MJ/kg. The composition depends on the temperature of the biomass decomposition. For example, for a temperature of 482 °C/926 °C, the composition is as follows (% V/V): 5.56/32.48 H2; 12.34/10.45 CH4; 33.50/35.25 CO; 3.03/1.07 C2H6; 0.71/2.43 C2H2. In total, flammable gases constituted 54.97% of the mixture for 482 °C or 81.68% for 926 °C [95]. The tar from gaseous pyrolysis products can condense at low temperatures and will lead to clogging or blockage in fuel lines, filters and engines [96]. Therefore, it must be controlled and removed.
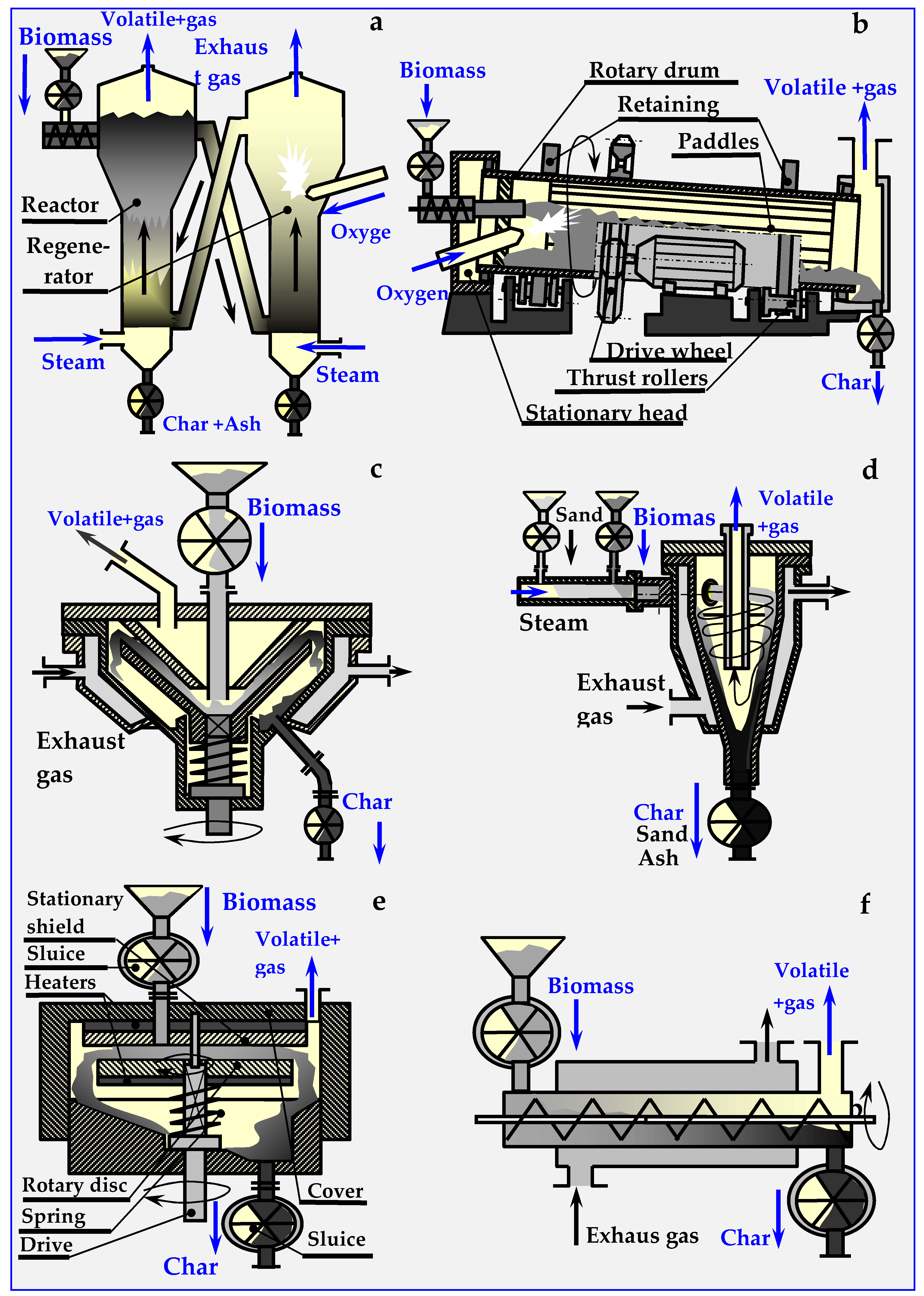
4.6. Types of Pyrolysis Reactors
4.6.1. Fluidized Pyro Duiven Lyzer
4.6.2. Rotary Kiln Pyrolyzers
4.6.3. Conical Rotary Pyrolyzer
4.6.4. Cyclone Pyrolyzer
4.6.5. Ablative Pyrolyzer
4.6.6. Auger or Screw Pyrolyzers
4.6.7. PyRos Pyrolyzer
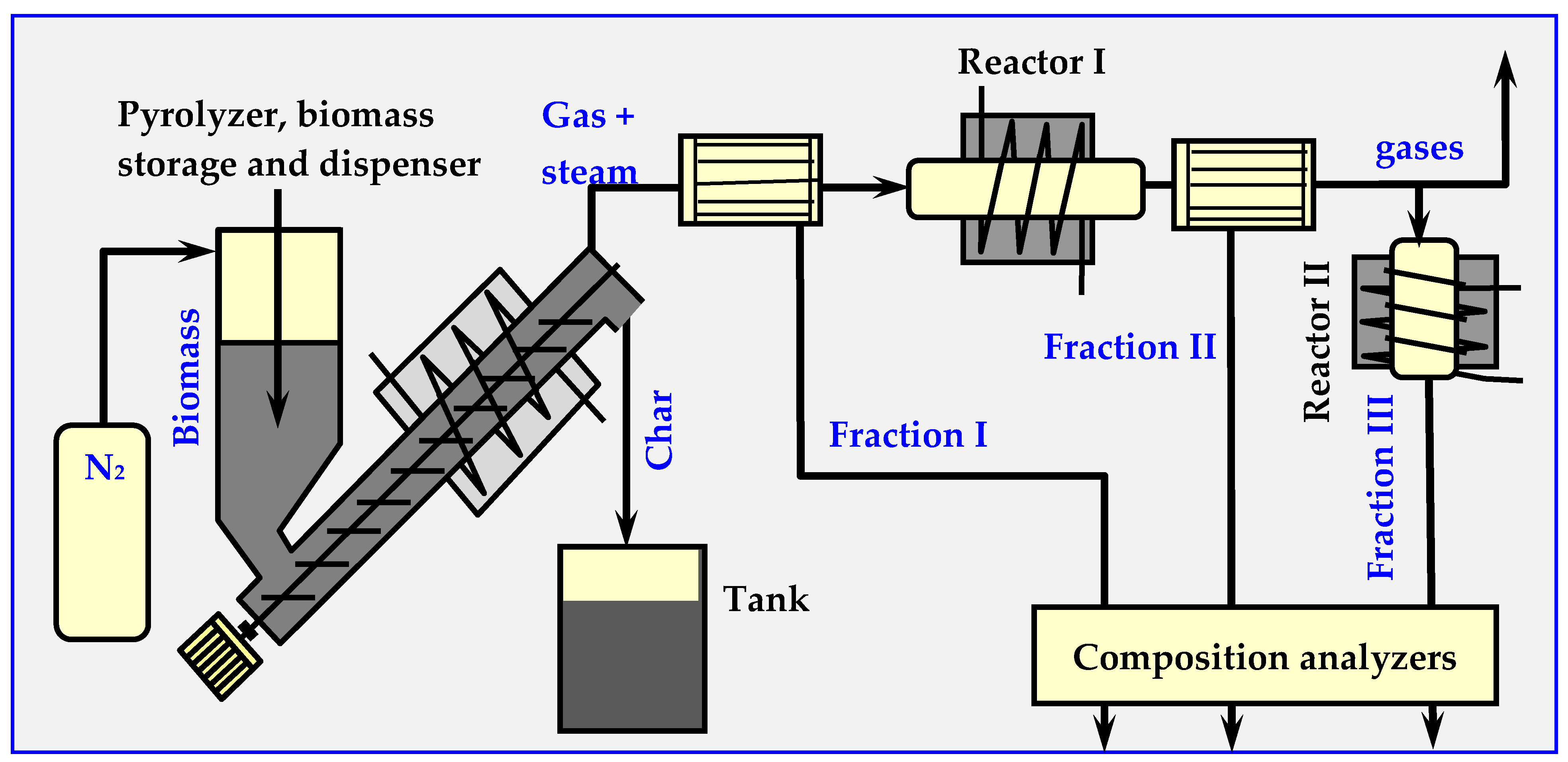
4.7. Examples of Pyrolytic Installations
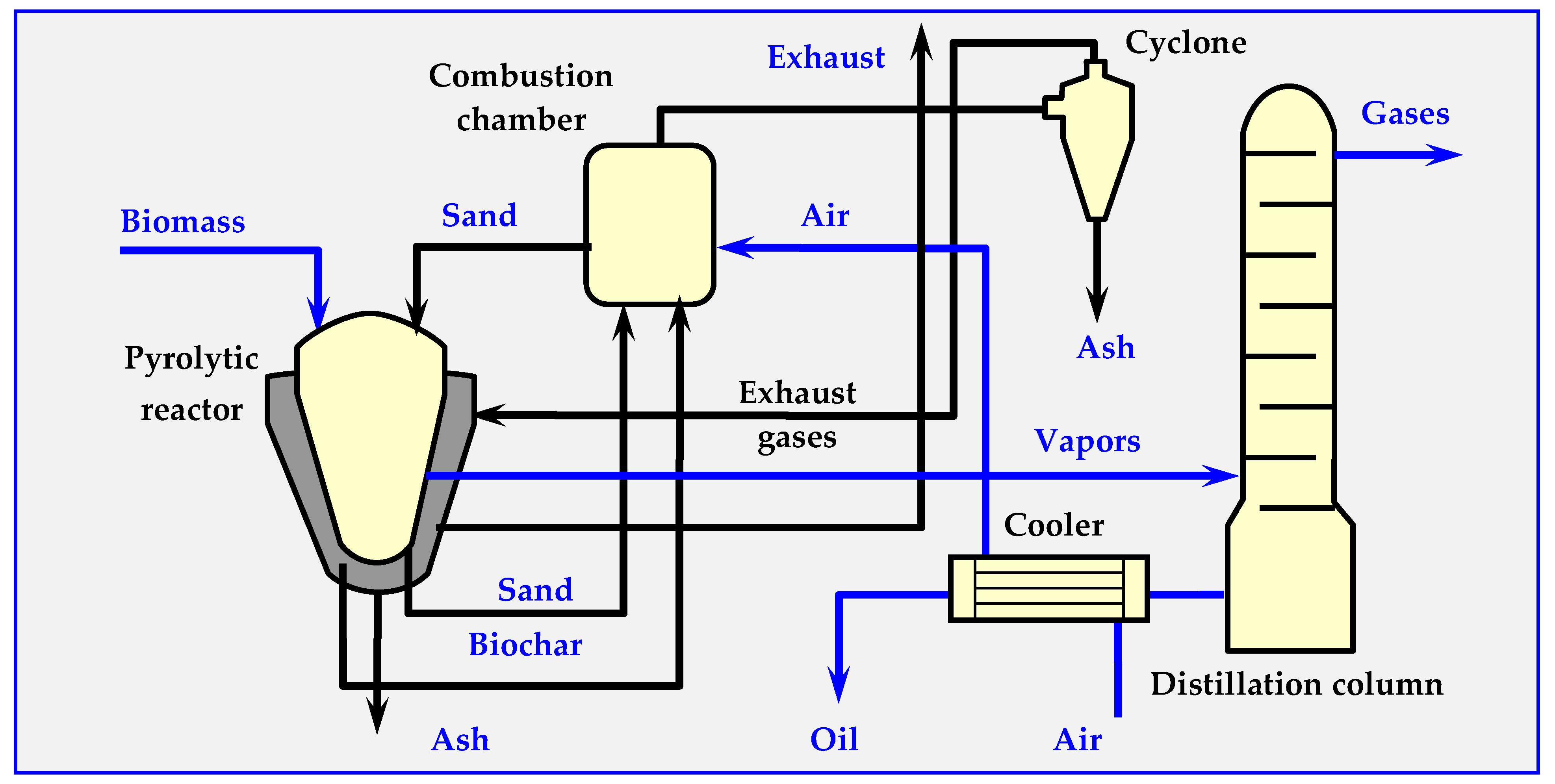
4.7.1. BTG Flash Pyrolysis Installation
4.7.2. Pyrocycling™ Technology
4.7.3. ENTECH Installation for Biomass Conversion
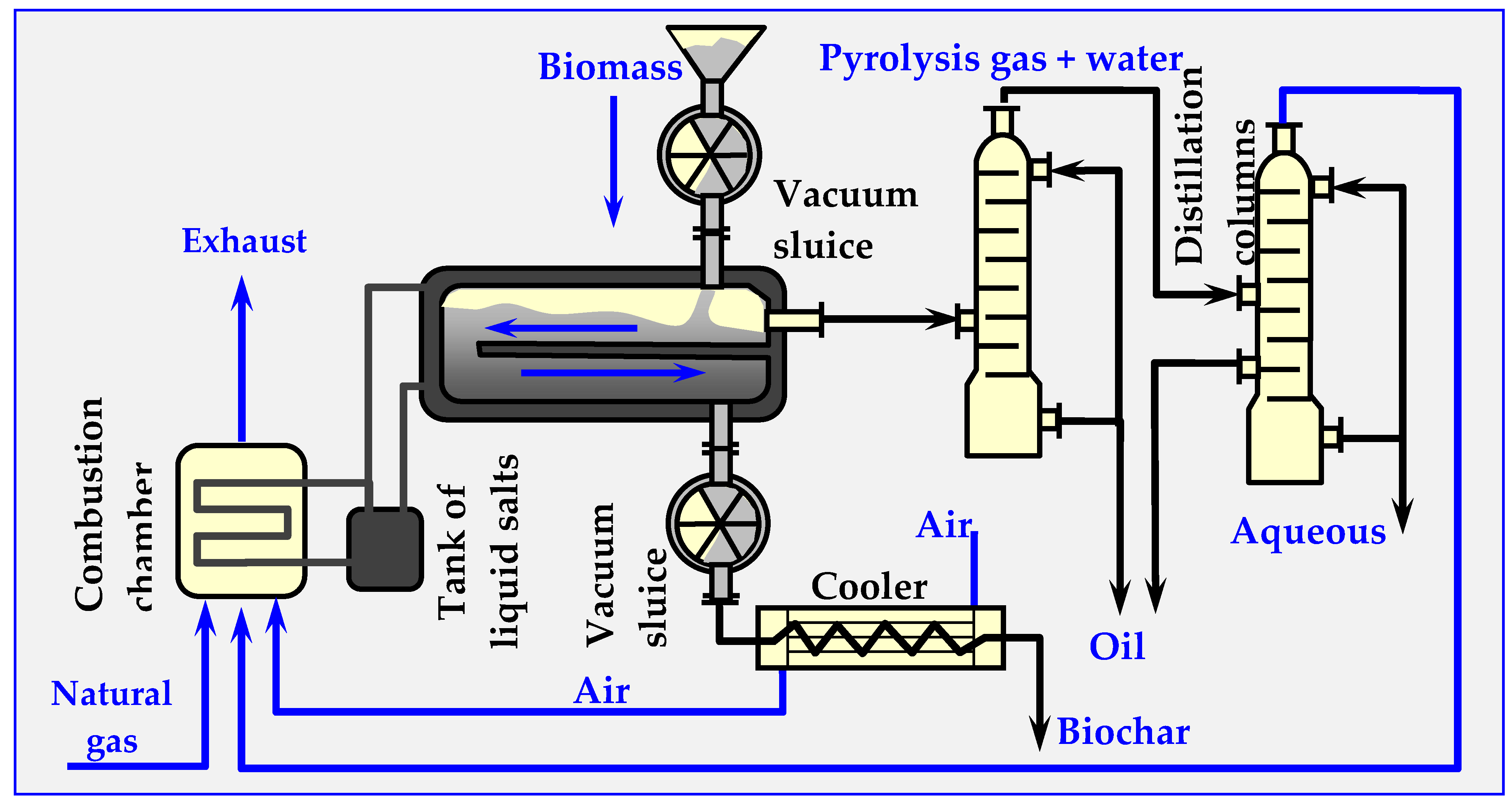
4.7.4. WGT Technology
4.7.5. Ragailler Technology
- -
- Process gas for ecological energy recovery (70%–80% of the energy potential of waste).
- -
- Coke breeze, i.e., coal with mineral substances contained in waste (20%–30% of the energy potential of waste).
- -
- Basalt-like aggregate from melted sorbent and mineral substances contained in waste (30%–50% is glass), or boiler slag if the coke breeze is co-burned with fine coal.
- -
- Ferrous and non-ferrous metals.
4.7.6. HD-PAWA-THERM Technology
4.8. Summary of Pyrolytic Decomposition
5. Biomass Gasification
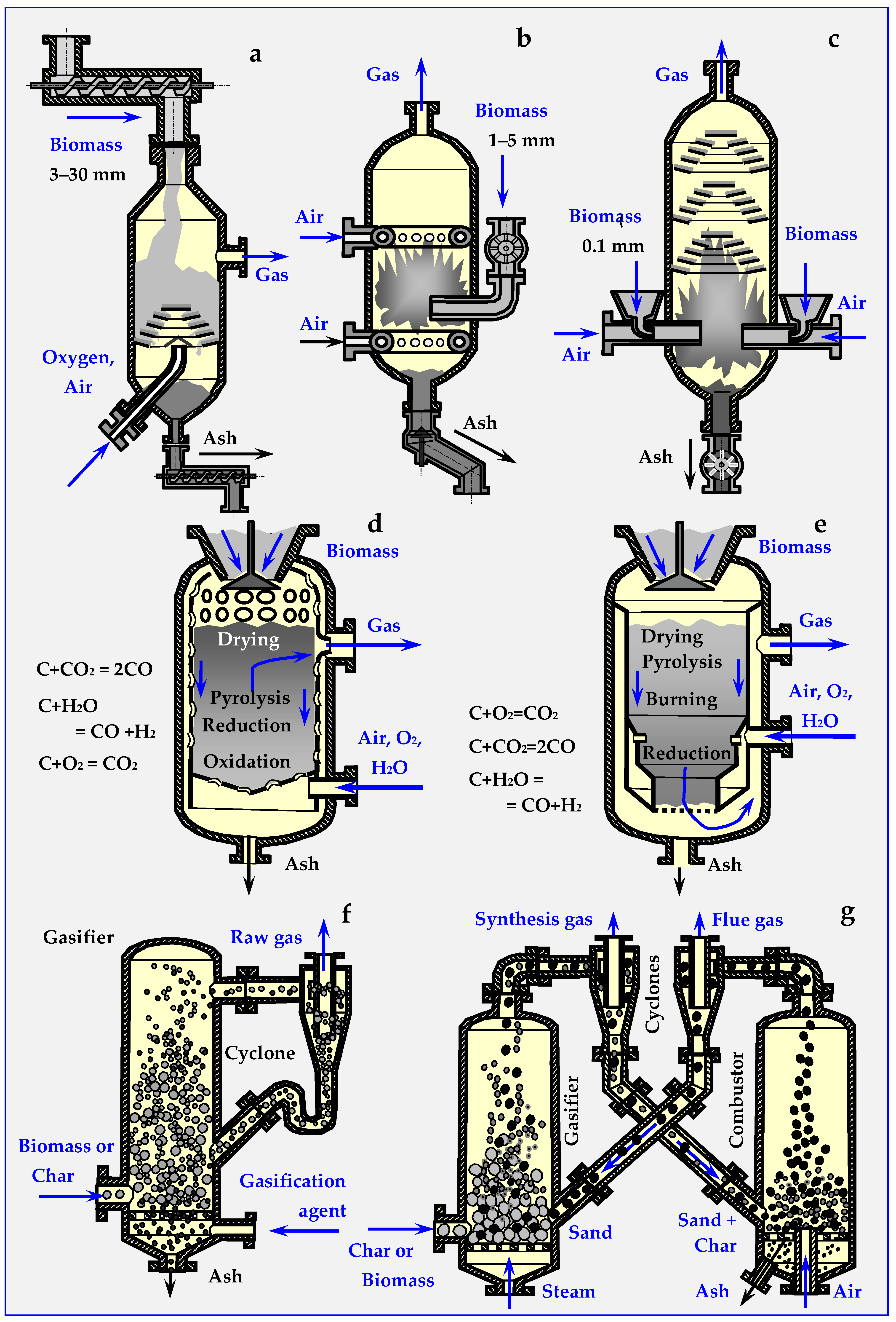
5.1. Biomass Gasification Methods
5.1.1. Indirect Gasification
5.1.2. Direct (Specific) Gasification
5.2. Types of Biomass Gasification Devices
5.2.1. Countercurrent Gasifiers
5.2.2. Co-Current Gasifiers
5.3. Gasification of Biochar
5.4. Hermetic Dosing of Biomass
5.5. Experiments on Biomass Gasification
- -
- Lahti Kymijarvi (Finland)—160 MW power output, processing waste from wood, textile and peat industries.
- -
- Värnamo (Sweden)—launched in 1996, with 6 MW of electrical power output and 9 MW of thermal power output, 80% generator efficiency, and the price of electricity at 33 EUR/MWh, which, given the biomass price of 6 EUR/MWh, bodes well for the future of this technology.
- -
- In the United Kingdom, a willow-fired (Salix viminalis) power plant project is being implemented with a total output of 8 MW (target output is 35 MW); the willow and Misanthus sinensis plantation area is expected to reach 7000 ha, which, together with waste wood from forests, will provide 150–170 Tg/a of biomass.
- -
- A reactor producing gas with an energy value of 4.9 MJ/m3 is operating in Bulle, Switzerland; the installation has a power output of 55 kW and gasifies 60 kg/h of waste wood.
- -
- In Greve-in-Chianti (Italy), gasification of a biomass mixture (poplar, locust bean plant (Robinia pseudoacacia), olive pomace and other waste, mainly from vines) has been successfully achieved. Ultimately, an electricity output of 12 MW is expected.
- -
- The Swiss company Xylowatt designs and builds biomass gasification installations mainly for developing countries. In the years 1996–2001, India purchased six installations with outputs within the range of 80–2000 kW.
6. Biomass Gasification by Hydrothermal Reforming
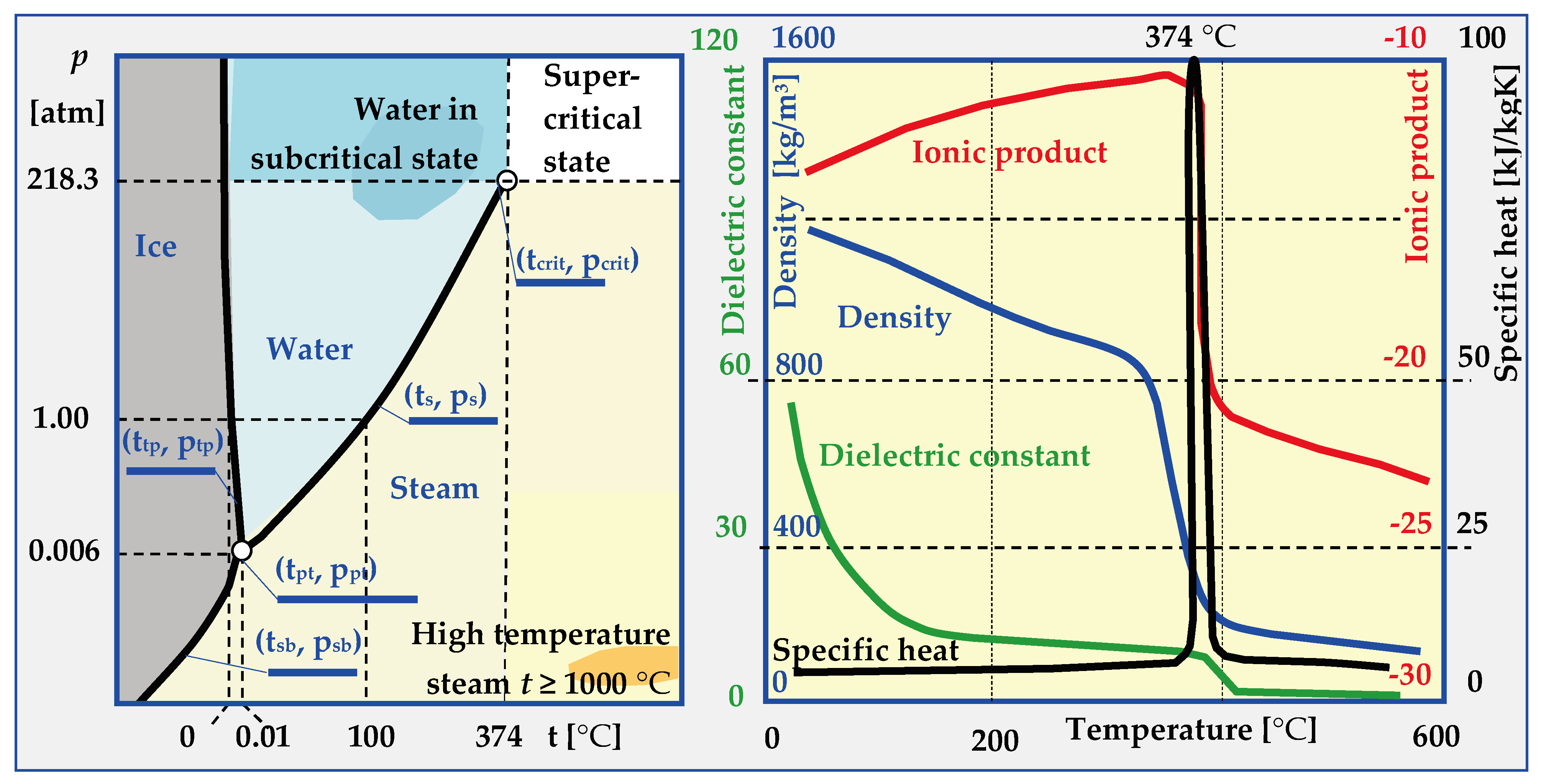
6.1. Physical States of Water
- -
- Normal, in which water is a solid (t < tpt and p > ptp or t < tsb and p > psb), liquid (ttp < t < ts and p > ps) or vapour (t > tsb and p < ppt or t > tpt and p < ps).
- -
- HCW (High Compressed Water), in which the parameters of hot compressed water are within the range: 250 °C < t < tcrit and p < pcrit.
- -
- Sub-critical when water or steam parameters are within the ranges: 250 °C < t < tcrit and p > pcrit (for water and t > tcrit and p < pcrit (for steam),
- -
- SCW (Supercritical Water), when (t > tcrit and p > pcrit) and, depending on the pseudo-critical temperature t*, water is in the form of a pseudo-liquid or pseudo-vapor.
- -
- HiTS (High Temperature Steam), when steam has a temperature of t ≥ 1000 °C, which is much higher than the critical temperature t >> tcrit., and the pressure is below critical p < pcrit.
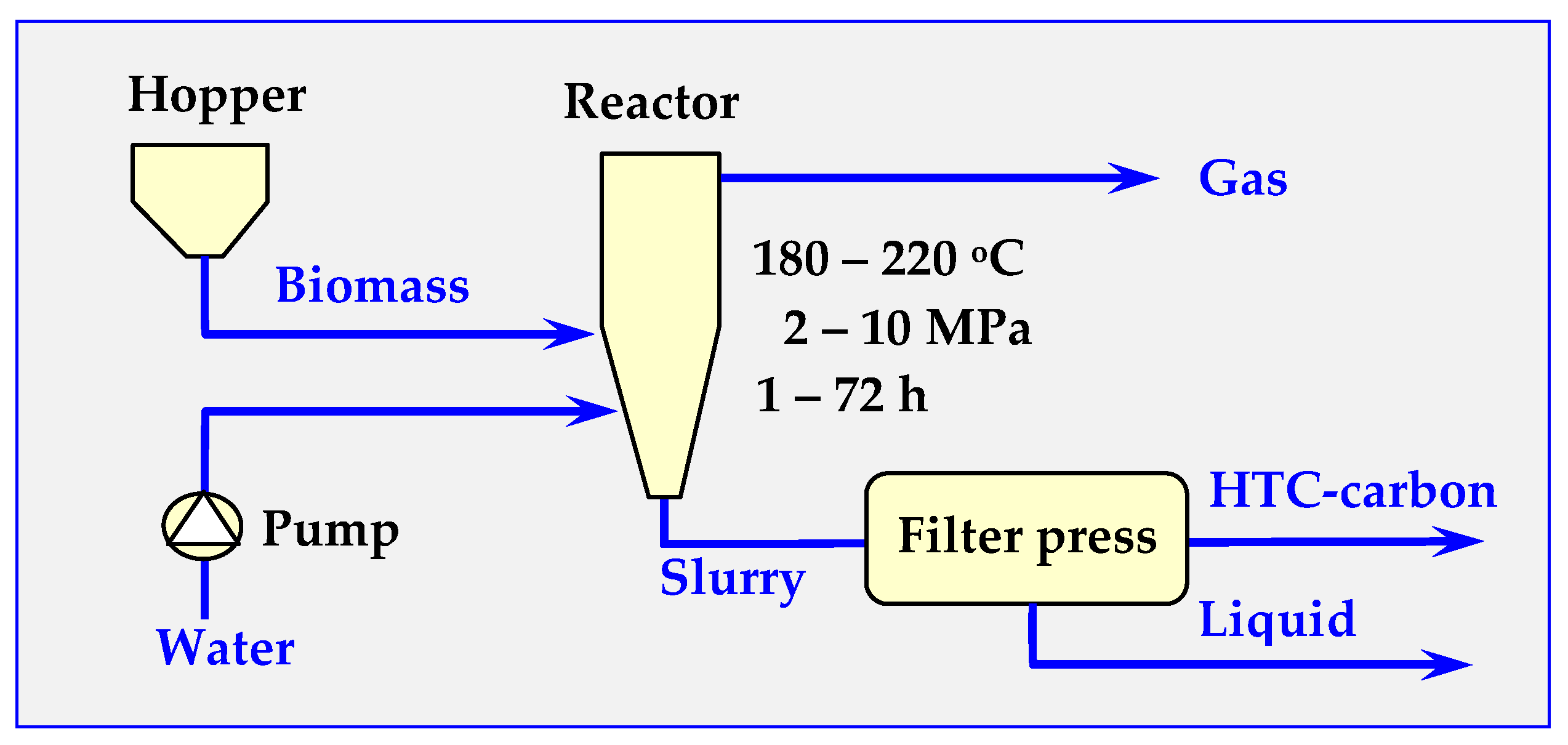
6.2. Hydrothermal Carbonization (HTC)
- An increase in C/N ratio (22.19–26.86),
- More alkyl C, aryl C and carbonyl C,
- Lower H/C (0.98–1.22) and O/C ratios (0.13–0.38),
- Less O-alky C, carboxylic C.
6.3. Water Properties in Supercritical State
6.4. SCW as a Biomass Gasification Medium
- -
- Higher efficiency: for example, 80% humidity biomass gasification increases for SCWG from η = 10% to η > 70%,
- -
- Pitch (tar) content in gas products remains trace, tarry compounds (aromatic, polycyclic phenol compounds) completely dissolve in SCW and undergo reforming, while the remaining compounds pass into the aqueous fraction and do not pollute the waste gas.
- -
- The amount of residual biochar is negligible, and biochar also transfers into the aqueous fraction,
- -
- Hydrogen content in final gas products is higher and CO content is lower than in traditional gasification. Additionally, hydrogen is already compressed, which facilitates its further practical use,
- -
- Heteroatoms and halogens, such as S, N, Cl, which may occur in biomass during SCWG gasification transfer to the aqueous fraction, where their neutralization is much simpler and cheaper than removal from the exhaust gas.
6.5. Oxidizing Properties of Supercritical Water
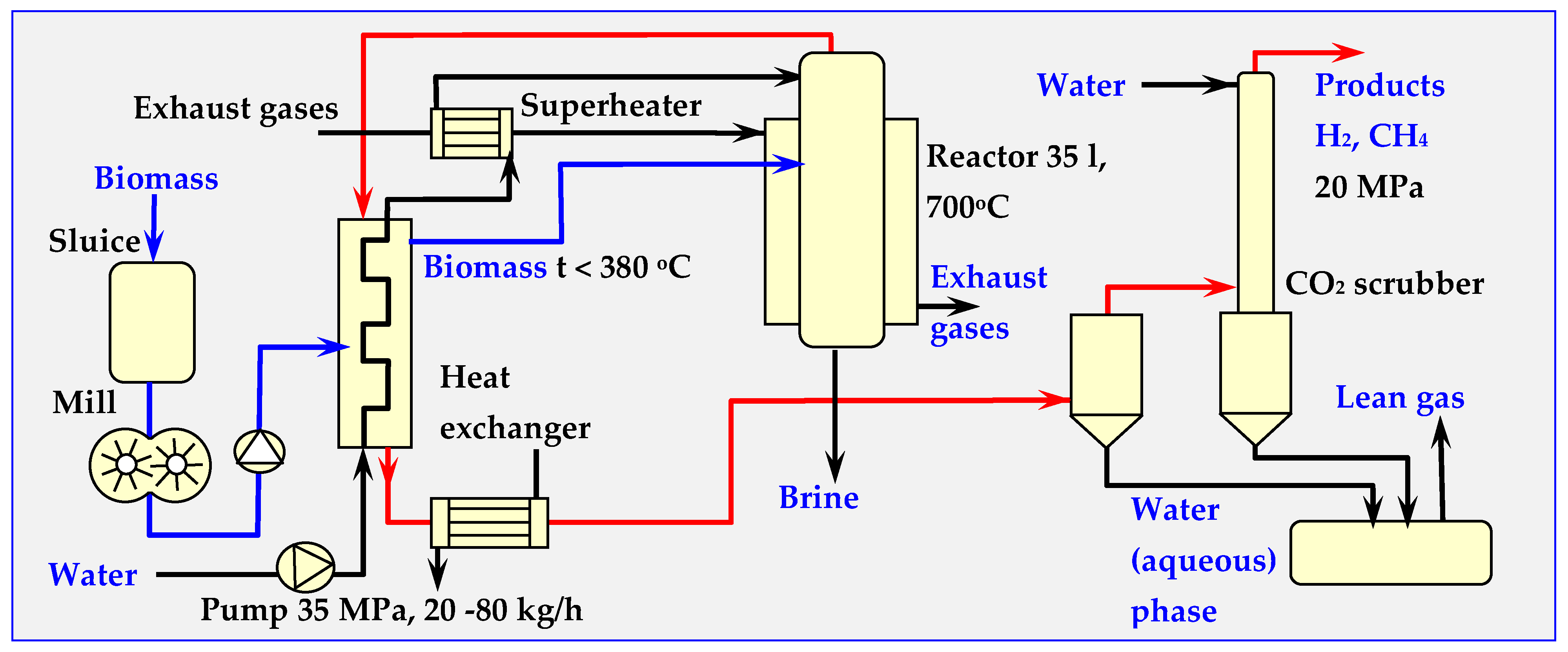
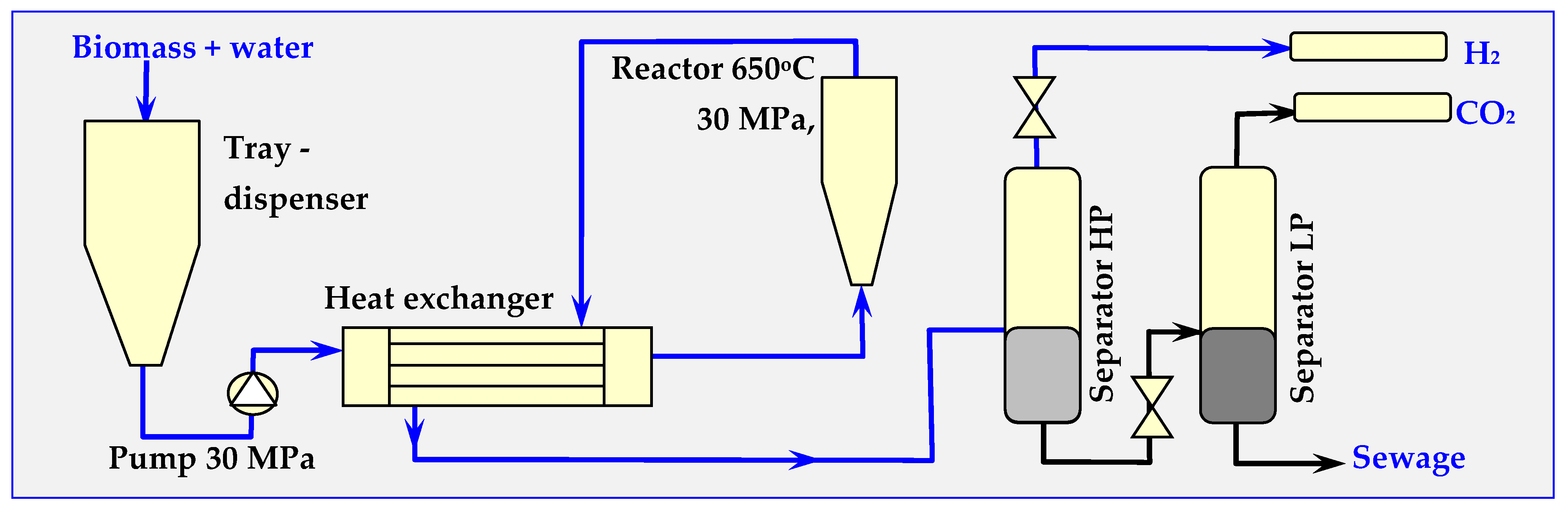
6.6. Examples of Using SCW for Biomass Gasification
6.7. SCWG Reactors for Biomass Gasification
- -
- Autoclaves,
- -
- Steel tubular reactors,
- -
- Mixers,
- -
- Quartz capillaries,
- -
- Reactors with a fluidized bed [73].
7. Biomass Gasification with High Temperature Steam
- -
- Uniform distribution of temperature field in the gasification chamber,
- -
- Lower fuel consumption,
- -
- Lower emissions,
- -
- Lack of nitrogen in gaseous products [187],
- -
- Greater hydrogen production efficiency, as demonstrated by the following reactions:Cm Hn + (m/2) O2 → m CO + (n/2)H2 (HiTAC),Cm Hn + m H2O → m CO + (m + n/2) H2 (HiTS).
The Future of SCWG Technology
8. Summary
Author Contributions
Funding
Conflicts of Interest
References
- Jensen, T.B. Purifying White Charcoal; Sort of Coal: Copenhagen, Denmark, May 2009; Available online: http://sortofcoal.com (accessed on 20 April 2020).
- Lewandowski, W.M.; Radziemska, E.; Ryms, M.; Ostrowski, P. Modern methods of thermochemical biomass conversion into gas, liquid and solid fuels. Ecol. Chem. Eng. S 2011, 18, 39–47. [Google Scholar]
- Bridgwater, A.V. Biomass Pyrolysis—A Guide to UK Capabilities; Aston University Bioenergy Research Group: Aston, UK, May 2011; pp. 1–26. [Google Scholar]
- Hitzl, M.; Corma Canós, A.; Pomares Garcia, F.; Renz, M. The hydrothermal carbonization (HTC) plant as a decentral biorefinery for wet biomass. Catal. Today 2015, 257, 154–159. [Google Scholar] [CrossRef]
- Bergman, P.C.A. Combined Torrefaction and Pelletisation: The TOP Process; ECN-C—05-073; 2005; Available online: https://publications.tno.nl/publication/34628560/S7JA61/c05073.pdf (accessed on 20 April 2020).
- Aho, A.; Kumar, N.; Eränen, K.; Salmi, T.; Hupa, M.; Murzin, D.Y. Catalytic pyrolysis of woody biomass in a fluidized bed reactor: Influence of the zeolite structure. Fuel 2008, 87, 2493–2501. [Google Scholar] [CrossRef]
- Zhang, X.; Lei, H.; Chen, S.; Wu, J. Catalytic co-pyrolysis of lignocellulosic biomass with polymers: A critical review. Green Chem. 2016, 18, 4145–4169. [Google Scholar] [CrossRef]
- Bulushev, D.A.; Ross, J.R.H. Catalysis for to fuels via pyrolysis and gasification: A review. Catal. Today 2011, 171, 1–13. [Google Scholar] [CrossRef]
- Bridgwater, A.V. The production of biofuels and renewable chemicals by fast pyrolysis of biomass. Int. J. Glob. Energy Issues 2007, 27, 160–203. [Google Scholar] [CrossRef]
- Carlson, T.R.; Cheng, Y.-T.; Jae, J.; Huber, G.H. Production of green aromatics and olefins by catalytic fast pyrolysis of wood sawdust. Energy Environ. Sci. 2011, 4, 145–161. [Google Scholar] [CrossRef]
- Kabir, G.; Hameed, B.H. Recent progress on catalytic pyrolysis of lignocellulosic biomass to high-grade bio-oil and bio-chemicals. Renew. Sustain. Energy Rev. 2017, 70, 945–967. [Google Scholar] [CrossRef]
- Bridgwater, A.V. Production of high-grade fuels and chemicals from catalytic pyrolysis of biomass. Catal. Today 1996, 29, 285–295. [Google Scholar] [CrossRef]
- Wang, D.; Czernik, S.; Montane, D.; Mann, M.; Chornet, E. Biomass to hydrogen via fast pyrolysis and catalytic steam reforming of the pyrolysis oil or its fractions. Ind. Eng. Chem. Res. 1997, 36, 1507–1518. [Google Scholar] [CrossRef]
- Cortazar, M.; Lopez, G.; Alvarez, J.; Amutio, M.; Bilbao, J.; Olazar, M. Behaviour of primary catalysts in the biomass steam gasification in a fountain confined spouted bed. Fuel 2019, 253, 1446–1456. [Google Scholar] [CrossRef]
- Hua, X.; Gholizadeh, M. Biomass pyrolysis: A review of the process development and challenges from initial researches up to the commercialisation stage. J. Energy Chem. 2019, 39, 109–143. [Google Scholar] [CrossRef]
- Reed, T.B.; Golden, A.D. Handbook of Biomass Downdraft Gasifier Engine System. In Solar Technical Information Program, Solar Energy Research Institute; U.S. GPO: Golden, CO, USA, 1988. [Google Scholar]
- Lewandowski, W.M.; Ryms, M. Biofuels—Renewable, Pro-Ecological Energy Sources; WNT: Warszawa, Poland, 2013. [Google Scholar]
- Shafizadeh, F. Pyrolysis and combustion of cellulosic materials. Adv. Carbohydr. Chem. 1968, 23, 419. [Google Scholar] [CrossRef]
- Shafizadeh, F. Pyrolytic Reactions and Products of Biomass, w Fundamentals of Thermochemical Biomass Conversion; Overend, B.P., Milne, T.A., Mudge, L.K., Eds.; Elsevier Applied Science Publishers: New York, NY, USA, 1985; pp. 183–218. [Google Scholar] [CrossRef]
- Antal, M.J., Jr. Biomass Pyrolysis: A Review of the Literature Part 1 – Carbohydrate Pyrolysis. In Advances in Solar Energy; Springer: Boston, MA, USA, 1985; pp. 61–111. [Google Scholar]
- Kilzer, F.J.; Broido, A. Speculations on the nature of cellulose pyrolysis. Pyrodynamics 1965, 2, 151. [Google Scholar]
- Piskorz, J.; Radlein, G.; Scott, D.S.; Czernik, S. Liquid products from the fast pyrolysis of wood ans cellulose. In Proceedings of the Research in Thermochemical Biomass Conversion, Phenix, AZ, USA, 2–6 May 1988; Bridgwater, A.V., Kuester, J.L., Eds.; Elsevier Applied Science Publishers: London, UK; New York, NY, USA, 1988; p. 557. [Google Scholar]
- Shen, D.K.; Gu, S. The mechanism for thermal decomposition of cellulose and its main products. Bioresour. Technol. 2009, 100, 6496–6504. [Google Scholar] [CrossRef]
- Evans, R.J.; Milne, T.A. Molecular characterization of the pyrolysis of biomass fundamentals. Energy Fuels 1987, 1, 123–137. [Google Scholar] [CrossRef]
- Evans, R.J.; Milne, T.A. Applied mechanistic studies of biomass pyrolysis. In Proceedings of the 1985 Biomass Thermochemical Conversion Contractors’ Meeting, Minneapolis, MN, USA, 15–16 October 1985; p. 57. [Google Scholar]
- Milne, T.A. Pyrolysis thermal behaviour of biomass below 600 °C. In A Survey of Biomass Gasification; SERI TR-33-239; Solar Energy Research Institute: Golden, CO, USA, July 1979; Volume II, pp. 95–132. Available online: https://www.nrel.gov/docs/legosti/old/239-2.pdf (accessed on 20 April 2020).
- Solres, E.J.; Elder, T.J. Pyrolysis. In Organic Chemicals from Biomass; Goldstein, I.S., Ed.; CRC Press: Boca Raton, FL, USA, 1981; pp. 63–100. [Google Scholar]
- Tang, W.K. Effect of inorganic salts on pyrolysis of wood, alpha cellulose and lignin. In US Forest Service Research Paper; U.S. Department of Agriculture: Madison, WI, USA, 1967; p. FPL71. Available online: https://www.fpl.fs.fed.us/documnts/fplrp/fplrp71.pdf (accessed on 20 April 2020).
- Amtal, M.J., Jr. Biomass pyrolysis: Review of the literature Part I—Lignocellulose Pyrolysis. In Advances in Solar Energy; Boer, K.W., Duffie, J.A., Eds.; Plenum Press: New York, NY, USA, 1985; Volume 2, p. 175. [Google Scholar]
- Diebold, J.P. Proceedings of the Specialists’ Workshop on the Fast Pyrolysis of Biomass, Copper Mountain, CO, USA; Solar Energy Research Institute: Golden, CO, USA, October 1980; SERI/CP-622-1096. [Google Scholar]
- Thrän, D.; Janet, W.; Kay, S.; Jaap, K.; Jaap, K.; Michiel, C.; Jörg, M.; Collins, N.; Jaap, K.; Eija, A.; et al. Moving torrefaction towards market introduction—Technical improvements and economic-environmental assessment along the overall torrefaction supply chain through the SECTOR project. Biomass Bioenergy 2016, 89, 184–200. [Google Scholar] [CrossRef]
- Tumuluru, J.S.; Sokhansanj, S.; Wright, C.T.; Hess, J.R.; Boardman, R.D. A Review on Biomass Torrefaction Process and Product Properties. In Proceedings of the S-1041 Symposium on Thermochemical Conversion, Oklahoma State University, Stillwater, OK, USA, 2 August 2011. [Google Scholar]
- Kumar, L.; Koukoulas, A.A.; Mani, S.; Satyavolu, J. Integrating Torrefaction in the Wood Pellet Industry: A Critical Review. Energy Fuels 2017, 31, 37–54. [Google Scholar] [CrossRef]
- van der Stelt, M.J.C. Chemistry and Reaction Kinetics of Biowaste Torrefaction; Technische Universiteit Eindhoven: Eindhoven, The Netherlands, 2011. [Google Scholar] [CrossRef]
- Wild, M. Update on the Status of Torrefaction as Biomass Upgrading Technology. In Proceedings of the IEA Bioenergy Conference, Berlin, Germany, 27–29 October 2015. [Google Scholar]
- Bridgwater, A.V. Biomass Pyrolysis; Aston University Bioenergy Research Group IEA Bioenergy: York, UK, 12 October 2010; pp. 1–44. [Google Scholar]
- Cheng, X.; Huang, Z.; Wang, Z.; Ma, C.; Chen, S. A novel on-site wheat straw pretreatment method: Enclosed torrefaction. Bioresour. Technol. 2019, 281, 48–55. [Google Scholar] [CrossRef]
- Chen, W.-H.; Peng, J.; Bi, X.T. A state-of-the-art review of biomass torrefaction, densification and applications. Renew. Sustain. Energy Rev. 2015, 44, 847–866. [Google Scholar] [CrossRef]
- Zinchik, S. Paddle Mixer-Extrusion Reactor for Torrefaction and Pyrolysis. Ph.D. Thesis, Michigan Technological University, Houghton, MI, USA, 2019. Available online: htps://digitalcommons.mtu.edu/etdr/906 (accessed on 20 April 2020).
- Kumar, P.; Barrett, D.; Delwiche, M.; Stroeve, P. Methods for Pretreatment of Lignocellulosic Biomass for Efficient Hydrolysis and Biofuel Production. Ind. Eng. Chem. Res. 2009, 48, 3713–3729. [Google Scholar] [CrossRef]
- Boskovic, A. Biomass Torrefaction—Grindability and Dust Explosibility; Dalhousie University Halifax: Halifax, NS, Canada, April 2015; Available online: http://hdl.handle.net/10222/56761 (accessed on 20 April 2020).
- Tumuluru, J.S. A review on biomass torrefaction proces and product properties for Energy applications. Ind. Biotechnol. 2011, 7, 348–401. [Google Scholar] [CrossRef]
- Sierra, R.; Smith, A.; Granda, C.; Holtzapple, M.T. Producing fuels and chemicals from lignocellulosic biomass. Chem. Eng. Process. 2008, 104, 10–18. [Google Scholar]
- Acharya, B.; Dutta, A.; Minaret, J. Review on comparative study of dry and wet torrefaction. Sustain. Energy Technol. Assess. 2015, 12, 26–37. [Google Scholar] [CrossRef]
- Kotowski, W.; Konopka, E. Miejsce biomasy drzewnej w procesach pozyskiwania energii ze źródeł odnawialnych. Ekoenergetyka 2006, 7, 532–537. Available online: https://www.cire.pl/pliki/2/elektroenergetyka_nr_06_07_e1.pdf (accessed on 20 April 2020).
- Bridgwater, A. Thermal Biomass conversion and utilization Biomass information system. In Directorate-General XII, Science, Research and Development B-1049; European Commission: Brussels, Belgium, 1996. [Google Scholar]
- Kizuka, R.; Ishii, K.; Sato, M.; Fujiyama, A. Characteristics of wood pellets mixed with torrefed rice straw as a biomass fuel. Int. J. Energy Environ. Eng. 2019, 10, 357–365. [Google Scholar] [CrossRef]
- Medic, D. Investigation of Torrefaction Process Parameters and Characterization of Torrefied Biomass. Master’s Dissertation Theses, Iowa State University Capstones, Ames, IA, USA, 2012; p. 12403. [Google Scholar]
- Kong, L.; Li, G.; Zhang, B.; He, W.; Wang, H. Hydrogen Production from Biomass Wastes by Hydrothermal Gasification, Energy Sources, Part A. Taylor Fr. Online 2008, 30, 1166–1178. [Google Scholar] [CrossRef]
- Shang, L.; Jesper, A.; Holm, J.K.; Søren, B.; Rui-zhi, Z.; Yong-hao, L.; Helge, E.; Henriksen, U.B. Intrinsic kinetics and devolatilization of wheat straw during torrefaction. J. Anal. Appl. Pyrolysis 2013, 100, 145–152. [Google Scholar] [CrossRef]
- Koppejan, J.; Sokhansanj, S.; Melin, S.; Madrali, S. Status overview of torrefaction technologies. In IEA Bioenergy Task 32 Report; IEA: Enschede, The Netherlands, December 2012. [Google Scholar]
- Rudolfsson, M.; Larsson, S.H.; Lestander, T.A. New tool for improved control of sub-process interactions in rotating ring die pelletizing of torrefied biomass. Appl. Energy 2017, 190, 835–840. [Google Scholar] [CrossRef]
- Strandberg, M. From Torrefaction to Gasification Pilot Scale Studies for Upgrading of Biomass; Umeå universitet: Umeå, Sweden, 2015; p. 58. [Google Scholar]
- Nam, S.B.; Park, Y.S.; Kim, D.J.; Gu, J.H. Torrefaction Reaction Characteristic of various Biomass Waste on Pilot Scale of Torrefaction Reaction System. Procedia Environ. Sci. 2016, 35, 890–894. [Google Scholar] [CrossRef]
- Strandberg, M.; Olofsson, I.; Linda, P.; Susanne, W.; Katarina, Å.; Anders, N. Effects of temperature and residence time on continuous torrefaction of spruce wood. Fuel Process. Technol. 2015, 134, 387–398. [Google Scholar] [CrossRef]
- Shang, L. Upgrading Fuel Properties of Biomass by Torrefaction. Ph.D. Thesis, Technical University of Denmark, Lyngby, Denmark, 2012. [Google Scholar]
- Available online: http://www.wyssmont.com/product_detail.php?section=Dryers&id=1 (accessed on 25 January 2020).
- Pawlak-Kruczek, H.; Krochmalny, K.; Mościcki, K.; Zgóra, J.; Czerep, M.; Ostrycharczyk, M.; Niedźwiecki, Ł. Torrefaction of Various Types of Biomass in Laboratory Scale, Batch-Wise Isothermal Rotary Reactor and Pilot Scale, Continuous Multi-Stage Tape Reactor. Inżynieria i Ochrona Środowiska (Eng. Prot. Environ.) 2017, 20, 457–472. [Google Scholar] [CrossRef]
- Biomass Technology Group (BTG). Available online: http://www.btgworld.com/en/rtd/technologies/torrefaction (accessed on 25 January 2020).
- Amutio, M.; Lopez, G.; Artetxe, M.; Elordi, G.; Olazar, M.; Bilbao, J. Influence of temperature on biomass pyrolysis in a conical spouted bed reactor. Resour. Conserv. Recycl. 2012, 59, 23–31. [Google Scholar] [CrossRef]
- Fernandez-Akarregi, A.R.; Makibar, J.; Lopez, G.; Amutio, M.; Olazar, M. Vaibhav Dhyani of a conical spouted bed reactor pilot plant (25 kg/h) for biomass fast pyrolysis. Fuel Process. Technol. 2013, 112, 48–56. [Google Scholar] [CrossRef]
- Amutio, M.; Lopez, G.; Aguado, R.; Artetxe, M.; Bilbao, J.; Olazar, M. Effect of vacuum on lignocellulosic biomass flash pyrolysis in a conical spouted bed reactor. Energy Fuels 2011, 25, 3950–3960. [Google Scholar] [CrossRef]
- Rasul, M.G.; Jahirul, M.I. Recent Developments in Biomass Pyrolysis for Bio-Fuel Production: Its Potential for Commercial Applications. In Proceedings of the Recent Researches in Environmental and Geological Sciences, 7th WSEAS International Conference on Energy & Environment (EE ’12), Kos Island, Greece, 14–17 July 2012; pp. 256–264, ISBN 978-1-61804-110-4. [Google Scholar]
- Kaasalainen, J.; Kallio-Könnö, M.; Isaksson, J. Large Scale CFB Gasification of Waste and Biomass. In Proceedings of the 12th International Conference on Fluidized Bed Technology, Krakow, Poland, 23–27 May 2017. [Google Scholar]
- Shafizadeh, F. Introduction to pyrolysis of biomass. J. Anal. Appl. Pyrolysis 1982, 3, 283–305. [Google Scholar] [CrossRef]
- Antal, M.J., Jr.; Croiset, E.; Dai, X.; DeAlmeida, C.; Mok, W.S.; Norberg, N.; Richard, J.; Majthoub, M. High-Yield Biomass Charcoal. Energy Fuels 1996, 10, 652. [Google Scholar] [CrossRef]
- Klass, D.L. Biomass for Renewable Energy. In Fuels, and Chemicals; Elsevier: Amsterdam, The Netherlands, 1998; p. 651. ISBN 978-0124109506. [Google Scholar] [CrossRef]
- Kim, D. Physico-Chemical Conversion of Lignocellulose: Inhibitor Effects and Detoxification Strategies: A mini review. Molecules 2018, 23, 309. [Google Scholar] [CrossRef]
- FAO. Simple Technologies for Charcoal Making. FAO Forestry Paper 41. 1987. Available online: http://www.fao.org/docrep/x5328e/x5328e00.htm#Contents (accessed on 20 November 2010).
- Garcia-Nunez, J.A.; Pelaez-Samaniego, M.R.; Garcia-Perez, M.E.; Fonts, I.; Abrego, J.; Westerhof, R.J.M.; Perez, M.G. Historical Developments of Pyrolysis Reactors: A Review. Energy Fuels 2017, 31, 5751–5775. [Google Scholar] [CrossRef]
- Kļaviņa, K.; Blumberga, D. A comparison of different charcoal production technology outputs. Environ. Technol. Res. 2015, 2, 137–140. [Google Scholar] [CrossRef]
- Ryms, M.; Januszewicz, K.; Kazimierski, P.; Łuczak, J.; Klugmann-Radziemska, E.; Lewandowski, W.M. Post-Pyrolytic Carbon as a Phase Change Materials (PCMs) Carrier for Application in Building Materials. Materials 2020, 13, 1268. [Google Scholar] [CrossRef]
- Basu, P. Biomass Gasification, Pyrolysis and Torrefaction, Practical Design and Theory; Elsevier: Amsterdam, The Netherlands, 2018. [Google Scholar] [CrossRef]
- ACM Simulation Program for Designing Complex Processes. Available online: www.aspentech.com (accessed on 20 April 2020).
- Calvo, L.F.; García, A.I.; Otero, M. An Experimental Investigation of Sewage Sludge Gasification in a Fluidized Bed Reactor. Sci. World J. 2013, 2013, 479403. [Google Scholar] [CrossRef] [PubMed]
- Gómez, C.; Manya, J.J.; Velo, E.; Puigjaner, L. Further Application of a Revisited Summative Model for Kinetics of Biomass Pyrolysis. Ind. Eng. Chem. Res. 2004, 43, 901. [Google Scholar] [CrossRef]
- Gómez, C.; Varhegyi, G.; Luis, P. Slow Pyrolysis of Woody Residues and an Herbaceous Biomass Crop: A Kinetic Study. Ind. Eng. Chem. Res. 2005, 44, 6650. [Google Scholar] [CrossRef]
- Mitta, N.R.; Gómez Díaz, C.J.; Velo, E.; Puigjaner, L. Modeling and Simulation of Biomass Pyrolysis as a First Step in a Gasification-Based System. Department of Chemical Engineering, Universitat Politècnica de Catalunya (UPC), 655d. 2009. Available online: http://aiche.confex.com/aiche/2006/techprogram/P58269.HTM (accessed on 20 April 2020).
- Antal, M.J., Jr.; Varhegyi, G. Cellulose Pyrolysis Kinetics: The Current State of Knowledge. Ind. Eng. Chem. Res. 1995, 34, 703. [Google Scholar] [CrossRef]
- Blasi, C.D. Comparison of semi-global mechanisms for primary pyrolysis of lgnocellulosic fuels. J. Anal. Appl. Pyrolysis 1998, 47, 43–64. [Google Scholar] [CrossRef]
- Broido, A.; Weinstein, M. Low-Temperature Isothermal Pyrolysis of Cellulose. In Proceedings of the 3rd International Conference on Thermal Analysis; Wiedemann, Ed.; Birkhauser Verlag: Basel, Switzerland, 1971; p. 285. [Google Scholar]
- Agrawal, R.K. Kinetics of reactions involved in pvrolvsis of cellulose II. The modified kilzer-bioid model. Can. J. Chem. Eng. 1988, 66, 413–418. [Google Scholar] [CrossRef]
- Shafizadeh, F.; Chin, P.P.S. Thermal Deterioration of Wood. ACS Symp. Ser. 1977, 43, 57–81. [Google Scholar] [CrossRef]
- Bradbury, A.G.W.; Sakai, Y.; Shafizadeh, F. A kinetic model for pyrolysis of cellulose. J. Appl. Polym. Sci. 1979, 23, 3271. [Google Scholar] [CrossRef]
- Shafizadeh, F. The chemistry of solid wood. In Advances in Chemistry; Rowell, R., Ed.; Series 207; American Chemical Society: Washington, DC, USA, 1984. [Google Scholar]
- Varhegyi, G.; Jakab, E.; Antal, M.J. Is the Broido-Shafizadeh Model for Cellulose Pyrolysis True? Energy Fuels 1994, 8, 1345–1352. [Google Scholar] [CrossRef]
- Koufopanos, C.A.; Maschio, G.; Lucchesi, A. Kinetic modeling of the pyrolysis of biomasscomponents. Can. J. Chem. Eng. 1989, 67, 75–84. [Google Scholar] [CrossRef]
- Chan, W.R.; Kelbon, M.; Krieger, B.B. Modelling and Experimental Verification of Physical and Chemical Processes during Pyrolysis of a Large Biomass Particle. Fuel 1985, 64, 1505. [Google Scholar] [CrossRef]
- Thurner, F.; Mann, U. Kinetic investigation of wood pyrolysis. Ind. Eng. Chem. Process. Des. Dev. 1981, 20, 482–488. [Google Scholar] [CrossRef]
- Font, R.; Marcilla, A.; Verdu, E.; Devesa, J. Kinetics of the Pyrolysis of Almond Shells and Almond Shells Impregnated with CoCl2 in a Fluidized Bed Reactor and in a Pyroprobe100. Ind. Eng. Chem. Res. 1990, 29, 1846–1855. [Google Scholar] [CrossRef]
- Porada, S. The reactions of formation of selected gas products during coal pyrolysis. Fuel 2004, 83, 1191–1196. [Google Scholar] [CrossRef]
- Porada, S. A comparison of basket willow and coal hydrogasification and pyrolysis. Fuel Process. Technol. 2009, 90, 717–721. [Google Scholar] [CrossRef]
- Fisher, T.; Hajaligol, M.; Waymack, B.; Kellogg, D. Pyrolysis behavior and kinetics of biomass derived materials. J. Anal. Appl. Pyrolysis 2002, 62, 331–349. [Google Scholar] [CrossRef]
- Morf, P.; Hasler, P.; Nussbaumer, T. Mechanisms and kinetic of homogeneous secondary reactions of tar from continuous pyrolysis of wood chips. Fuel 2002, 81, 843–853. [Google Scholar] [CrossRef]
- Pilawski, M.; Grzybek, A.; Rogulska, M. Energetyczny recykling odpadów organicznych. Ekol. Tech. 2000, 8, 48–53. [Google Scholar]
- Han, J.; Kim, H. The reduction and control technology of tar during biomass gasification/pyrolysis: An overview. Renew. Sustain. Energy Rev. 2008, 12, 397–416. [Google Scholar] [CrossRef]
- Kowalik, P. Development of bioenergy sector in European Union. In Proceedings of the Oil &Fuels for Sustainable Development AUZO, Gdańsk, Poland, 8–11 September 2008. [Google Scholar]
- Popczyk, J. Development of bioenergy sector in the light of European energy and climate policies. In Proceedings of the Oil &Fuels for Sustainable Development AUZO, Gdańsk, Poland, 8–11 September 2008. [Google Scholar]
- Bałtycki Klaster Ekoenergetyczny. IMP PAN Gdańsk. Available online: http://www.bkee.pl (accessed on 20 April 2020).
- Kobyłecki, R.; Bis, Z.; Nowak, W. Paliwo z biomasy i paliw alternatywnych—Konwersja energii. Czysta Energ. 2005, 3, 23–25. [Google Scholar]
- Lewandowski, W.M.; Januszewicz, K.; Kosakowski, W. Efficiency and proportions of waste tyre pyrolysis products depending on the reactor type—A review. J. Anal. Appl. Pyrolysis 2019, 140, 25–53. [Google Scholar] [CrossRef]
- Butler, E.; Devlin, G.; Meier, D.; McDonnell, K. A review of recent laboratory research and commercial developments in fast pyrolysis and upgrading. Renew. Sustain. Energy Rev. 2011, 15, 4171–4186. [Google Scholar] [CrossRef]
- Bridgwater, A.V. A guide to fast pyrolysis of biomass for fuels and chemicals. In PyNe Guide 1; Bio-Energy Research Group, Aston University: Birmingham, UK, 1999; Available online: http://courses.washington.edu/pse104/images/newslet6.pdf (accessed on 20 April 2020).
- Ryms, M.; Januszewicz, K.; Lewandowski, W.M.; Klugmann-Radziemska, E. Pyrolysis proces of whole tires as a biomass energy recycling. Ecol. Chem. Eng. S 2013, 20, 93–107. [Google Scholar] [CrossRef]
- Amutio, M.; Lopez, G.; Aguado, R.; Bilbao, J.; Olazar, M. Biomass oxidative flash pyrolysis: Autothermal operation, yields and product properties. Energy Fuels 2012, 26, 1353–1362. [Google Scholar] [CrossRef]
- Mullen, C.A.; Boateng, A.A.; Goldberg, N.M.; Lima, I.M.; Laird, D.A.; Hicks, K.B. Bio-oil and bio-char production from corn cobs and stover by fast pyrolysis. Biomass Bioenergy 2010, 34, 67–74. [Google Scholar] [CrossRef]
- Bridgwater, A.V.; Peacocke, G.V.C. Fast pyrolysis processes for biomass. Renew. Sustain. Energy Rev. 2000, 4, 1–73. [Google Scholar] [CrossRef]
- Isahak, W.N.R.W.; Hisham, M.W.M.; AmbarYarmo, M.; Hin, T.Y. A review on bio-oil production from biomass by using pyrolysis method. Renew. Sustain. Energy Rev. 2012, 16, 5910–5923. [Google Scholar] [CrossRef]
- Briens, C.; Piskorz, J.; Berruti, F. Biomass valorization for fuel and chemicals production—A review. Int. J. Chem. React. Eng. 2008, 6, 1–49. [Google Scholar] [CrossRef]
- Craig, H.; Briens, C.; Berruti, F.; Chan, E. A Review of Short Residence Time Cracking Processes. IJCRE 2005, 3, R1. [Google Scholar] [CrossRef]
- Bridgwater, A.V. Review of fast pyrolysis of biomass and product upgrading. Biomass Bioenergy 2012, 38, 68–94. [Google Scholar] [CrossRef]
- Prins, W.; Wagenaar, B.M. Rewiew of rotating cone technology for flash pyrolysis of biomass. In Biomass Gasif Pyrolysis; Kaltschmitt, M.K., Bridgwater, A.V., Eds.; CPL Press: Stuttgart; Germany, 1997; pp. 316–326. [Google Scholar]
- Diebod, J.; Scahill, J. Ablative Fast Pyrolysis of Biomass in the Entrained-Flow Cyclonic Reactor at SERI. In Proceedings of the 14-th Biomass Thermochemical Conversion Contractor’s Review Meeting Arlington, Arlington, VA, USA, 22–25 June 1982. [Google Scholar]
- Peacocke, G.V.C.; Bridgwater, A.V. Design of a novel ablative pyrolysis reactor. Adv. Thermochem. Biomass Convers. 1993, 1134–1150. [Google Scholar] [CrossRef]
- Bramer, E.A.; Brem, G. A Novel Technology for Fast Pyrolysis of Biomass: PyRos reactor. In Proceedings of the 5th Biomass Conference of the Americas, Orlando, FA, USA, 17–21 September 2001. [Google Scholar]
- Dhyani, V.; Bhaskar, T. Biofuels: Alternative Feedstocks and Conversion Processes for the Production of Liquid and Gaseous Biofuels, 2nd ed.; A volume in Biomass, Biofuels, Biochemicals, (Biomass, Biofules, Biochemicals); Academic Press: Cambridge, MA, USA, 2019; pp. 217–244. [Google Scholar] [CrossRef]
- BTG Biomass Technology Group. Available online: www.btgworld.com (accessed on 20 April 2020).
- Eliasson, B.; Riemer, P.; Wokaun, A. Greenhouse Gas Control Technologies; Elsevier Science Book: Amsterdam, The Netherlands, 1999. [Google Scholar]
- Roy, C.; Morin, D.; Dubé, F. The biomass PyrocyclingTM process. In Biomass Gasification & Pyrolysis: State of the Art and Future Prospects; Kaltschmitt, M., Bridgwater, A.V., Eds.; CPL Press: London, UK, 1997; pp. 307–315. [Google Scholar]
- Roy, C.; Blanchette, D.; de Caumia, B.; Dubé, F.; Pinault, J.; Belanger, E.; Laprise, P. Industrial Scale Demonstration of the Pyrocycling™ Process for the Conversion of Biomass to Biofuels and Chemicals. In Proceedings of the 1st World Conference on Biomass for Energy and Industry, Sevilla, Spain, 5–9 June 2000; Kyritsis, S., Beenackers, A.A.C.M., Helm, P., Grassi, A., Chiaramonti, D., Eds.; James & James (Science Publishers) Ltd.: London, UK, 2001; Volume II, pp. 1032–1035. [Google Scholar]
- ENTECH Renewable Energy Technologies PTY Ltd.: Australia. Available online: http://www.entech-res.com (accessed on 20 April 2020).
- Reddy, P.J. Energy Recovery from Municipal Solid Waste by Thermal Conversion Technologies; CRC Press Book: Boca Raton, FL, USA, 2016. [Google Scholar] [CrossRef]
- RATech. Available online: http://www.ratech.com.pl (accessed on 20 April 2020).
- UC Prozesstechnik GmbH, Dillingen, Germany. Available online: www.ucgmbh.de (accessed on 20 April 2020).
- Westerhout, R.W.J.; Waanders, J.; Kuipers, J.A.M.; van Swaaij, W.P.M. Development of a Continuous Rotating Cone Reactor Pilot Plant for the Pyrolysis of Polyethene and Polypropene. Ind. Eng. Chem. Res. 1998, 37, 2316–2322. [Google Scholar] [CrossRef]
- Wagenaar, B.M.; Venderbosch, R.H.; Carrasco, J.; Strenziok, R.; d van der Aa, B.J. Rotating cone bio-oil production and applications. In Progress in Thermochemical Biomasss Conversion; Bridgwater, A.V., Ed.; Blackwell Science: London, UK, 2001; pp. 1268–1280. [Google Scholar]
- Roy, C.; Chaala, A.; Darmstadt, H. The vacuum pyrolysis of used tires. End-uses for oil and carbon black products. J. Anal. Appl. Pyrolysis 1999, 51, 201–221. [Google Scholar] [CrossRef]
- Roy, C.; Blanchette, D.; Caumia, B. Horizontal Moving Bed Reactor. Patant WO 1998034996A1, 13 August 1998. [Google Scholar]
- Rabe, R.C. A Model for the Vacuum Pyrolysis of Biomass. Master’s Thesis, Department of Process Engineering, The University of Stellenbosh, Stellenbosch, South Africa, December 2005. Available online: https://scholar.sun.ac.za/bitstream/handle/10019.1/1675/rabe_model_2005.pdf (accessed on 20 April 2020).
- Ragailler, T. Piec Obrotowy. Urząd Patentowy RP. Patent No 192415 PL., 24 August 2000. [Google Scholar]
- HD-PAWA-THERM® Process for the Decentralized Recycling of Municipal Sewage Sludge by Generating a High-Calorific Pyrolysis Gas. Available online: http://www.ucgmbh.de/download/hdpawatherm.pdf (accessed on 20 April 2020).
- Nikitin, N.I. The Chemistry of Cellulose and Wood; (translated in 1966 from Russian by J. Schmorak, Israel Program for Scientific Translations, Jerusalem, Israel); Academy of Sciences of the USSR, Institute of High Molecular Compounds: Moscow-Leningrad, Russia, 1962. [Google Scholar]
- Reed, T.B.; Das, A. Handbook of Biomass Downdraft Gasifier Engine Systems. In Solar Energy Research Institute under the U.S., Department of Energy Solar Technical Information Program; The Biomass Foundation Press: Golden, CO, USA, 1988. [Google Scholar]
- Bain, R.L. Biomass Gasification Overview. In Proceedings of the Presentation in National Renewable Energy Laboratory, Golden, CO, USA, 28 January 2004. [Google Scholar]
- Geurds, M. Biomass gasification technologies status and applications. In Proceedings of the CTIT Conference, Hanoi, Vietnam, 18–20 September 2006. [Google Scholar]
- Bukar, A.A.; Oumarou, M.B.; Tela, B.M.; Eljummah, A.M. Assessment of Biomass Gasification: A Review of Basic Design Considerations. Am. J. Energy Res. 2019, 7, 1–14. [Google Scholar] [CrossRef]
- Stassen, H.E. Small-Scale Biomass Gasifiers for Heat and Power: A Global Review, World Bank Technical Papers; Energy Series: Washington, DC, USA, 1995; pp. 1–88. [Google Scholar]
- Beheshti, S.M.; Ghassemi, H.; Shahsavan-Markadeh, R. Process simulation of biomass gasification in a bubbling fluidized bed reactor. Energy Convers. Manag. 2015, 94, 345–352. [Google Scholar] [CrossRef]
- Van Den Aarsen, F.G.; Susanto, H.; Beenackers, A.A.C.M.; Van Swaaij, W.P.M. Energy recovery by gasification of agricultural and forestry wastes in fluidized bed reactors and in moving bed reactors with internal recycle of pyrolysis gas, EUR 10012 EN. In Process Development and Reaktor Modeling; Commission of the European Communities Energy: Luxembourg, 1986; pp. 1–326. [Google Scholar]
- Bedi, E. W kierunku odnawialnych źródeł energii. In Nowe wiadomości dla Europy Środkowo-Wschodniej; Wyd., Z.G., Ed.; Polskiego Klubu Ekologicznego: Kraków, Poland, 1996; p. 12. [Google Scholar]
- Fish, J.D.; Hawn, D.C. Closed loop thermochemical energy transport based on CO2 reforming of methane: Balancing the reaction system. J. Sol. Energy Eng. 1987, 109, 215–220. [Google Scholar] [CrossRef]
- Johansson, I.B.; Kelly, H.; Reddy, A.K.N.; Williams, R.H. (Eds.) Renewable Energy—Sources for Fuels and Electricity; Island Press: Washington, DC, USA, 1993. [Google Scholar]
- Stelmach, S.; Wasielewski, R.; Figa, J. Zgazowanie biomasy—Przykłady nowych technologii. Arch. Gospod. Odpad. Ochr. Śr. 2008, 7, 9–29. Available online: http://ago.helion.pl (accessed on 20 April 2020).
- Higman, C.; van der Burgt, M.J. Gasification; Elsevier: Amsterdam, The Netherlands; GPP: Burlington, MA, USA, 2003. [Google Scholar]
- Paisley, M.A.; Overend, R.P. The SilvaGas® Process from Future Energy Reosurces—A Commercialization Success. In Proceedings of the 12th European Conference and Technology Exhibition on Biomass for Energy, Industry, and Climate Protection, Amsterdan, The Netherlands, 17–22 June 2002. [Google Scholar]
- Rauch, R.; Hrbek, J.; Hofbauer, H. Biomass gasification for synthesis gas production and applications of the syngas, Advanced Review. WIREs Energy Environ. 2013, 3, 343–362. [Google Scholar] [CrossRef]
- Radtke, K. ThyssenKrupp Uhde’s fluidised bed and entrained flow gasification technologies for biomass and coal. In Proceedings of the Presentation at SGC International Seminar on Gasification 2012, Stockholm, Sweden, 18–19 October 2012. [Google Scholar]
- Aysu, T.; Küçük, M.M. Biomass pyrolysis in a fixed-bed reactor: Effects of pyrolysis parameters on product yields and characterization of products. Energy 2014, 64, 1002–1025. [Google Scholar] [CrossRef]
- Roeck, D.R. Technology Overview: Circulating Fluidized-Bed Combustion. Final Report; EPA-600/7-82-051; US Environmental Protection Agency: Bedford, MA, USA, June 1982; pp. 1–55.
- Greil, C.; Hirschfelder, H. Biomass Integrated CFB Gasification Combined Cycle Plants. In Proceedings of the Paper presented at IChemE Conference, “Gasification: Gateway to a Cleaner Future”, Dresden, Germany, 23–24 September 1998. [Google Scholar]
- Greil, C.; Hirschfelder, J.; Turna, O.; Obermeier, T. Operating Results from Gasification of Waste Material and Biomass in Fixed Bed and Circulating Fluidized Bed Gasifiers. In Proceedings of the Paper presented at IChemE Conference, “Gasification: The Clean Choice for Carbon Management”, Noordwijk, The Netherlands, 8–10 April 2002. [Google Scholar]
- Mahishi, M. Theoretical and Experimental Investigation of Hydrogen Production by Gasification of Biomass. Ph.D. Thesis, University of Florida, Gainesville, FA, USA, 2006. [Google Scholar]
- Rezaiyan, J.; Cheremisinoff, N.P. Gasification Technologies A Primer for Engineers and Scientists; Taylor & Francis Group, LLC, CRC: Boca Raton, FL, USA, 2005. [Google Scholar]
- Rollinson, A.N.; Williams, O. Experiments on torrefied wood pellet: Study by gasification and characterization for waste biomass to energy applications. R. Soc. Opensci. 2016, 3, 150578. [Google Scholar] [CrossRef] [PubMed]
- Maurstad, O. An Overview of Coal based Integrated Gasification Combined Cycle (IGCC) Technology September 2005. MIT LFEE 2005-002 WP. Available online: https://sequestration.mit.edu/pdf/LFEE_2005-002_WP.pdf (accessed on 20 April 2020).
- Holt, N. EPRI, “Gasification process selection—Tradeoffs and ironies”. In Proceedings of the Gasification Technologies Conference, Washington, DC, USA, 3–6 October 2004. [Google Scholar]
- Licznerski, E.; Ryms, M. EC in Gueesing (Austria); Optima Invest: S.A. Gdańsk, Poland, 2007; Available online: http://live.pege.org/2005-wood/powerplant.htm (accessed on 20 April 2020).
- European BIO CHP. Available online: https://bioenergyeurope.org/ (accessed on 20 April 2020).
- Mäkelä, M.; Verónica, B.; Andrés, F. Hydrothermal carbonization of lignocellulosic biomass: Effect of process conditions on hydrochar properties. Appl. Energy 2015, 155, 576–584. [Google Scholar] [CrossRef]
- Lucian, M.; Maurizio, V.; Lihui, G.; Giovanni, P.; Goldfarb, J.L.; Luca, F. Impact of hydrothermal carbonization conditions on the formation of hydrochars and secondary chars from the organic fraction of municipal solid waste. Fuel 2018, 233, 257–268. [Google Scholar] [CrossRef]
- Herguido, J.; Corella, J.; Gonzalez-Saiz, J. Steam gasification of lignocellulosic residues in a fluidized bed at a small pilot scale: Effect of the type of feedstock. Ind. Eng. Chem. Res. 1992, 31, 1274–1282. [Google Scholar] [CrossRef]
- Amin, S.; Reid, R.C.; Modell, M. Reforming and decomposition of glucose in an aqueous phase. In Proceedings of the Intersociety Conference on Environmental System, San Francisco, CA, USA, 21–24 July 1975. 75-ENAs-21. [Google Scholar]
- Tester, J.W.; Holgate, H.R.; Armellini, F.J.; Webley, P.A.; Killilea, W.R.; Hong, G.T.; Barner, H.E. Supercritical oxidation technology process development and fundamental research. In Emerging Technologies for Hazardous Waste Management III; w Teddler, W.D., Pohland, F.G., Eds.; American Chemical Society Symposium Series; American Chemical Society: Washington, DC, USA, 1993; Volume 518, pp. 35–76. [Google Scholar]
- Yoshida, Y.; Kiyoshi, D.; Yukihiko, M.; Ryuji, M.; Dayin, L.; Hisashi, I.; Hiroshi, K. Comprehensive comparison of efficiency and CO2 emission between biomass energy conversion technologies—Position of supercritical water gasification biomass technologies. Biomass Bioenergy 2003, 25, 257–272. [Google Scholar] [CrossRef]
- Pińkowska, H. Water in Sub- and Supercritical State as a New Reaction Medium [in Polish]. WIEDZAinfo.pl. Available online: http://wiedzainfo.ue.wroc.pl/wyklady/686/woda_w_stanie_pod_i_nadkrytycznym_jako_nowe_medium_reakcyjne.html (accessed on 20 April 2020).
- Schmitt, C.C.; Renata, M.; Neves, R.C.; Daniel, R.; Axel, F.; Klaus, R.; Jan-Dierk, G.; Nicolaus, D. From agriculture residue to upgraded product: The thermochemical conversion of sugarcane bagasse for fuel and chemical products. Fuel Process. Technol. 2020, 197, 106199. [Google Scholar] [CrossRef]
- Biswas, B.; Kumar, J.; Bhaskar, T. Advanced Hydrothermal Liquefaction of Biomass for Bio-Oil Production. In Biofuels: Alternative Feedstocks and Conversion Processes for the Production of Liquid and Gaseous Biofuels (Second Edition). Biomass Biofuels Biochem. 2019, 245–266. [Google Scholar] [CrossRef]
- Kirtania, K. Thermochemical Conversion Processes for Waste Biorefinery in Waste Biorefinery. In Waste Biorefinery: Potential and Perspectives; Bhaskar, T., Pandey, A., Mohan, S.V., Lee, D.J., Khanal, S.K., Eds.; Elsevier: Amsterdam, Netherland, 2018; pp. 129–156. [Google Scholar] [CrossRef]
- Smith, A.M.; Whittaker, C.; Shield, I.; Ross, A.B. The potential for production of high quality bio-coal from early harvested Miscanthus by hydrothermal carbonization. Fuel 2018, 220, 546–557. [Google Scholar] [CrossRef]
- Ahmed, I.I.; Nipattummakul, N.; Gupta, A.K. Characteristics of syngas from co-gasification of polyethylene and woodchips. Appl. Energy 2011, 88, 165–174. [Google Scholar] [CrossRef]
- Nizamuddina, S.; Balochb, H.A.; Griffina, G.J.; Mubarakc, N.M.; Bhuttob, A.W.; Abrod, R.; Mazarib, S.A.; Si Alie, B. An overview of effect of process parameters on hydrothermal carbonization of biomass. Renew. Sustain. Energy Rev. 2017, 73, 1289–1299. [Google Scholar] [CrossRef]
- Volpe, M.; Fiori, L.; Volpe, R.; Messineo, A. Upgrading of olive tree trimmings residue as biofuel by hydrothermal carbonization and torrefaction: A comparative study. Chem. Eng. Trans. 2016, 50, 13–18. [Google Scholar] [CrossRef]
- Volpe, M.; Wüst, D.; Merzari, F.; Lucian, M.; Andreottola, G.; Kruse, A.; Fiori, L. One stage olive mill waste streams valorisation via hydrothermal carbonization. Waste Manag. 2018, 80, 224–234. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Qimei, L.; Ruidong, H.; Xiaorong, Z.; Guitong, L. Hydrochar production from watermelon peel by hydrothermal carbonization. Bioresour. Technol. 2017, 241, 236–243. [Google Scholar] [CrossRef] [PubMed]
- Pecchi, M.; Patuzzi, F.; Benedetti, V.; Di Maggio, R.; Baratieri, M. Thermodynamics of hydrothermal carbonization: Assessment of the heat release profile and process enthalpy change. Fuel Process. Technol. 2020, 197, 106206. [Google Scholar] [CrossRef]
- Lynam, J.G.; Coronella, C.J.; Yan, W.; Reza, M.T.; Vasquez, V.R. Acetic acid and lithium chloride effects on hydrothermal carbonization of lignocellulosic biomass. Bioresour. Technol. 2011, 102, 6192–6199. [Google Scholar] [CrossRef]
- Mašek, O. Biochar in thermal and thermochemical biorefineries—Production of biochar as a coproduct. Handbook of Biofuels Production (Second Edition). Process. Technol. 2016, 655–671. [Google Scholar] [CrossRef]
- Al-Salem, S.M.; Lettieri, P.; Baeyens, J. Recycling and recovery routes of plastic solid waste (PSW): A review. Waste Manag. 2009, 29, 2625–2643. [Google Scholar] [CrossRef]
- Anderson, J.; Furusjo, E.; Wetterlund, E.; Lundgren, J.; Landalv, I. Co-gasification of black liquor and pyrolysis oil: Evaluation of blend ratios and methanol production capacities. Energy Convers. Manag. 2016, 110, 240–248. [Google Scholar] [CrossRef]
- Andersson, J.; Lundgren, J. Techno-economic analysis of ammonia production via integrated biomass gasification. Appl. Energy 2014, 130, 484–490. [Google Scholar] [CrossRef]
- Malhotra, A. Thermodynamic Properties of Supercritical Steam. Supercritical Steam Tables. Copyright © 2009 by Ashok Malhotra, Published by SteamCenter.com. Available online: http://www.lulu.com/shop/ashok-malhotra/thermodynamic-properties-of-supercritical-steam/paperback/product-304448.html (accessed on 20 April 2020).
- Kruse, A. Hydrothermal biomass gasification. J. Supercrit. Fluids 2009, 47, 391–399. [Google Scholar] [CrossRef]
- Boukis, N.; Galla, U.; Műller, H.; Dinjus, E. Biomass gasification in supercritical water experimental progress achieved with the VERENA pilot plant. In Proceedings of the 15th European Biomass Conference & Exhibition, Berlin, Germany, 7–11 May 2007. [Google Scholar]
- Mozaffarian, M.; Deurwaarder, E.P.; Kersten, S.R.A. Green Gas (SNG) Production by Supercritical Gasification of Biomass; The Novem project number is: 268-03-04-02-006; Dutch Ministry of Economic Affairs: Petten, The Netherlands, November 2004.
- Matsumura, Y.; Minowa, T.; Potic, B.; Minowa, T.; Kersten, S.R.A.; Prins, W.; de Beld, B.; Elliott, D.C.; Neuenschwander, G.G.; Andrea, K. Biomass gasification in near- and super-critical water: Status and prospects. Biomass Bioenergy 2005, 29, 269–292. [Google Scholar] [CrossRef]
- Hydrogen-Rich Fuel Gas from Supercritical Water Gasification of Wine Grape Residues and Greenhouse Rest Biomass, Programme: FP5-Energy, Environment, Sustainable Development, Including: Bauer Kompost (D),- Wiesloch Winzerkeller (D),- Feluwa Pumpen (D),- Callaghan Engineering (IRL),- Sparqle International (NL),- Composteringsbedrijf Zuid-Holland (NL),- Forschungszentrum Karlsruhe (D),- Biomass Technology Group (NL),- Promikron (NL), European Commission, Research & Innovation. 2011. Available online: https://cordis.europa.eu/project/id/ENK5-CT-2001-30010 (accessed on 20 April 2020).
- Gupta, A.K. Emissions Reduction and Efficiency Improvements in Power Generation and Industrial Processes; WREC 09: Bangkok, Thailand, 2009. [Google Scholar]
- Balu, E.; Lee, U.; Chung, J.N. High temperature steam gasification of woody biomass—A combined experimental and mathematical modeling approach. Int. J. Hydrogen Energy 2015, 40, 14104–14115. [Google Scholar] [CrossRef]
- Yoshikawa, K. Production of Useful Fuels and Electricity from Biomass and Waste Resource. In Proceedings of the VII International Conference “FUEL FROM WASTE 2009”, Szczyrk, Poland, 21–23 October 2009. [Google Scholar]


























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Yield [cm3/g] | ||||||||
|---|---|---|---|---|---|---|---|---|
| CH4 | C2H6 | C2H4 | C3H8 | C3H6 | H2 | CO | CO2 | |
| Willow | 15.83 | 1.09 | 0.88 | 0.28 | 0.38 | 143.56 | 76.96 | 52.28 |
| Coal | 33.76 | 4.00 | 1.05 | 1.05 | 1.12 | 234.76 | 29.75 | 29.75 |
| Mixture | 20.18 | 1.86 | 0.68 | 0.48 | 0.61 | 173.26 | 57.46 | 45.29 |
| Process | Reactions | Enthalpy kJ/g mol | |
|---|---|---|---|
| 300 K | 1000 K | ||
| Methanation | CO + 3 H2 = CH4 + H2O CO + 4 H2 = CH4 + 2 H2O | −205 −167 | −226 −192 |
| Methanolysis | CO + 2 H2 = CH3OH CO2 + 3 H2 = CH3OH + H2O | −92 −50 | −105 −71 |
| Slow pyrolysis | 0.17 C6H10O5 = C + 0.85 H2O | −81 | −80 |
| Hydrogasification | CO + H2O = CO2 + H2 | −42 | −33 |
| Properties | Pyrolysis | ||
|---|---|---|---|
| Flash | Slow | ||
| Moisture content % | 20 | 14.6 | |
| pH - | 2.5 | 2.0 | |
| Specific weight kg/dm3 | 1.21 | 1.195 | |
| C % | for anhydrous | 56.4 | approx. 61.9 |
| H % | 6.2 | 6.0 | |
| N % | 0.2 | 1.05 | |
| S % | <0.01 | 0.03 | |
| Ash % | 0.1 | 1.5 | |
| O % | 37.1 | 29.5 | |
| C/H ratio - | 9.1 | 10.3 | |
| Calorific value MJ/kg | 23 | 26.3 | |
| Dynamic viscosity at 40 °C cP | 51 | 300 | |
| Kinematic viscosity at 25/40 °C cSt | 233/134 | - | |
| Solidification temperature °C | −23 | 27 | |
| Technology | Producer | Material | Performance | Temp. |
|---|---|---|---|---|
| kg/h | °C | |||
| BTG Flash Pyrolyze [117] | BTG Biomass Technology Group | Biomass | 250, 5000 (at start) | 500 |
| Pyrocycling Process [118,119] | Pyrovac Group Inc., Ecosun b.v. [120] | Biomass | 3500 | 475 |
| ENTECH Pyrolytic Gasification System | ENTECH Renewable Energy Technologies PTY Ltd. [121] | Biomass and organic waste | Series 200–36,000 | 500 |
| Waste Gas Technology | Waste Gas Technology Ltd. [122] | Sewage sludge | 500 | 750–850 |
| Dry distillation using the Ragailler method | RATech [123] | Municipal waste, biomass | b.d. | 450–560 |
| HD-PAWA-THERM | UC Prozesstechnik GmbH [124] | Sewage sludge | b.d. | 600–700 |
| Process | Temp. | H2 | CO | CO2 | CnH2n+2 |
|---|---|---|---|---|---|
| °C | % mol. | % mol. | % mol. | % mol. | |
| Dehydration | 155–200 | 0.0 | 30.5 | 68.0 | 2.0 |
| Carbon oxides are formed (oxidation) | 200–280 | 0.2 | 30.5 | 66.5 | 3.3 |
| Hydrocarbons CnH2n+2 appear | 280–380 | 5.5 | 20.5 | 35.5 | 36.6 |
| The amount of hydrocarbons increases | 380–500 | 7.5 | 12.3 | 31.5 | 48.7 |
| Dissociation to hydrogen | 500–700 | 48.7 | 24.5 | 12.2 | 20.4 |
| Further increase in hydrogen content | 700–900 | 80.7 | 9.6 | 0.4 | 8.7 |
| Normal Liquid | HCW State | Supercritical State | Dry Saturated Steam | |||
|---|---|---|---|---|---|---|
| Temperature | °C | 25 | 250 | 400 | 400 | 150 |
| Pressure | MPa | 0.1 | 4 | 25 | 50 | 0.1 |
| Density | kg/m3 | 997 | 799 | 170 | 580 | 0.52 |
| Dynamic viscosity | mPa | 0.89 | 0.11 | 0.03 | 0.07 | 0.01 |
| Dielectric constant | - | 78.5 | 27.1 | 5.9 | 10.5 | 1.00 |
| Thermal conductivity | W/mK | 0.607 | 0.618 | 0.330 (p = 30 MPa) | 0.029 | |
| Prandtl number Pr | - | 6.13 | 0.85 | 3.33 (p = 30 MPa) | 0.97 | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lewandowski, W.M.; Ryms, M.; Kosakowski, W. Thermal Biomass Conversion: A Review. Processes 2020, 8, 516. https://doi.org/10.3390/pr8050516
Lewandowski WM, Ryms M, Kosakowski W. Thermal Biomass Conversion: A Review. Processes. 2020; 8(5):516. https://doi.org/10.3390/pr8050516
Chicago/Turabian StyleLewandowski, Witold M., Michał Ryms, and Wojciech Kosakowski. 2020. "Thermal Biomass Conversion: A Review" Processes 8, no. 5: 516. https://doi.org/10.3390/pr8050516
APA StyleLewandowski, W. M., Ryms, M., & Kosakowski, W. (2020). Thermal Biomass Conversion: A Review. Processes, 8(5), 516. https://doi.org/10.3390/pr8050516