Review of Sulfuric Acid Decomposition Processes for Sulfur-Based Thermochemical Hydrogen Production Cycles



Abstract
1. Introduction
2. Sulfur-Based Thermochemical Cycles
2.1. Main Sulfur-Based Thermochemical Cycles
2.1.1. Hybrid Sulfur Cycle
2.1.2. Sulfur Iodine Cycle
2.1.3. Sulfur Bromine Cycle
2.1.4. Sulfur Ammonia Cycle
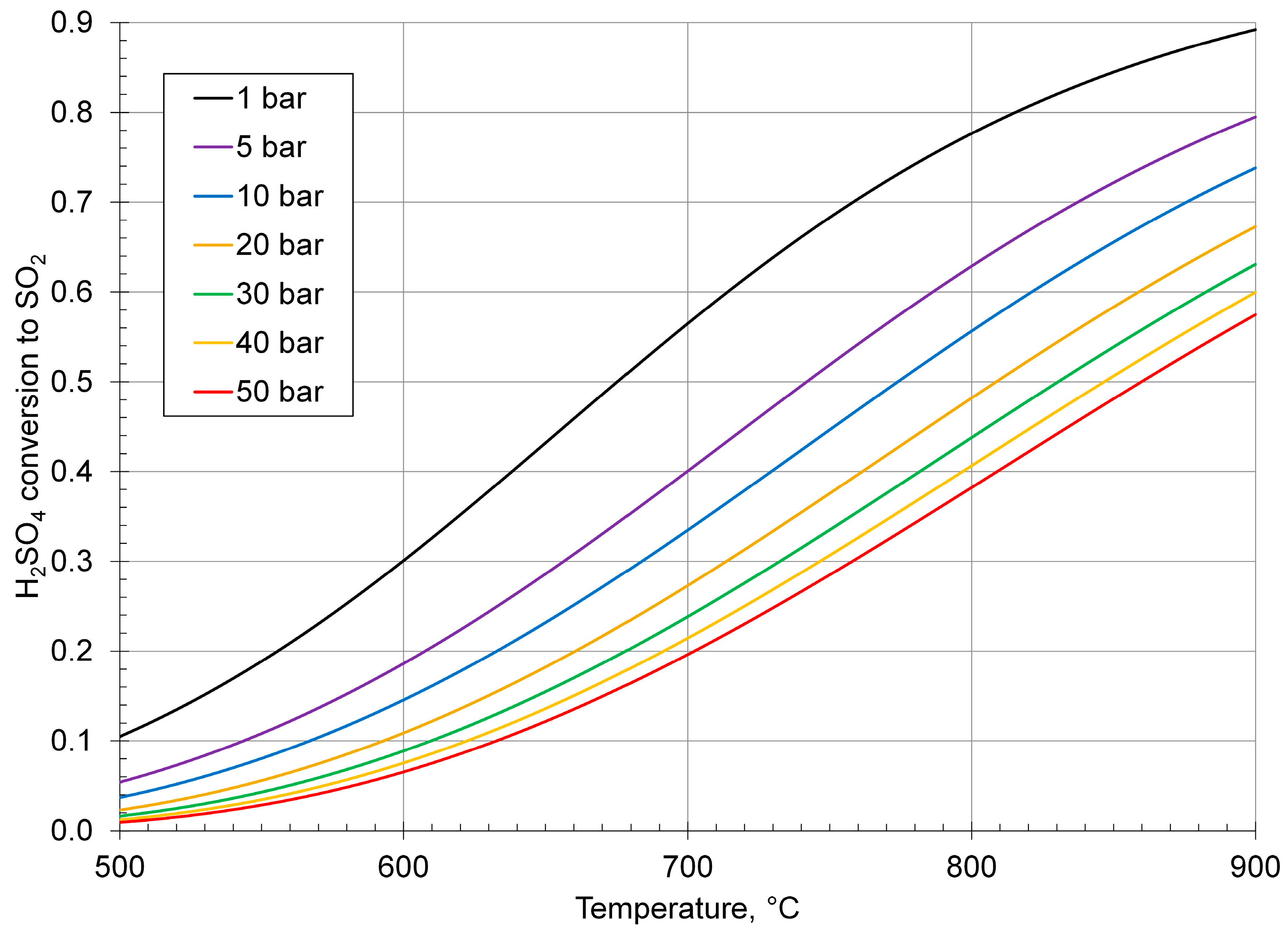
3. Sulfuric Acid Thermal Decomposition
3.1. High Temperature Sulfuric Acid Decomposition
3.1.1. Constitutive Materials
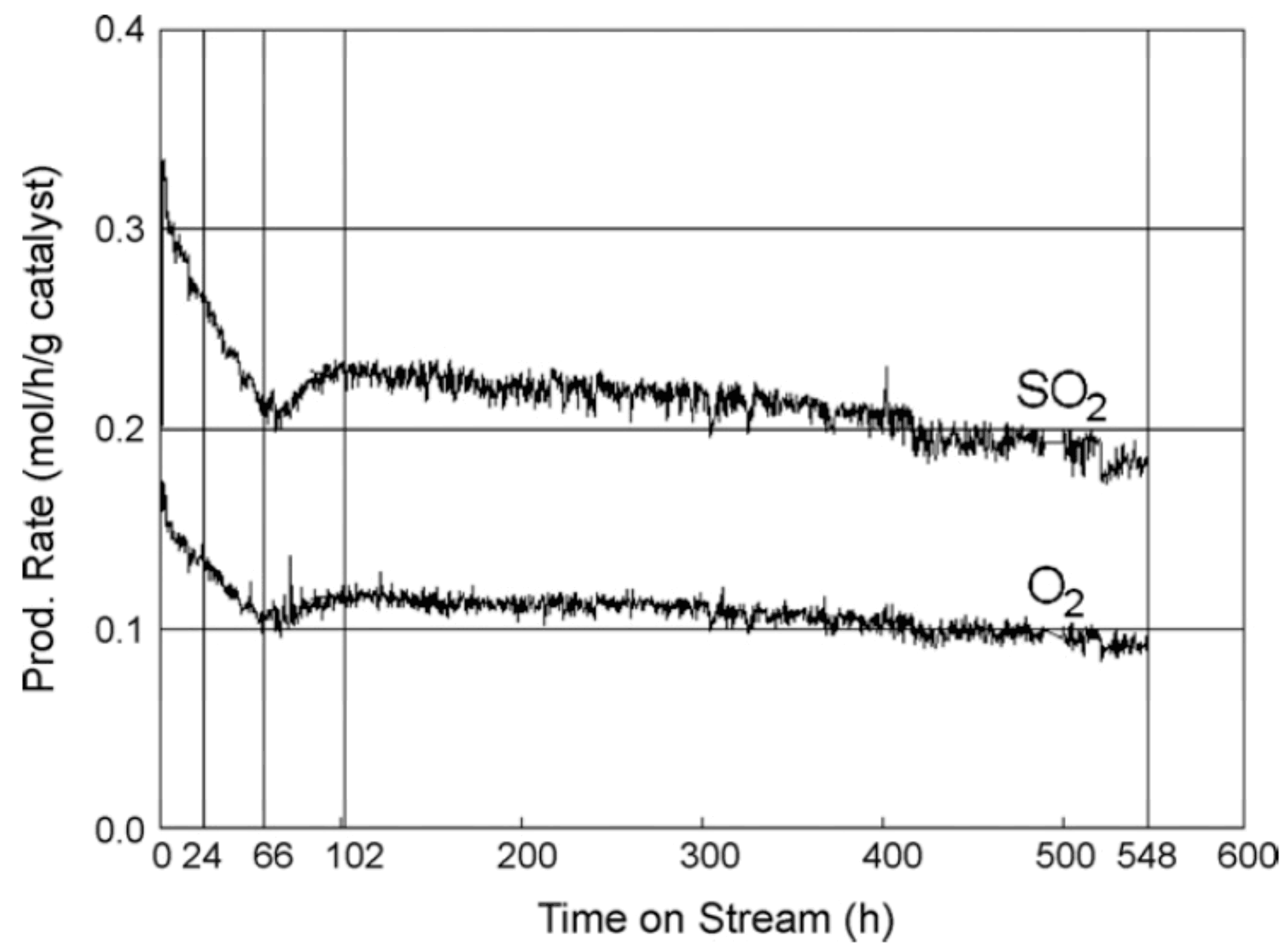
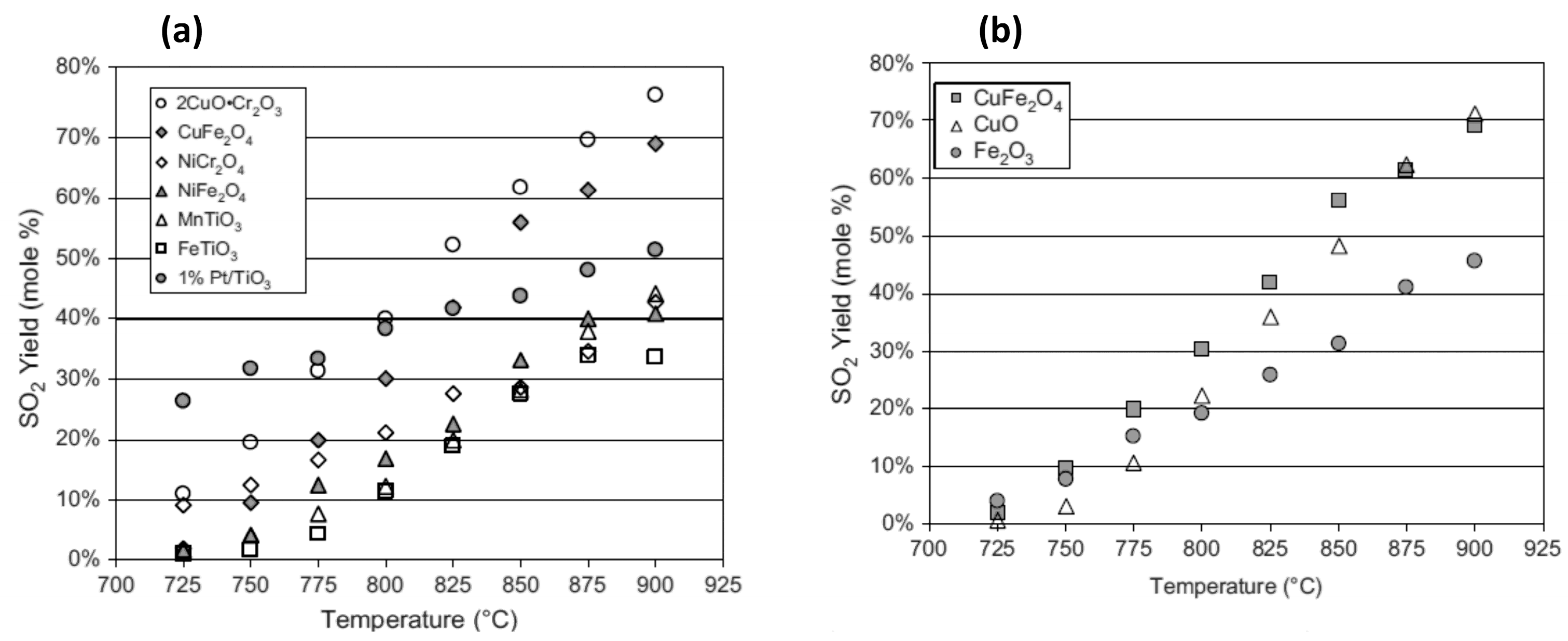
3.1.2. Catalysts
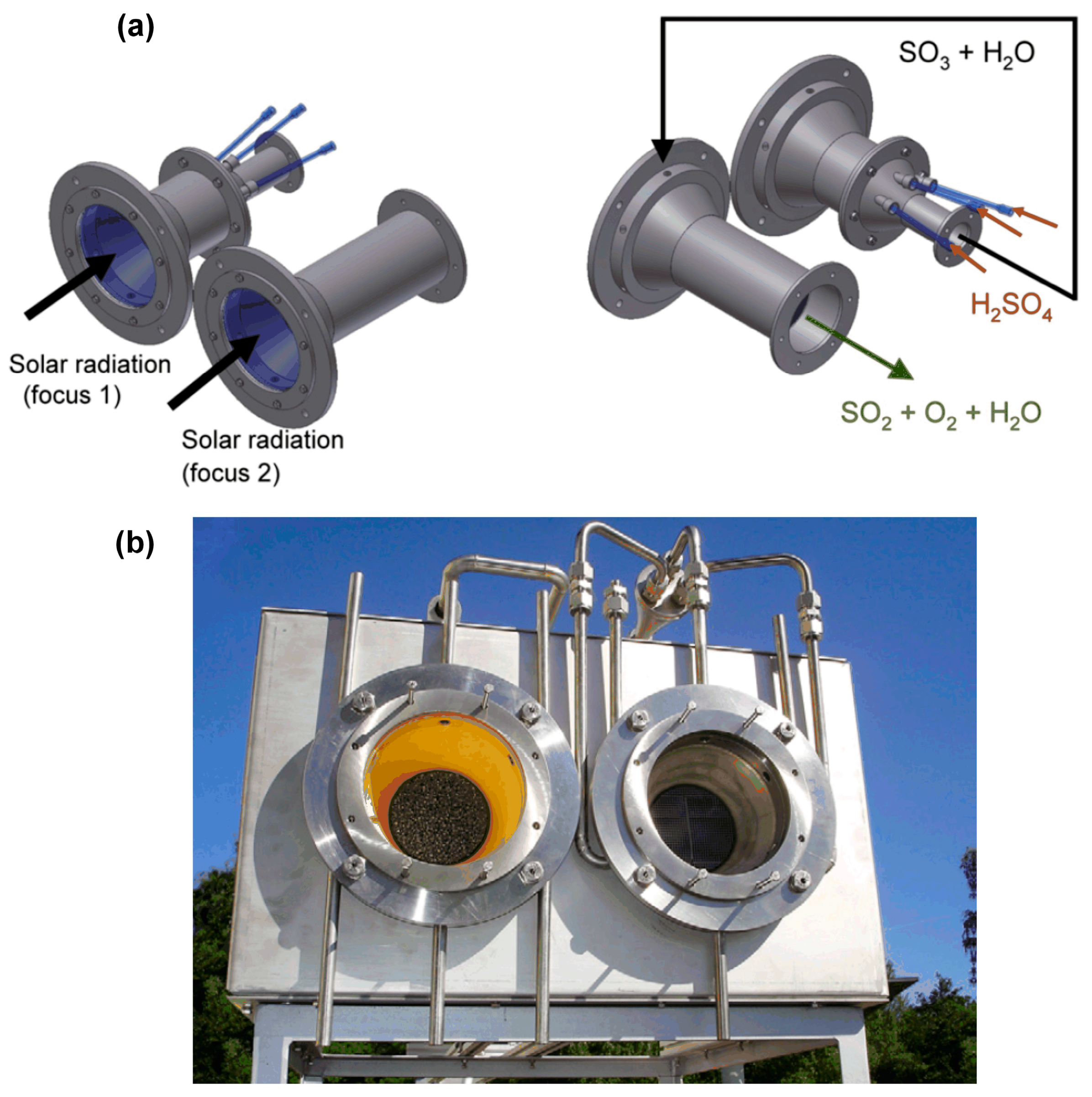
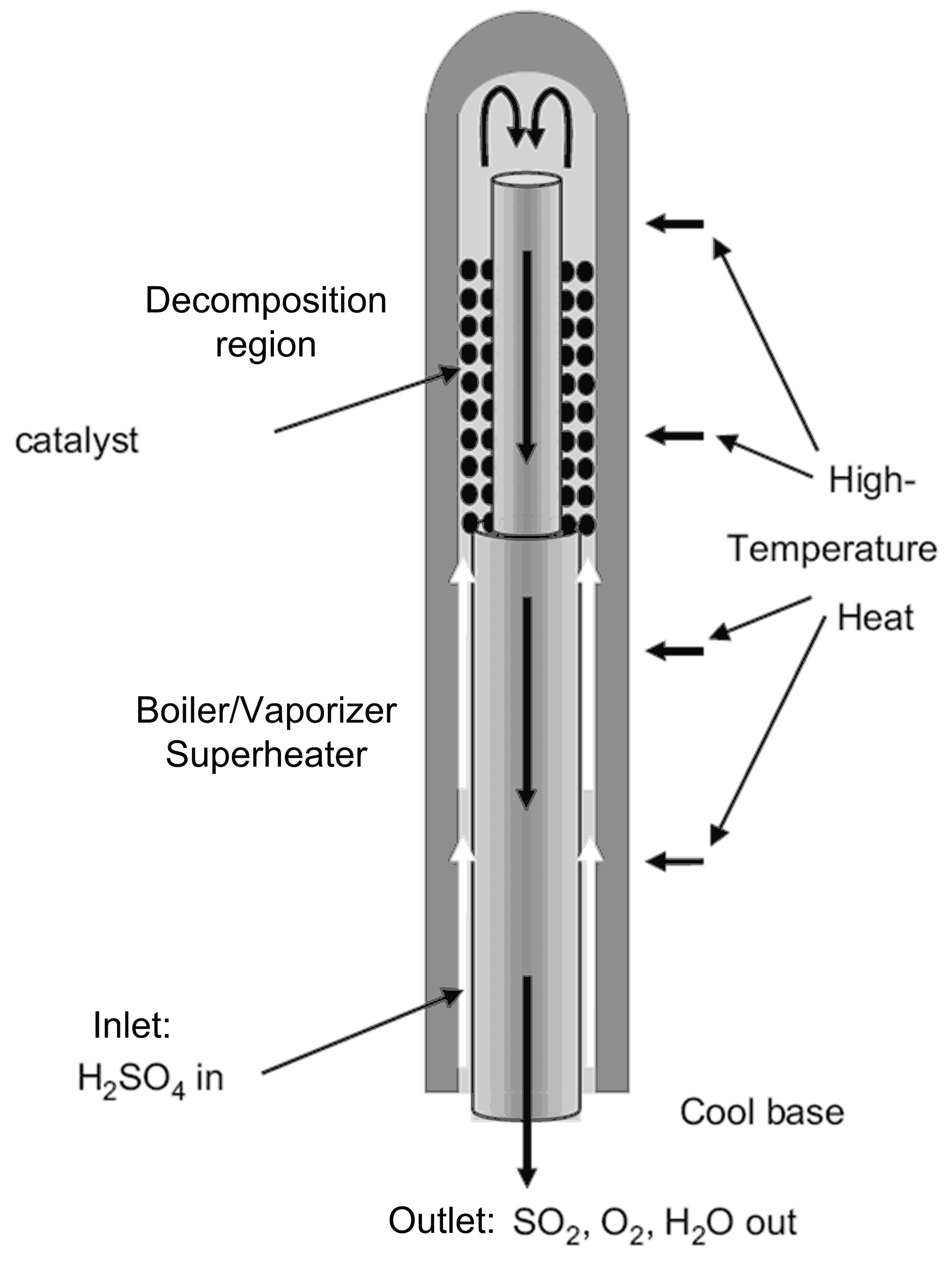
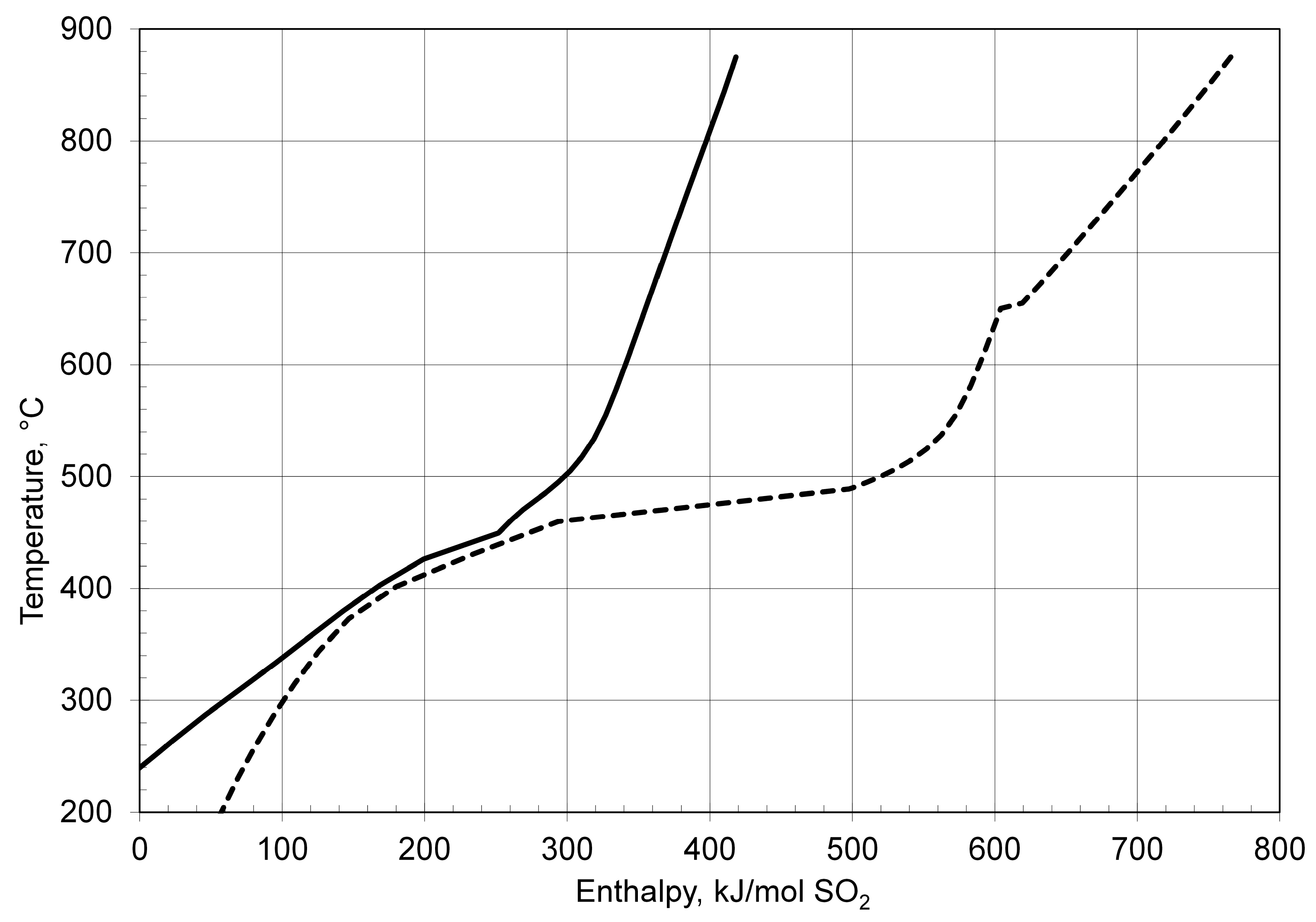
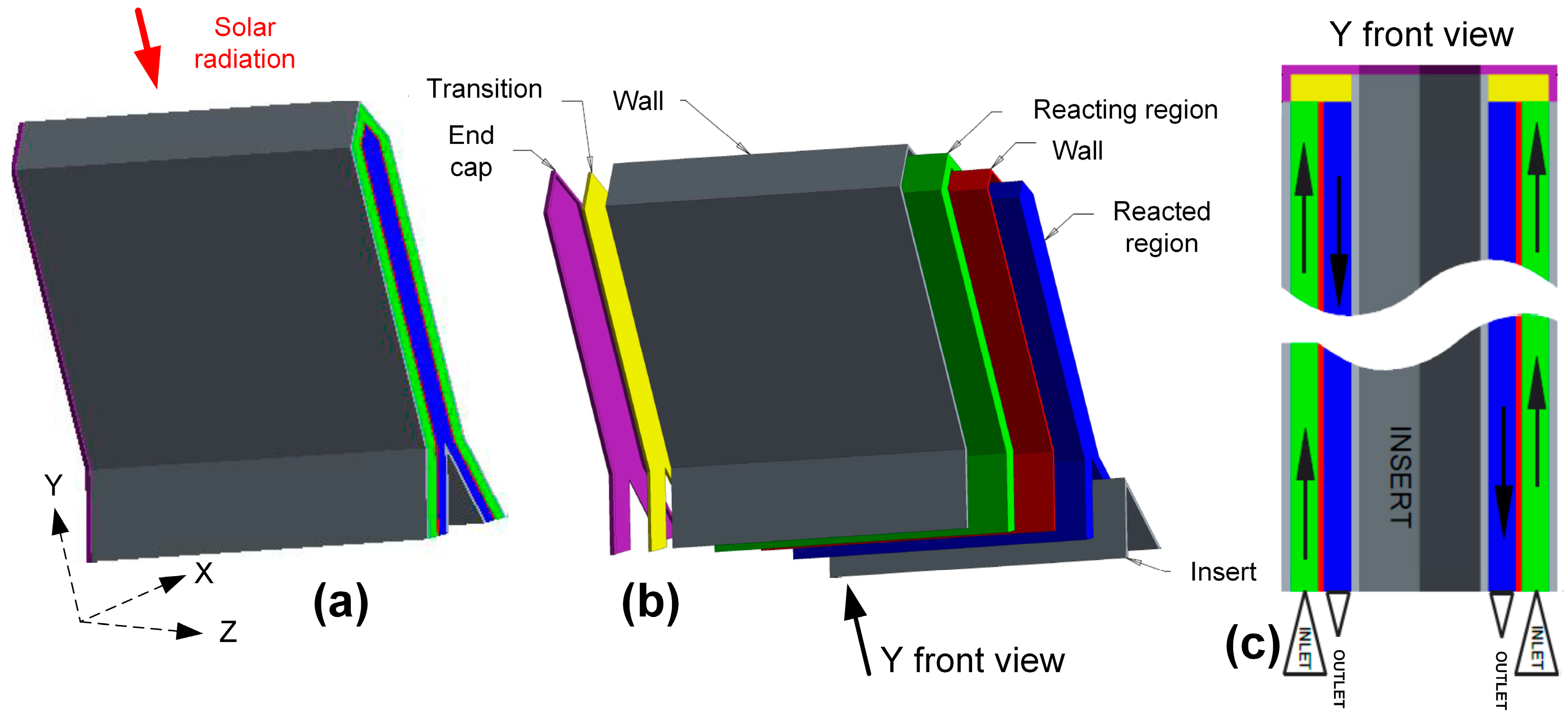
3.1.3. Reactor Concepts
3.2. Sulfuric Acid Concentration
3.3. Separation of Oxygen and Sulfur Dioxide
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Nomenclature
| HyS | Hybrid Sulfur |
| SI | Sulfur Iodine |
| SRNL | Savannah River National Laboratory |
| SNL | Sandia National Laboratory |
| PEM | Proton exchange membrane |
| LHV | Low heating value (120 MJ/kgH2) |
| DOE | U.S. Department of Energy |
| HFTO | DOE Hydrogen Fuel Cell Technology Office |
| DLR | German Aerospace Center |
| eNRTL | Electrolyte Non-Random Two Liquids |
| CFD | Computational Fluid Dynamic |
| s-PBI | Sulfonated polybenzimidazole |
References
- Pivovar, B.; Rustagi, N.; Satyapal, S. Hydrogen at Scale (H2@Scale): Key to a clean.; economic.; and sustainable energy system. Electrochem. Soc. Interface 2018, 27, 47–52. [Google Scholar] [CrossRef]
- Perkins, C.; Weimer, A.W. Likely near-term solar-thermal water splitting technologies. Int. J. Hydrogen Energy 2004, 29, 1587–1599. [Google Scholar] [CrossRef]
- Buttler, A.; Spliethoff, H. Current status of water electrolysis for energy storage.; grid balancing and sector coupling via power-to-gas and power-to-liquids: A review. Renew. Sustain. Energy Rev. 2018, 82, 2440–2454. [Google Scholar] [CrossRef]
- Schmidt, O.; Gambhir, A.; Staffell, I.; Hawkes, A.; Nelson, J.; Few, S. Future cost and performance of water electrolysis: An expert elicitation study. Int. J. Hydrogen Energy 2017, 42, 30470–30492. [Google Scholar] [CrossRef]
- Felgenhauer, M.; Hamacher, T. State-of-the-art of commercial electrolyzers and on-site hydrogen generation for logistic vehicles in South Carolina. Int. J. Hydrogen Energy 2015, 40, 2084–2090. [Google Scholar] [CrossRef]
- Stoots, C.M.; O’Brien, J.E.; Condie, K.G.; Hartvigsen, J.J. High-temperature electrolysis for large-scale hydrogen production from nuclear energy–experimental investigations. Int. J. Hydrogen Energy 2010, 35, 4861–4870. [Google Scholar] [CrossRef]
- Mougin, J. Hydrogen production by high-temperature steam electrolysis. In Compendium of Hydrogen Energy; Woodhead Publishing: Cambridge, UK, 2015; pp. 225–253. [Google Scholar]
- Funk, J.E. Thermochemical hydrogen production: Past and present. Int. J. Hydrogen Energy 2001, 26, 185–190. [Google Scholar] [CrossRef]
- Dincer, I.; Acar, C. Review and evaluation of hydrogen production methods for better sustainability. Int. J. Hydrogen Energy 2015, 40, 11094–11111. [Google Scholar] [CrossRef]
- Steinfeld, A. Solar thermochemical production of hydrogen––A review. Solar. Energy 2005, 78, 603–615. [Google Scholar] [CrossRef]
- Beghi, G.E. A decade of research on thermochemical hydrogen at the joint research centre Ispra. Int. J. Hydrogen Energy 1986, 11, 761–771. [Google Scholar] [CrossRef]
- Kodama, T.; Gokon, N. Thermochemical cycles for high-temperature solar hydrogen production. Chem. Rev. 2007, 107, 4048–4077. [Google Scholar] [CrossRef] [PubMed]
- Brown, L.C.; Besenbruch, G.E.; Lentsch, R.D.; Schultz, K.R.; Funk, J.F.; Pickard, P.S.; Marshall, A.C.; Showalter, S.K. High Efficiency Generation of Hydrogen Fuels Using Nuclear Power; Final Technical Report from General Atomics Corp. to US DOE. GA-A24285; General Atomics: San Diego, CA, USA, 2003. [Google Scholar]
- Abanades, S.; Charvin, P.; Flamant, G.; Neveu, P. Screening of water-splitting thermochemical cycles potentially attractive for hydrogen production by concentrated solar energy. Energy 2006, 31, 2805–2822. [Google Scholar] [CrossRef]
- Russell, J.; Porter, J. Production of Hydrogen from Water; General Atomics Report GA–A12889; General Atomics: San Diego, CA, USA, 1974. [Google Scholar]
- Norman, J.H.; Besenbruch, G.; O’keefe, D.R. Thermochemical Water-Splitting for Hydrogen Production; Final Report General Atomic Co PB-82-208174; General Atomics: San Diego, CA, USA, 1981. [Google Scholar]
- Perret, R. Solar Thermochemical Hydrogen Production Research (STCH); Sandia National Lab.(SNL-CA): Livermore, CA, USA, 2011. Available online: https://www1.eere.energy.gov/hydrogenandfuelcells/pdfs/solar_thermo_h2. (accessed on 24 September 2020).
- Nuclear Hydrogen R&D Plan. DOE Fuel Cell Technology Office. 2004. Available online: https://www.energy.gov/sites/prod/files/2015/01/f19/fcto_nuclear_h2_r%26d_plan.pdf (accessed on 12 February 2019).
- Yalcin, S. A review of nuclear hydrogen production. Int. J. Hydrogen Energy 1989, 14, 551–561. [Google Scholar] [CrossRef]
- Brecher, L.E.; Spewock, S.; Warde, C.J. The westinghouse sulfur cycle for the thermochemical decomposition of water. Int. J. Hydrogen Energy 1977, 2, 7–15. [Google Scholar] [CrossRef]
- Henderson, A.D.; Taylor, A. The US Department of Energy research and development programme on hydrogen production using nuclear energy. Int. J. Nucl. Hydrog. Prod. Appl. 2006, 1, 51–56. [Google Scholar] [CrossRef]
- Corgnale, C.; Summers, W.A. Solar hydrogen production by the Hybrid Sulfur process. Int. J. Hydrogen Energy 2011, 36, 11604–11619. [Google Scholar] [CrossRef]
- Elvington, M.C.; Colón-Mercado, H.; McCatty, S.; Stone, S.G.; Hobbs, D.T. Evaluation of proton-conducting membranes for use in a sulfur dioxide depolarized electrolyzer. J. Power Sources 2010, 195, 2823–2829. [Google Scholar] [CrossRef]
- Staser, J.A.; Gorensek, M.B.; Weidner, J.W. Quantifying individual potential contributions of the hybrid sulfur electrolyzer. J. Electrochem. Soc. 2010, 157, B952–B958. [Google Scholar] [CrossRef]
- Steimke, J.L.; Steeper, T.J.; Cólon-Mercado, H.R.; Gorensek, M.B. Development and testing of a PEM SO2-depolarized electrolyzer and an operating method that prevents sulfur accumulation. Int. J. Hydrogen Energy 2015, 40, 13281–13294. [Google Scholar] [CrossRef]
- Weidner, J.W. High Performance Electrolyzers for Hybrid Thermochemical Cycles; University of South Carolina Research Foundation (No. DOE/ID/14752); University of South Carolina: Columbia, SC, USA, 2009. [Google Scholar]
- Garrick, T.R.; Wilkins, C.H.; Pingitore, A.T.; Mehlhoff, J.; Gulledge, A.; Benicewicz, B.C.; Weidner, J.W. Characterizing voltage losses in an SO2 depolarized electrolyzer using sulfonated polybenzimidazole membranes. J. Electrochem. Soc. 2017, 164, F1591–F1595. [Google Scholar] [CrossRef]
- Garrick, T.R.; Gulledge, A.; Staser, J.A.; Benicewicz, B.; Weidner, J.W. Polybenzimidazole membranes for hydrogen production in the hybrid sulfur electrolyzer. ECS Trans. 2015, 66, 31–40. [Google Scholar] [CrossRef]
- Gorensek, M.B.; Corgnale, C.; Staser, J.A.; Weidner, J.W. Solar Thermochemical Hydrogen (STCH) Processes. Electrochem. Soc. Interface 2018, 27, 53–56. [Google Scholar] [CrossRef]
- Gorensek, M.B.; Summers, W.A. Hybrid sulfur flowsheets using PEM electrolysis and a bayonet decomposition reactor. Int. J. Hydrogen Energy 2009, 34, 4097–4114. [Google Scholar] [CrossRef]
- Colón-Mercado, H.R.; Hobbs, D.T. Catalyst evaluation for a sulfur dioxide-depolarized electrolyzer. Electrochem. Commun. 2007, 9, 2649–2653. [Google Scholar] [CrossRef]
- Meekins, B.H.; Thompson, A.B.; Gopal, V.; Mehrabadi, B.A.; Elvington, M.C.; Ganesan, P.; Newhouse-lllige, T.A.; Shepard, A.W.; Scipioni, L.; Greer, J.A.; et al. In-situ and ex-situ comparison of the electrochemical oxidation of SO2 on carbon supported Pt and Au catalysts. Int. J. Hydrogen Energy 2020, 45, 1940–1947. [Google Scholar] [CrossRef]
- Niehoff, A.G.; Botero, N.B.; Acharya, A.; Thomey, D.; Roeb, M.; Sattler, C.; Pitz-Paal, R. Process modelling and heat management of the solar hybrid sulfur cycle. Int. J. Hydrogen Energy 2015, 40, 4461–4473. [Google Scholar] [CrossRef]
- Corgnale, C.; Gorensek, M.; Summers, W.A. Solar Hybrid Sulfur Cycle Water-Splitting Process; SRNL-STI-2015e00546; Revision 0; Savannah River National Laboratory: Aiken, SC, USA, 2015. [Google Scholar]
- Gorensek, M.B.; Corgnale, C.; Summers, W.A. Development of the hybrid sulfur cycle for use with concentrated solar heat. I. Conceptual design. Int. J. Hydrogen Energy 2017, 42, 20939–20954. [Google Scholar] [CrossRef]
- Gorensek, M.; Summers, W.; Boltrunis, C.; Lahoda, E.; Allen, D.; Greyvenstein, R. Hybrid Sulfur Process Reference Design and Cost Analysis; Report No. SRNL-L1200-2008-00002; Savannah River National Laboratory: Aiken, SC, USA, 2009. [Google Scholar]
- Summers, W.A.; Buckner, M.R.; Gorensek, M.B. The hybrid sulfur cycle for nuclear hydrogen production. In Proceedings of the GLOBAL 2005, Tsukuba, Japan, 9–13 October 2005. [Google Scholar]
- Hinkley, J.T.; O’Brien, J.A.; Fell, C.J.; Lindquist, S.E. Prospects for solar only operation of the hybrid sulphur cycle for hydrogen production. Int. J. Hydrogen Energy 2011, 36, 11596–11603. [Google Scholar] [CrossRef]
- Kromer, M.; Roth, K.; Takata, R.; Chin, P. Support for Cost Analyses on Solar-Driven High Temperature Thermochemical Water-Splitting Cycles. Final Report to DOE DE-DT0000951. 2011. Available online: https://www.energy.gov/sites/prod/files/2014/03/f11/solar_thermo_h2_cost.pdf (accessed on 23 September 2020).
- U.S. Department of Energy HydroGEN Program. Available online: https://www.h2awsm.org/ (accessed on 8 August 2020).
- O’keefe, D.; Allen, C.; Besenbruch, G.; Brown, L.; Norman, J.; Sharp, R.; McCorkle, K. Preliminary results from bench-scale testing of a sulfur-iodine thermochemical water-splitting cycle. Int. J. Hydrogen Energy 1982, 7, 381–392. [Google Scholar] [CrossRef]
- Cerri, G.; Salvini, C.; Corgnale, C.; Giovannelli, A.; Manzano, D.D.L.; Martinez, A.O.; le Duigou, A.; Borgard, J.-M.; Mansilla, C. Sulfur–Iodine plant for large scale hydrogen production by nuclear power. Int. J. Hydrogen Energy 2010, 35, 4002–4014. [Google Scholar] [CrossRef]
- International Atomic Energy Agency. Hydrogen Production Using Nuclear Energy; IAEA Nuclear Energy Series No. NP-T-4.2; International Atomic Energy Agency: Vienna, Austria, 2013. [Google Scholar]
- LeDuigou, A.; Borgard, J.M.; Larousse, B.; Doizi, D.; Allen, R.; Ewan, B.C.; Priestman, G.H.; Elder, R.; Devonshire, R.; Ramos, V.; et al. HYTHEC: An EC funded search for a long term massive hydrogen production route using solar and nuclear technologies. Int. J. Hydrogen Energy 2007, 32, 1517–1529. [Google Scholar]
- Onuki, K.; Kubo, S.; Terada, A.; Sakaba, N.; Hino, R. Thermochemical water-splitting cycle using iodine and sulfur. Energy Environ. Sci. 2009, 2, 491–497. [Google Scholar] [CrossRef]
- Mysels, K.J. Method of Extracting Iodine from Liquid Mixtures of Iodine, Water and Hydrogen Iodide. U.S. Patent 4176169, 27 November 1979. [Google Scholar]
- Cho, W.C.; Park, C.S.; Kang, K.S.; Kim, C.H.; Bae, K.K. Conceptual design of sulfur–iodine hydrogen production cycle of Korea Institute of Energy Research. Nucl. Eng. Des. 2009, 239, 501–507. [Google Scholar] [CrossRef]
- Guo, H.; Kasahara, S.; Tanaka, N.; Onuki, K. Energy requirement of HI separation from HI–I2–H2O mixture using electro-electrodialysis and distillation. Int. J. Hydrogen Energy 2012, 37, 13971–13982. [Google Scholar] [CrossRef]
- Berndhaeuser, C.; Knoche, K.F. Experimental investigations of thermal HI decomposition from H2O-HIx-I2 solutions. Int. J. Hydrogen Energy 1994, 19, 239–244. [Google Scholar] [CrossRef]
- Murphy, J.E., IV; O’Connell, J.P. Process simulations of HI decomposition via reactive distillation in the sulfur–iodine cycle for hydrogen manufacture. Int. J. Hydrogen Energy 2012, 37, 4002–4011. [Google Scholar] [CrossRef]
- Norman, J.H.; Mysels, K.J.; Sharp, R.; Williamson, D. Studies of the sulfur-iodine thermochemical water-splitting cycle. Int. J. Hydrogen Energy 1982, 7, 545–556. [Google Scholar] [CrossRef]
- Leybros, J.; Gilardi, T.; Saturnin, A.; Mansilla, C.; Carles, P. Plant sizing and evaluation of hydrogen production costs from advanced processes coupled to a nuclear heat source. Part I: Sulphur–iodine cycle. Int. J. Hydrogen Energy 2010, 35, 1008–1018. [Google Scholar] [CrossRef]
- van Velzen, D.; Langenkamp, H.; Schutz, G.; Lalonde, D.; Flamm, J.; Fiebelmann, P. Development and design of a continuous laboratory scale plant for hydrogen production by the Mark-13 cycle. Int. J. Hydrogen Energy 1980, 5, 131–139. [Google Scholar] [CrossRef]
- van Velzen, D.; Langenkamp, H. Status report on the operation of the bench-scale plant for hydrogen production by the mark-13 process. Int. J. Hydrogen Energy 1982, 7, 629–636. [Google Scholar] [CrossRef]
- T-Raissi, A.; Muradov, N.; Huang, C.; Adebiyi, O. Hydrogen from solar via light-assisted high-temperature water splitting cycles. J. Solar. Energy Eng. 2007, 129, 184–189. [Google Scholar] [CrossRef]
- Kalyva, A.E.; Vagia, E.C.; Konstandopoulos, A.G.; Srinivasa, A.R.; T-Raissi, A.; Muradov, N.; Kakosimos, K.E. Investigation of the solar hybrid photo-thermochemical sulfur-ammonia water splitting cycle for hydrogen production. Chem. Eng. Trans. 2015, 45, 361–366. [Google Scholar]
- Littlefield, J.; Wang, M.; Brown, L.C.; Herz, R.K.; Talbot, J.B. Process modeling and thermochemical experimental analysis of a solar sulfur ammonia hydrogen production cycle. Energy Procedia 2012, 29, 616–623. [Google Scholar] [CrossRef]
- Kaur, H.; Wang, M.; Gorensek, M.B.; Chen, C.C. Thermodynamic modeling of the hybrid sulfur (HyS) cycle for hydrogen production. Fluid Phase Equilibria 2018, 460, 175–188. [Google Scholar] [CrossRef]
- Wong, B.; Trester, P.W. Materials Development for Sulfur-Iodine Thermochemical Hydrogen; General Atomics Report GA-A25750; General Atomics: San Diego, CA, USA, 2007; Chapter 4. [Google Scholar]
- Wong, B.; Buckingham, R.T.; Brown, L.C.; Russ, B.E.; Besenbruch, G.E.; Kaiparambil, A.; Roy, A. Construction materials development in sulfur–iodine thermochemical water-splitting process for hydrogen production. Int. J. Hydrogen Energy 2007, 32, 497–504. [Google Scholar] [CrossRef]
- Kubo, S.; Futakawa, M.; Ioka, I.; Onuki, K.; Yamaguchi, A. Corrosion resistance of structural materials in high-temperature aqueous sulfuric acids in thermochemical water-splitting iodine–sulfur process. Int. J. Hydrogen Energy 2013, 38, 6577–6585. [Google Scholar] [CrossRef]
- Karagiannakis, G.; Agrafiotis, C.C.; Pagkoura, C.; Konstandopoulos, A.G.; Thomey, D.; de Oliveira, L.; Roeb, M.; Sattler, C. Hydrogen production via sulfur-based thermochemical cycles: Part 3: Durability and post-characterization of silicon carbide honeycomb substrates coated with metal oxide-based candidate catalysts for the sulfuric acid decomposition step. Int. J. Hydrogen Energy 2012, 37, 8190–8203. [Google Scholar] [CrossRef]
- Roeb, M.; Neises, M.; Monnerie, N.; Call, F.; Simon, H.; Sattler, C.; Schmücker, M.; Pitz-Paal, R. Materials-related aspects of thermochemical water and carbon dioxide splitting: A review. Materials 2012, 5, 2015–2054. [Google Scholar] [CrossRef]
- O’keefe, D.R.; Norman, J.H.; Williamson, D.G. Catalysis research in thermochemical water-splitting processes. Catal. Rev. Sci. Eng. 1980, 22, 325–369. [Google Scholar] [CrossRef]
- Tagawa, H.; Endo, T. Catalytic decomposition of sulfuric acid using metal oxides as the oxygen generating reaction in thermochemical water splitting process. Int. J. Hydrogen Energy 1989, 14, 11–17. [Google Scholar] [CrossRef]
- Schultz, K.; Sink, C.; Pickard, P.; Herring, S.; O’Brien, J.; Buckingham, B.; Summers, W.; Lewis, M. Status of the US nuclear hydrogen initiative. In Proceedings of the ICAPP Conference, Bangkok, Thailand, 20–25 June 2007. Paper 7530:13-8. [Google Scholar]
- Ginosar, D.M.; Petkovic, L.M.; Glenn, A.W.; Burch, K.C. Stability of supported platinum sulfuric acid decomposition catalysts for use in thermochemical water splitting cycles. Int. J Hydrogen Energy 2007, 32, 482–488. [Google Scholar] [CrossRef]
- Petkovic, L.M.; Ginosar, D.M.; Rollins, H.W.; Burch, K.C.; Pinhero, P.J.; Farrell, H.H. Pt/TiO2 (rutile) catalysts for sulfuric acid decomposition in sulfur-based thermochemical water-splitting cycles. Appl. Catal. A Gen. 2008, 338, 27–36. [Google Scholar] [CrossRef]
- Ginosar, D.M.; Rollins, H.W.; Petkovic, L.M.; Burch, K.C.; Rush, M.J. High-temperature sulfuric acid decomposition over complex metal oxide catalysts. Int. J. Hydrogen Energy 2009, 34, 4065–4073. [Google Scholar] [CrossRef]
- Karagiannakis, G.; Agrafiotis, C.C.; Zygogianni, A.; Pagkoura, C.; Konstandopoulos, A.G. Hydrogen production via sulfur-based thermochemical cycles: Part 1: Synthesis and evaluation of metal oxide-based candidate catalyst powders for the sulfuric acid decomposition step. Int. J. Hydrogen Energy 2011, 36, 2831–2844. [Google Scholar] [CrossRef]
- Giaconia, A.; Sau, S.; Felici, C.; Tarquini, P.; Karagiannakis, G.; Pagkoura, C.; Agrafiotis, C.; Konstandopoulos, A.G.; Thomey, D.; de Oliveira, L.; et al. Hydrogen production via sulfur-based thermochemical cycles: Part 2: Performance evaluation of Fe2O3-based catalysts for the sulfuric acid decomposition step. Int. J. Hydrogen Energy 2011, 36, 6496–6509. [Google Scholar] [CrossRef]
- Corgnale, C. High temperature reactor catalyst material development for low cost and efficient solar driven sulfur-based processes. In Proceedings of the DOE Annual Merit Review Presentation, Crystal City, VV, USA, 19–21 May 2019. [Google Scholar]
- Noglik, A.; Roeb, M.; Rzepczyk, T.; Hinkley, J.; Sattler, C.; Pitz-Paal, R. Solar thermochemical generation of hydrogen: Development of a receiver reactor for the decomposition of sulfuric acid. J. Solar. Energy Eng. 2009, 131, 011003-1. [Google Scholar] [CrossRef]
- Thomey, D.; de Oliveira, L.; Sack, J.-P.; Roeb, M.; Sattler, C. Development and test of a solar reactor for decomposition of sulphuric acid in thermochemical hydrogen production. Int. J. Hydrogen Energy 2012, 37, 16615–16622. [Google Scholar] [CrossRef]
- Moore, R.; Pickard, P.; Parma, E.; Vernon, M.; Gelbard, F. Integrated Boiler.; Superheater and Decomposer for Sulphuric Acid Decomposition. Sandia Corp. U.S. Patent No. 7645437 B1, 12 January 2010. [Google Scholar]
- Russ, B. Sulfur Iodine Process Summary for the Hydrogen Technology Down-Selection. Available online: https://art.inl.gov/NGNP/Subcontractors%20Documents/General%20Atomics/Sulfur%20Iodine%20Process%20Summary%20for%20the%20Hydrogen%20Technology%20Down-Selection.pdf (accessed on 20 September 2020).
- Sattler, C.; Roeb, M.; Agrafiotis, C.; Thomey, D. Solar hydrogen production via sulphur based thermochemical water-splitting. Solar Energy 2017, 156, 30–47. [Google Scholar] [CrossRef]
- Gorensek, M.B.; Edwards, T.B. Energy efficiency limits for a recuperative bayonet sulfuric acid decomposition reactor for sulfur cycle thermochemical hydrogen production. Ind. Eng. Chem. Res. 2009, 48, 7232–7245. [Google Scholar] [CrossRef]
- Corgnale, C.; Shimpalee, S.; Gorensek, M.; Satjaritanun, P.; Weidner, J.; Summers, W. Numerical modeling of a bayonet heat exchanger-based reactor for sulfuric acid decomposition in thermochemical hydrogen production processes. Int. J. Hydrogen Energy 2017, 42, 2046. [Google Scholar] [CrossRef]
- Corgnale, C.; Ma, Z.; Shimpalee, S. Modeling of a direct solar receiver reactor for decomposition of sulfuric acid in thermochemical hydrogen production cycles. Int. J. Hydrogen Energy 2019, 44, 27237–27247. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Process | Conditions | Candidate Materials | Compatibility–Corrosion Results | Comments |
|---|---|---|---|---|
| H2SO4 vaporization | 350–550 °C H2O + SO3, other contaminants | Structural: Incoloy 800 H AL610, high Si steel SiC, Si3N4 Hastelloy G, C-276 | 800 H, 800 HT.High Si steel (SiO2) < 5 mm/year. SiC ≈ no corrosion in 1000 h test at 75–79% acid. C-276 ≈ 1 mm/year at 476 h. | Coated materials (Pt) cost issue. Ceramics promising, but fabrication and joining issues. Dry wall boiler with ceramics may be an option. |
| H2SO4 decomposition | 550–950 °C H2O, H2SO4, SO3, SO2, O2 | Structural: Incoloy 800 HT, Incoloy 800 H (with aluminide coatings), AL610 Ceramics, Pt or Au coatings on superalloy structural materials | Incoloy, Inconel bare–2–4 mg/cm2 in 1000 h at 900 °C. Aluminide coatings—approximately 1 mg/cm2 in 1000 h at 900 °C. Intergranular corrosion observed for 800 H. Noble metal coatings may provide corrosion protection. | Incoloy 800 HT may address integranular corrosion. C-SiC composites should be examined. Pt coatings may serve the function of catalyst and reduce corrosion. Corrosion benefits of noble metal coatings must be demonstrated. |
| Catalyst | Onset of Failure Temperature (K) | Failure Mode |
|---|---|---|
| 0.5% Pt/Al2O3 | 890 | Al2(SO4)3 poisoning |
| Fe2O3/Al2O3 | 1000 | Sulfate formation |
| V2O5/Al2O3 | 910 | Sulfate formation, volatile |
| Cr2O3/Al2O3 | 1070 | Sulfate formation, volatile |
| CuO/Al2O3 | 950 | Sulfate formation |
| 0.1% Pd/Al2O3 | 970 | Al2(SO4)3 poisoning |
| MnO2/Al2O3 | 1120 | Sulfate formation |
| CoO/Al2O3 | 1140 | Sulfate formation |
| NiO/Al2O3 | 1160 | Sulfate formation |
| 0.1% Pt/Al2O3 | 950 | Al2(SO4)3 poisoning |
| Al2O3 | 1250 | Poor catalyst |
| CuO/SiO2 | 1010 | CuSiO3 formation? |
| 0.5% Pt/SiO2 | 850 | Gradual temperature cut-off |
| CeO2 | 1180 | Poor catalyst |
| 0.08% Pt/TiO2 (surface) | 800 | Gradual temperature cut-off |
| 0.1 % Pt/TiO2 | 900 | Gradual temperature cut-off |
| Pd/TiO2 | 1090 | Initially better, sulfation? |
| Fe2O3/TiO2 | 1090 | Unknown |
| CuO/TiO2 | 1000 | Sulfate formation |
| 0.08% Pt/TiO2 | 790 | Gradual temperature cut-off |
| TiO2 | 1140 | Poor catalyst |
| Pt/ZrO2 (surface) | 830 | Substrate sulfation |
| Fe2O3/ZrO2 | 1020 | Sulfate formation |
| CuO/ZrO2 | 970 | Sulfate formation |
| ZrO2 | 1130 | Poor catalyst |
| Nb2O5/BaSO4 | 1140 | Poor catalyst |
| CuO/BaSO4 | 1050 | Sulfate formation |
| Fe2O3/BaSO4 | 980 | Sulfate formation |
| U3O8/BaSQ4 | 1070 | Sulfate formation |
| BaSO4 | 1250 | Poor catalyst |
| 0.07% Pt/BaSO4-TiO2 | 780 | Gradual temperature cut-off |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Corgnale, C.; Gorensek, M.B.; Summers, W.A. Review of Sulfuric Acid Decomposition Processes for Sulfur-Based Thermochemical Hydrogen Production Cycles. Processes 2020, 8, 1383. https://doi.org/10.3390/pr8111383
Corgnale C, Gorensek MB, Summers WA. Review of Sulfuric Acid Decomposition Processes for Sulfur-Based Thermochemical Hydrogen Production Cycles. Processes. 2020; 8(11):1383. https://doi.org/10.3390/pr8111383
Chicago/Turabian StyleCorgnale, Claudio, Maximilian B. Gorensek, and William A. Summers. 2020. "Review of Sulfuric Acid Decomposition Processes for Sulfur-Based Thermochemical Hydrogen Production Cycles" Processes 8, no. 11: 1383. https://doi.org/10.3390/pr8111383
APA StyleCorgnale, C., Gorensek, M. B., & Summers, W. A. (2020). Review of Sulfuric Acid Decomposition Processes for Sulfur-Based Thermochemical Hydrogen Production Cycles. Processes, 8(11), 1383. https://doi.org/10.3390/pr8111383