Formation and Evolution Mechanism for Carbonaceous Deposits on the Surface of a Coking Chamber


Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Preparation
2.2. Measurement and Characterization
3. Results and Discussion
3.1. Elemental Composition
3.2. Morphology
3.3. X-ray Diffraction (XRD)
3.4. Fourier Transform Infrared Spectrometer (FTIR)
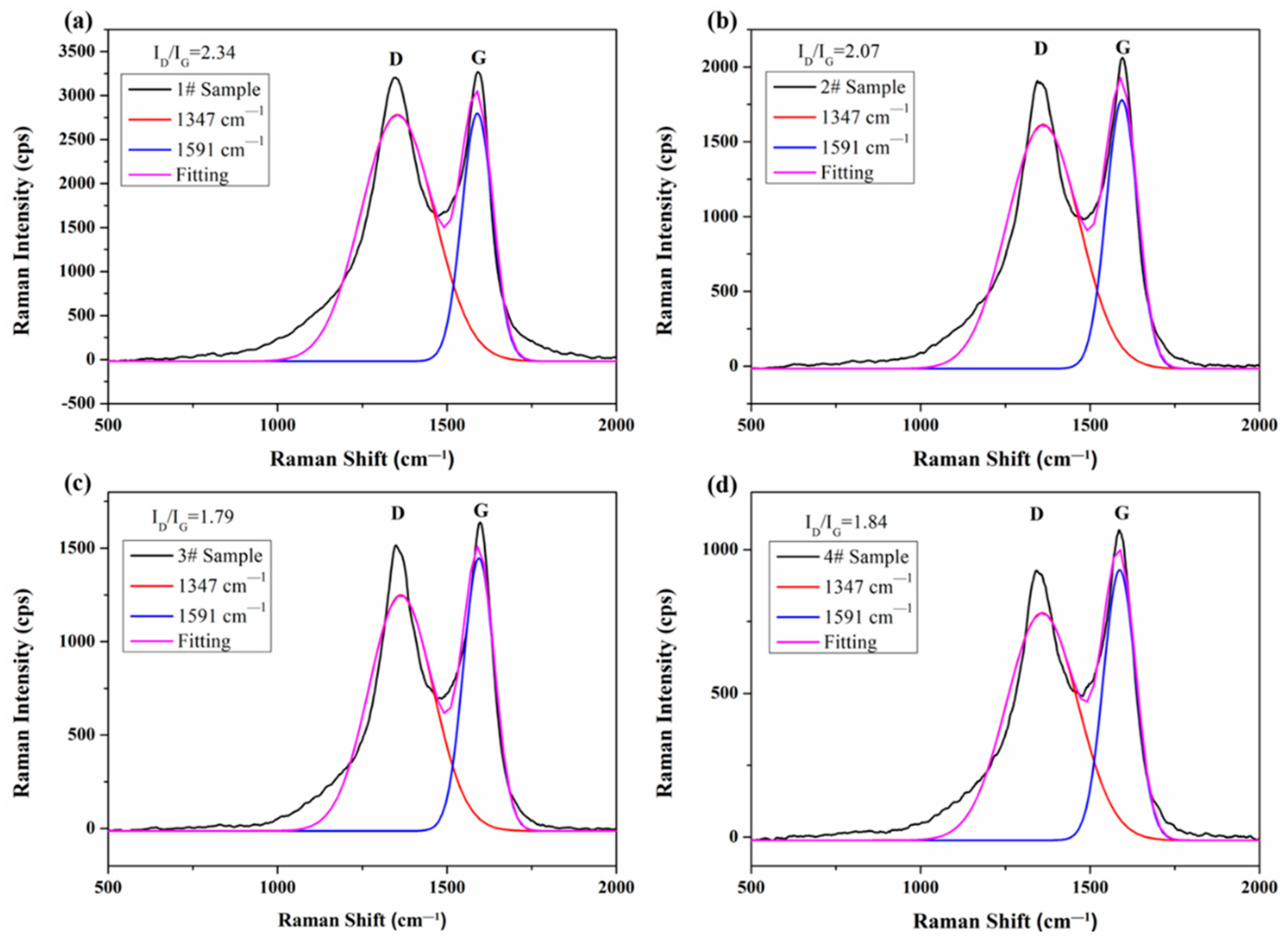
3.5. Raman Spectroscopy
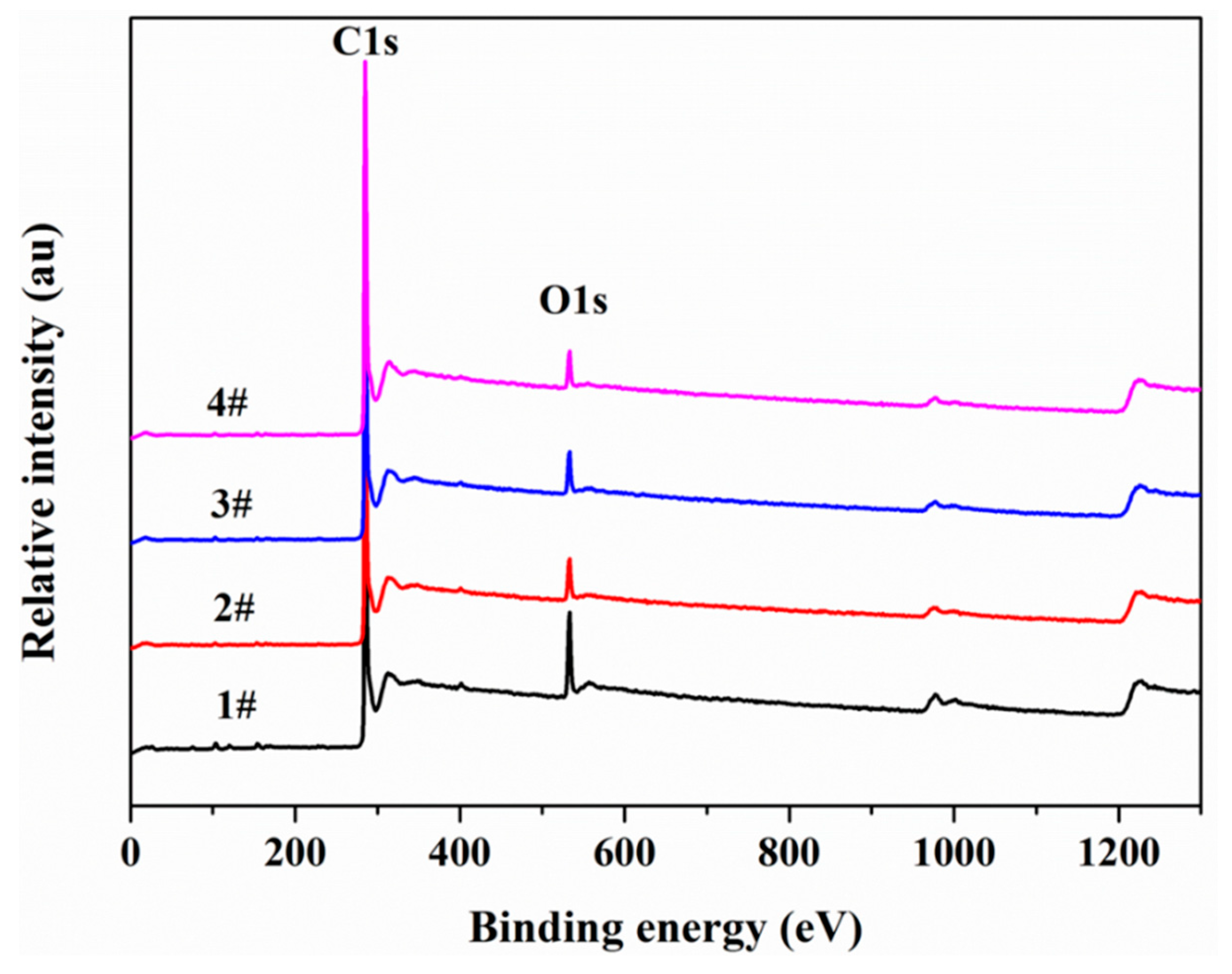
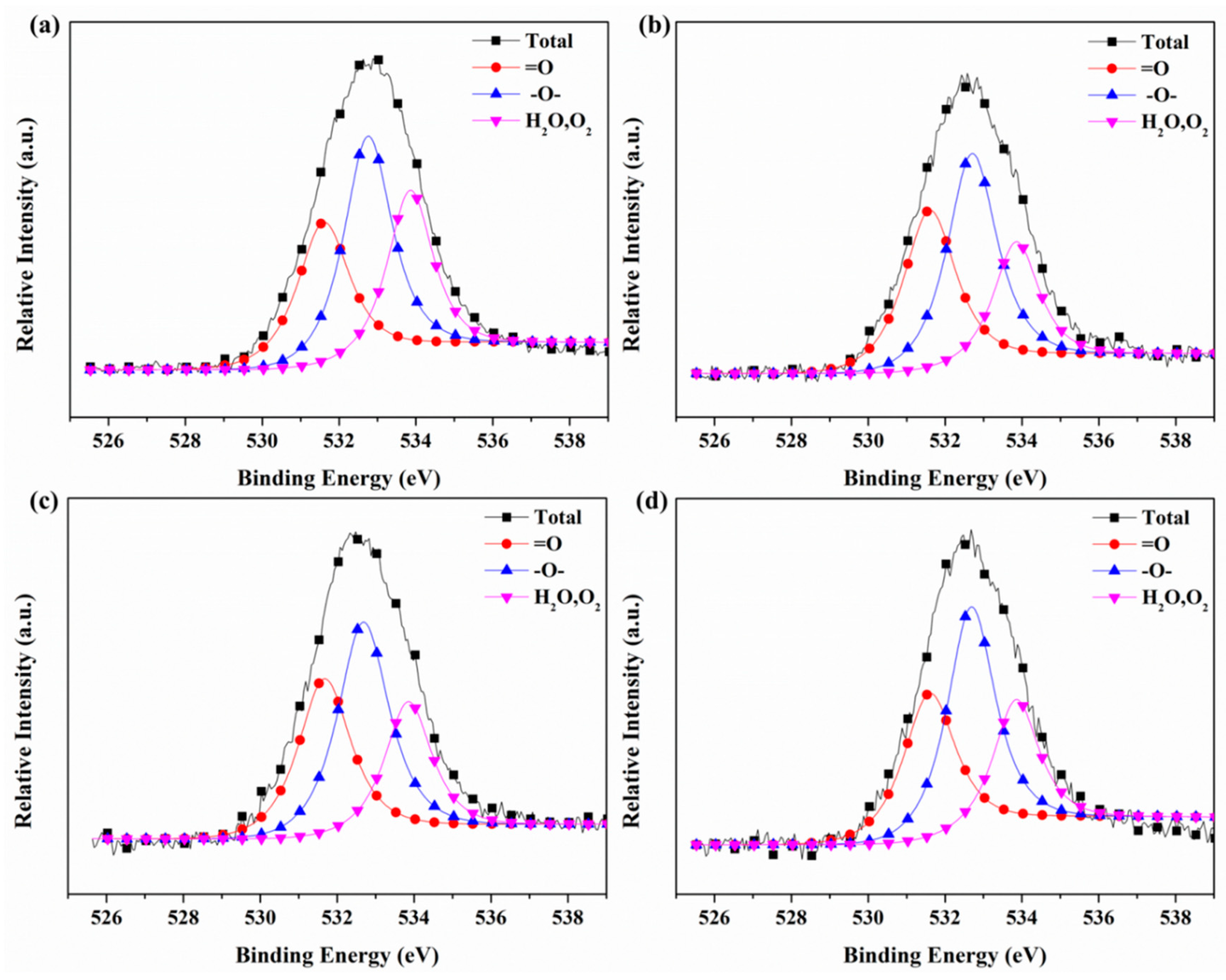
3.6. X-ray Photoelectron Spectroscopy (XPS)
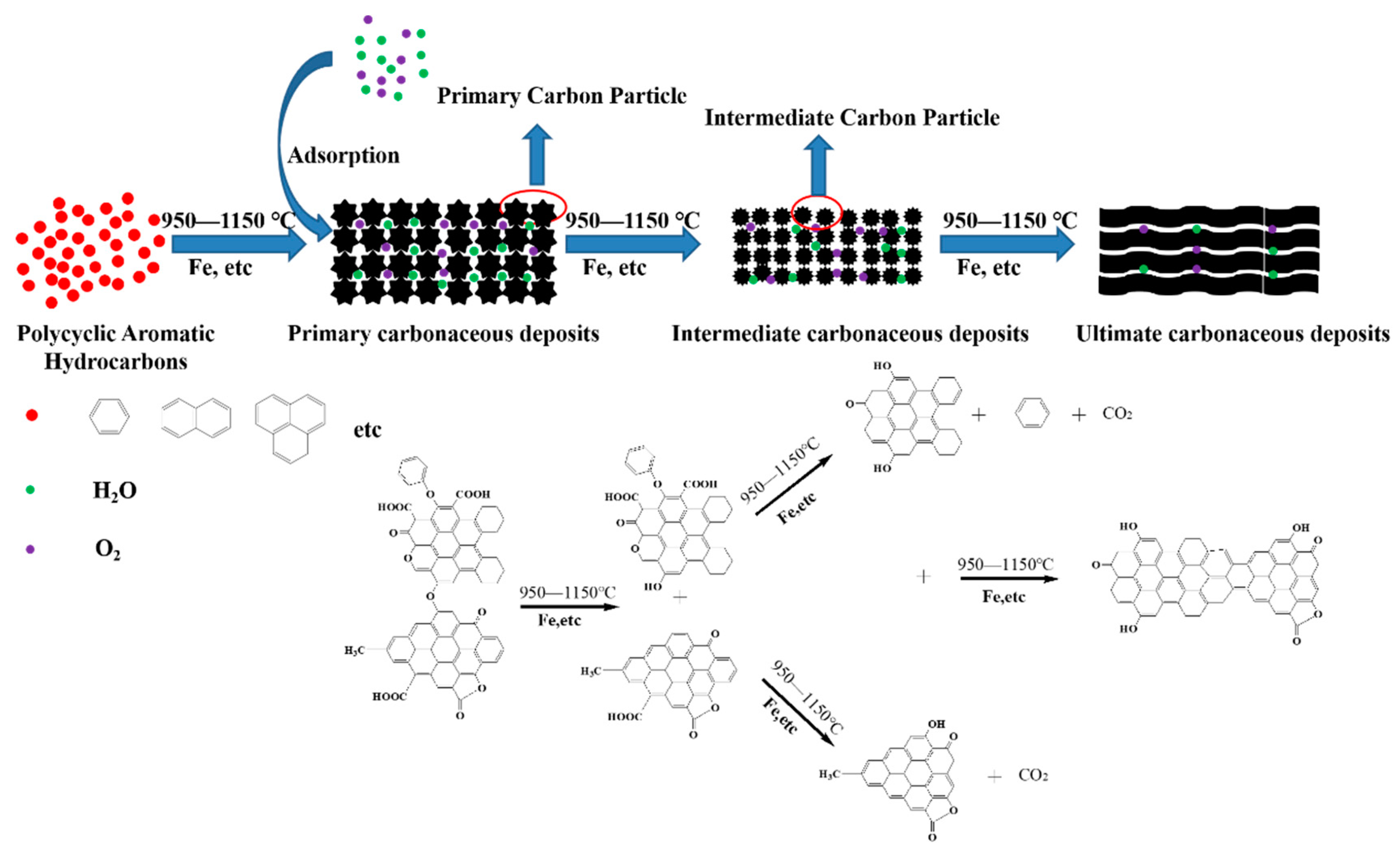
3.7. Mechanism Consideration
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Li, J.; Zhou, Y.; Simayi, M.; Deng, Y.Y.; Xie, S.D. Spatial-temporal variations and reduction potentials of volatile organic compound emissions from the coking industry in China. J. Clean. Prod. 2019, 214, 224–235. [Google Scholar] [CrossRef]
- Bai, X.F.; Ding, H.; Lian, J.J.; Ma, D.; Yang, X.Y.; Sun, N.X.; Xue, W.L.; Chang, Y.J. Coal production in China: Past, present, and future projections. Int. Geol. Rev. 2017, 60, 535–547. [Google Scholar] [CrossRef]
- Li, J.Y.; Ma, X.X.; Liu, H.; Zhang, X.Y. Life cycle assessment and economic analysis of methanol production from coke oven gas compared with coal and natural gas routes. J. Clean. Prod. 2018, 185, 299–308. [Google Scholar] [CrossRef]
- Terui, K.; Matsui, T.; Fukada, K.; Dohi, Y. Influence of Temperature Distribution in Combustion Chamber on Coke Cake Discharging Behavior. ISIJ Int. 2018, 58, 633–641. [Google Scholar] [CrossRef]
- Sobolewski, A.; Fitko, H. Individual regulation system of raw gas pressure in coke oven chambers. Przem. Chem. 2014, 93, 2115–2120. [Google Scholar]
- Zhang, R.D.; Gao, F.R. Multivariable decoupling predictive functional control with non-zero-pole cancellation and state weighting: Application on chamber pressure in a coke furnace. Chem. Eng. Sci. 2013, 94, 30–43. [Google Scholar] [CrossRef]
- Deng, J.; Zhao, J.Y.; Xiao, Y.; Zhang, Y.N.; Huang, A.C.; Shu, C.M. Thermal analysis of the pyrolysis and oxidation behaviour of 1/3 coking coal. J. Therm. Anal. Calorim. 2017, 129, 1779–1786. [Google Scholar] [CrossRef]
- Wu, Y.; Li, Y.; Jin, L.; Hu, H. Integrated process of coal pyrolysis with steam reforming of ethane for improving tar yield. Energy Fuels 2018, 32, 12268–12276. [Google Scholar] [CrossRef]
- Pretorius, G.N.; Bunt, J.R.; Gräbner, M.; Neomagus, H.; Waanders, F.B.; Everson, R.C.; Strydom, C.A. Evaluation and prediction of slow pyrolysis products derived from coals of different rank. J. Anal. Appl. Pyrolysis 2017, 128, 156–167. [Google Scholar] [CrossRef]
- Fidalgo, B.; van Niekerk, D.; Millan, M. The effect of syngas on tar quality and quantity in pyrolysis of a typical South African inertinite-rich coal. Fuel 2014, 134, 90–96. [Google Scholar] [CrossRef]
- Chen, X.; Zheng, D.; Guo, J.; Liu, J.; Ji, P. Energy analysis for low-rank coal based process system to co-produce semicoke, syngas and light oil. Energy 2013, 52, 279–288. [Google Scholar] [CrossRef]
- Marcin, S.; Ludwik, K. Evaluation of the energy consumption in coal coking by using a method for reconcilation of substance and energy balances. Przem. Chem. 2014, 93, 681–685. [Google Scholar]
- Uebo, K.; Kunimasa, H.; Suyama, S. Carbon deposition in a coke oven chamber at high productivity operation. Tetsu Hagane 2004, 90, 721–727. [Google Scholar] [CrossRef]
- Furusawa, A.; Nakagawa, T.; Maeno, Y.; Komaki, I. Influence of coal moisture control on carbon deposition in the coke oven chamber. ISIJ Int. 1998, 38, 1320–1325. [Google Scholar] [CrossRef][Green Version]
- Dumay, D.; Gaillet, J.P.; Krebs, V.; Furdin, G.; Mareché, J.F. Formation and reduction of carbon deposits in coke ovens. Rev. Metall. Cah. Inf. Tech. 1994, 91, 1109–1116. [Google Scholar]
- Krebs, V.; Furdin, G.; Mareché, J.; Dumay, D. Effects of coal moisture content on carbon deposition in coke ovens. Fuel 1996, 75, 979–986. [Google Scholar] [CrossRef]
- Zymla, V.; Honnart, F. Coke oven carbon deposits growth and their burning off. ISIJ Int. 2007, 47, 1422–1431. [Google Scholar] [CrossRef]
- Nakagava, T.; Kudo, T.; Kamada, Y.; Suzuki, T.; Komaki, I. Control of Carbon Deposition in the Free Space of Coke Oven Chamber by Injecting Atomized Water. Tetsu Hagane 2002, 88, 386–392. [Google Scholar] [CrossRef]
- Ando, T.; Kasaoka, S.; Onozawa, T.; Nakai, S. Coating Composition for Carbonization Chamber for Coke Oven and Application Method. U.S. Patent 6,165,923, 26 December 2000. [Google Scholar]
- Yu, J.W.; Qiao, K.; Zhu, B.; Cai, X.; Liu, D.W.; Li, M.J.; Yuan, X.M. Structural research of activated carbon fibers during a novel phosphoric acid reactivation process assisted by sonication. Funct. Mater. Lett. 2018, 11, 1850066. [Google Scholar] [CrossRef]
- Liu, C.; Xiao, N.; Wang, Y.W.; Li, H.Q.; Wang, G.; Dong, Q.; Bai, J.P.; Xiao, J.; Qiu, J.S. Carbon clusters decorated hard carbon nanofibers as high-rate anode material for lithium-ion batteries. Fuel Process. Technol. 2018, 180, 173–179. [Google Scholar] [CrossRef]
- Gao, R.T.; Liu, B.Y.; Xu, Z.M. Fabrication of magnetic zeolite coated with carbon fiber using pyrolysis products from waste printed circuit boards. J. Clean. Prod. 2019, 231, 1149–1157. [Google Scholar] [CrossRef]
- Zheng, J.W.; Song, F.F.; Che, S.L.; Li, W.C.; Ying, Y.; Yu, J.; Qiao, L. One step synthesis process for fabricating NiFe2O4 nanoparticle loaded porous carbon spheres by ultrasonic spray pyrolysis. Adv. Powder Technol. 2018, 29, 1474–1480. [Google Scholar] [CrossRef]
- Sonibare, O.O.; Haeger, T.; Foley, S.F. Structural characterization of Nigerian coals by X-ray diffraction, Raman and FTIR spectroscopy. Energy 2010, 35, 5347–5353. [Google Scholar] [CrossRef]
- Chutia, S.; Narzari, R.; Bordoloi, N.; Saikia, R.; Gogoi, L.; Sut, D.; Bhuyan, N.; Kataki, R. Pyrolysis of dried black liquor solids and characterization of the bio-char and bio-oil. Mater. Today Proc. 2018, 5, 13193–23202. [Google Scholar] [CrossRef]
- Wang, K.; Deng, J.; Zhang, Y.N.; Wang, C.P. Kinetics and mechanisms of coal oxidation mass gain phenomenon by TG–FTIR and in situ IR analysis. J. Therm. Anal. Calorim. 2018, 132, 591–598. [Google Scholar] [CrossRef]
- Wang, S.G.; Gao, R.; Zhou, K.Q. The influence of cerium dioxide functionalized reduced graphene oxide on reducing fire hazards of thermoplastic polyurethane nanocomposites. J. Colloid Interface Sci. 2019, 536, 127–134. [Google Scholar] [CrossRef]
- Tang, G.; Zhang, R.; Wang, X.; Wang, B.; Song, L.; Hu, Y.; Gong, X. Enhancement of flame retardant performance of bio-based polylactic acid composites with the incorporation of aluminum hypophosphite andexpanded graphite. J. Macromol. Sci. Part A 2013, 50, 255–269. [Google Scholar] [CrossRef]
- Krawczyk, P. Effect of ozone treatment on properties of expanded graphite. Chem. Eng. J. 2011, 172, 1096–1102. [Google Scholar] [CrossRef]
- Bourbigot, S.; Le Bras, M.; Delobel, R.; Gengembre, L. XPS study of an intumescent coating: II. Application to the ammonium polyphosphate/pentaerythritol/ethylenic terpolymer fire retardant system with and without synergistic agent. Appl. Surf. Sci. 1997, 120, 15–29. [Google Scholar] [CrossRef]
- Zhang, P.; Hu, Y.; Song, L.; Ni, J.X.; Xing, W.Y.; Wang, J. Effect of expanded graphite on properties of high-density polyethylene/paraffin composite with intumescent flame retardant as a shape-stabilized phase change material. Sol. Energy Mater. Sol. Cells 2010, 94, 360–365. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Composition | Content (wt%) | Standard Error (wt%) |
|---|---|---|
| SO2 | 34.51 | 0.24 |
| SiO2 | 30.54 | 0.23 |
| Al2O3 | 19.22 | 0.20 |
| Fe2O3 | 5.60 | 0.11 |
| CaO | 1.74 | 0.07 |
| ZnO | 1.44 | 0.06 |
| Cr2O3 | 1.15 | 0.05 |
| MnO | 1.15 | 0.05 |
| Cl | 1.05 | 0.05 |
| MgO | 0.725 | 0.036 |
| TiO2 | 0.686 | 0.034 |
| Na2O | 0.595 | 0.037 |
| K2O | 0.591 | 0.029 |
| P2O5 | 0.574 | 0.029 |
| Sample | 2θ002 | 2θ100 | d002/nm | FWHM002/nm | La/nm | FWHM100/nm | Lc/nm | N |
|---|---|---|---|---|---|---|---|---|
| #1 | 25.92 | 42.98 | 0.3437 | 0.855 | 30.81 | 0.567 | 9.97 | 30 |
| #2 | 25.76 | 42.80 | 0.3458 | 0.865 | 27.84 | 0.627 | 9.85 | 29 |
| #3 | 25.68 | 42.64 | 0.3468 | 0.953 | 35.18 | 0.496 | 8.94 | 27 |
| #4 | 25.58 | 42.97 | 0.3482 | 0.965 | 36.47 | 0.479 | 8.82 | 26 |
| Sample | C (%) | O (%) | N | Al | Si | Ca | Fe | S | P | C/O | C/N |
|---|---|---|---|---|---|---|---|---|---|---|---|
| #1 | 82.41 | 10.91 | 2.20 | 1.12 | 1.73 | 0.50 | 0.37 | 0.54 | 0.21 | 7.55 | 37.46 |
| #2 | 89.43 | 6.20 | 1.49 | 0.76 | 0.91 | 0.27 | 0.28 | 0.43 | 0.22 | 14.42 | 60.02 |
| #3 | 89.69 | 6.32 | 1.17 | 0.77 | 0.72 | 0.30 | 0.31 | 0.46 | 0.26 | 14.19 | 76.66 |
| #4 | 91.51 | 5.14 | 0.76 | 0.85 | 0.62 | 0.39 | 0.25 | 0.23 | 0.24 | 17.80 | 120.39 |
| Sample | C–C/C–H | C–O/C–N | C=O/C=N |
|---|---|---|---|
| 284.7 eV | 285.5 eV | 287.4 eV | |
| #1 | 69.73% | 21.68% | 8.60% |
| #2 | 64.59% | 25.87% | 9.54% |
| #3 | 66.60% | 24.51% | 8.89% |
| #4 | 69.46% | 21.67% | 8.87% |
| Sample | =O | –O– | O2/H2O |
|---|---|---|---|
| 531.6 eV | 532.8 eV | 533.8 eV | |
| #1 | 30.10% | 42.10% | 27.80% |
| #2 | 35.16% | 43.39% | 21.44% |
| #3 | 33.48% | 43.19% | 23.33% |
| #4 | 32.43% | 44.89% | 22.68% |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, H.; Jin, B.; Wang, X.; Tang, G. Formation and Evolution Mechanism for Carbonaceous Deposits on the Surface of a Coking Chamber. Processes 2019, 7, 508. https://doi.org/10.3390/pr7080508
Wang H, Jin B, Wang X, Tang G. Formation and Evolution Mechanism for Carbonaceous Deposits on the Surface of a Coking Chamber. Processes. 2019; 7(8):508. https://doi.org/10.3390/pr7080508
Chicago/Turabian StyleWang, Hao, Baosheng Jin, Xiaojia Wang, and Gang Tang. 2019. "Formation and Evolution Mechanism for Carbonaceous Deposits on the Surface of a Coking Chamber" Processes 7, no. 8: 508. https://doi.org/10.3390/pr7080508
APA StyleWang, H., Jin, B., Wang, X., & Tang, G. (2019). Formation and Evolution Mechanism for Carbonaceous Deposits on the Surface of a Coking Chamber. Processes, 7(8), 508. https://doi.org/10.3390/pr7080508