Workflow for Data Analysis in Experimental and Computational Systems Biology: Using Python as ‘Glue’

 ,
,  , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Methods
2.1. Python Libraries and Applications
- numpy [13], a numerical processing library that supports multi-dimensional arrays;
- scipy [14], a scientific processing library providing advanced tools for data analysis, including regression, ODE solvers and integrators, linear algebra and statistical functions;
- pandas [15], a data and table manipulation library that offers similar functionality to spreadsheets such as Excel; and
- matplotlib [16], a plotting library with tools to display data in a variety of ways.
2.1.1. PySCeS
2.1.2. NMRPy
2.1.3. Jupyter Notebook as Software Platform
2.2. Experimental Protocols
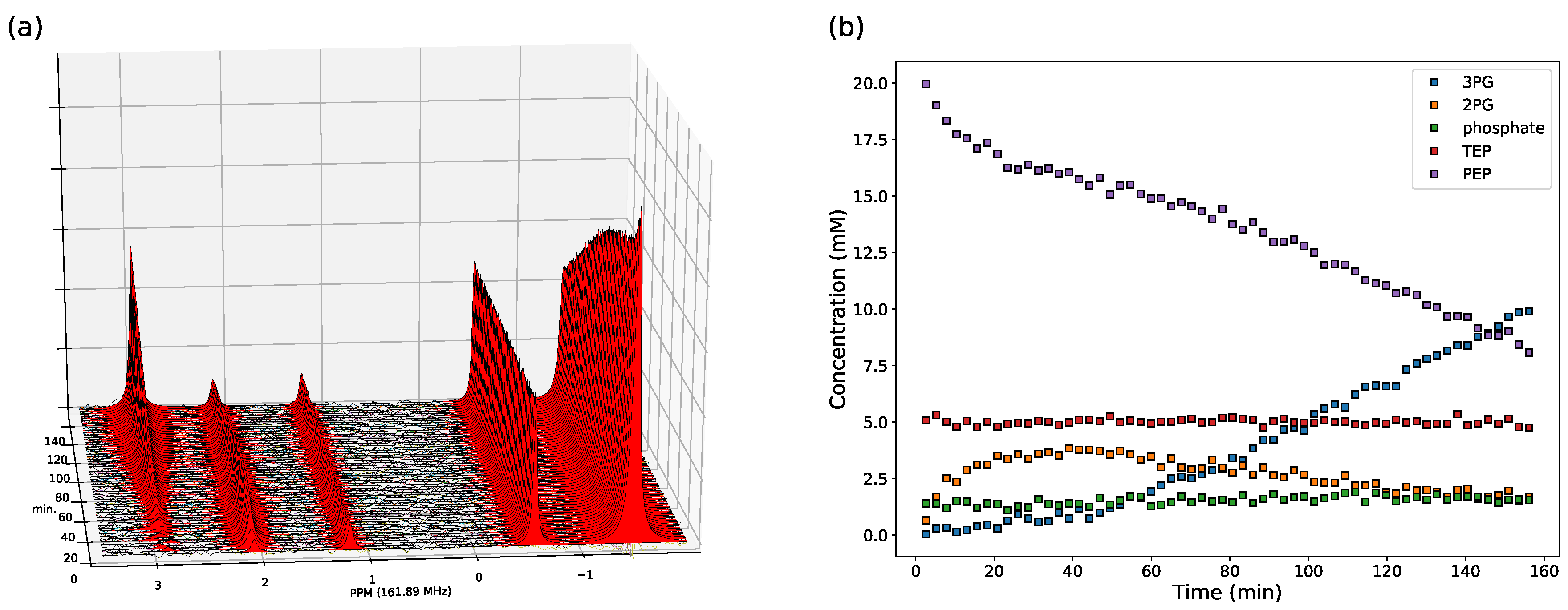
2.2.1. NMR Spectroscopy Assays
2.2.2. Spectrophotometric Assays
2.2.3. Fitting Experimental Data to Obtain Kinetic Parameters
2.2.4. Validation Data
3. Results
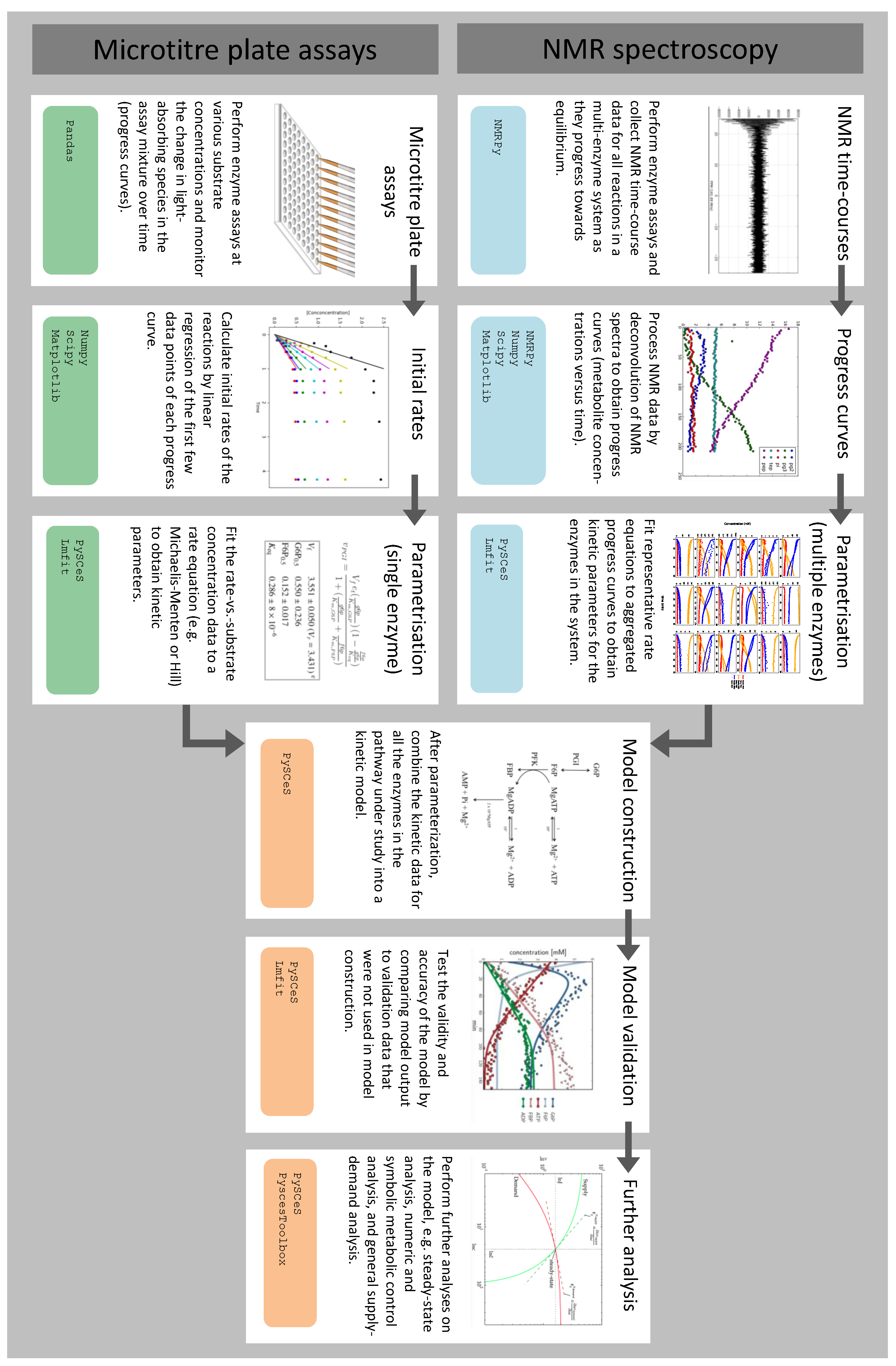
3.1. Summary of Workflow for Kinetic Model Construction
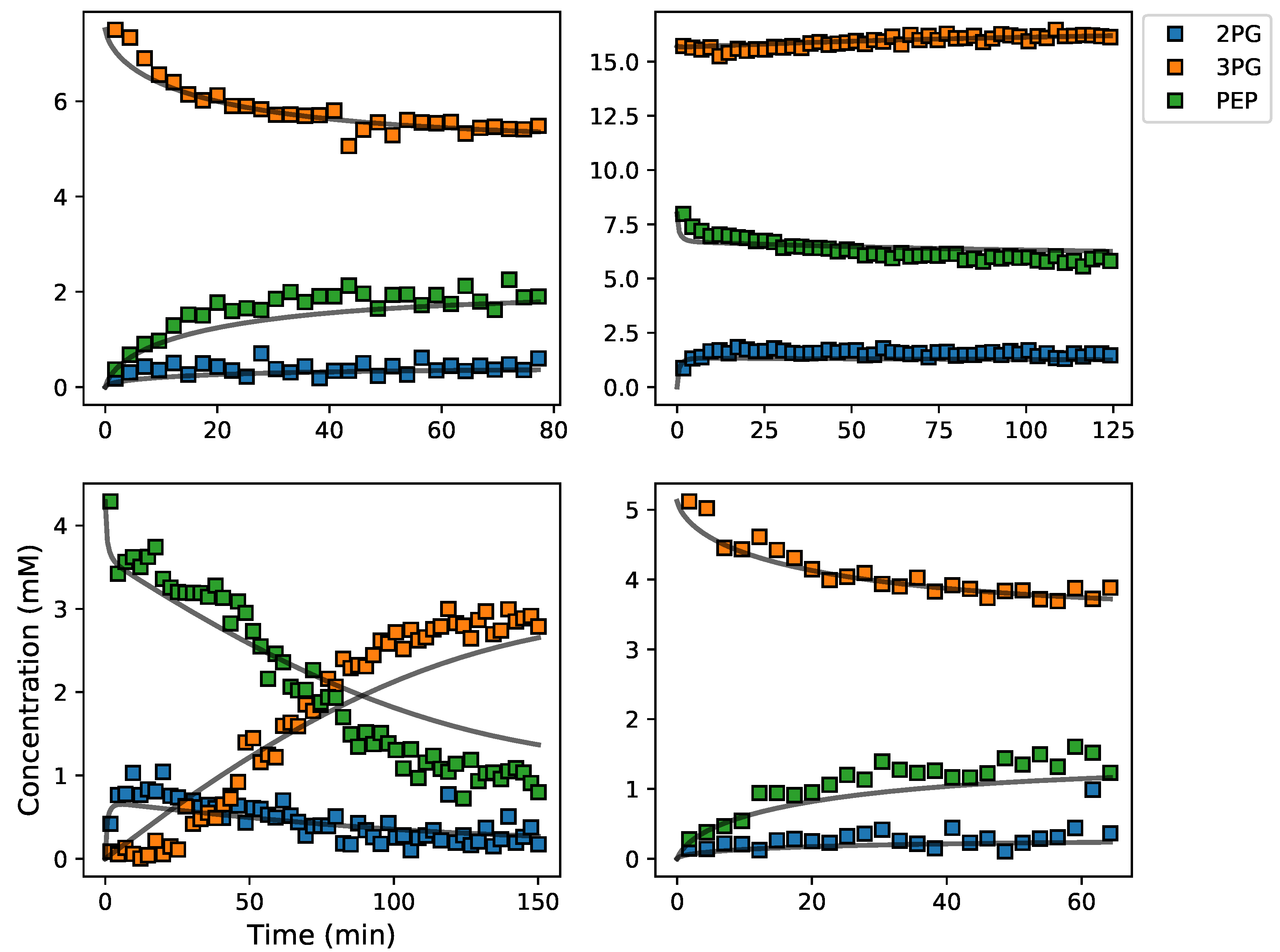
3.2. Enzyme Kinetics from NMR Spectroscopy
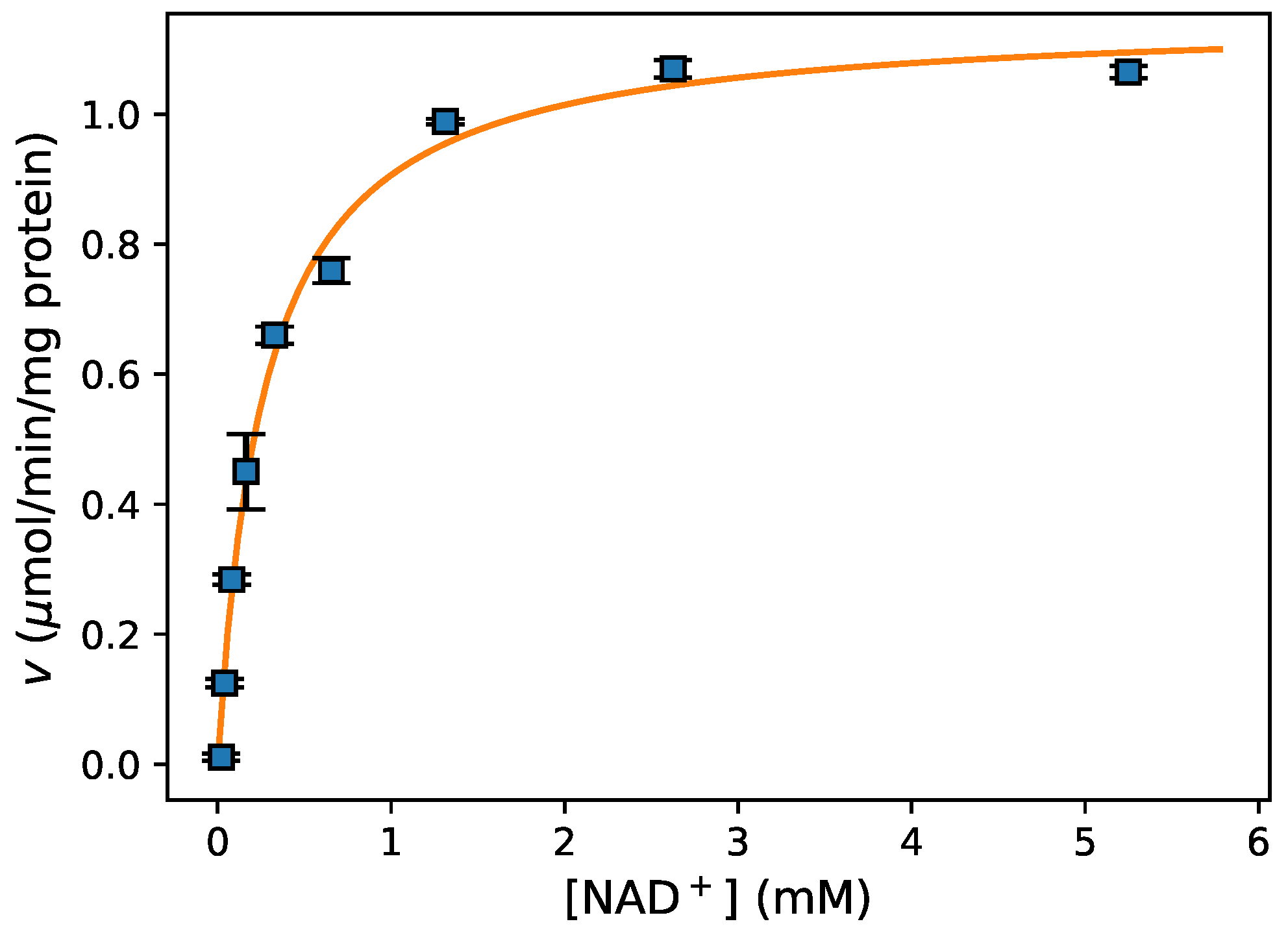
3.3. Enzyme Kinetics from Spectrophotometric Assays
- Importing data New dataframes were created in pandas from a variety of input formats, including Excel and CSV. Several preprocessing and customisation methods were used (e.g., for the conversion of date/time fields into a format that can be used by Python), as near-perfect tabulated data are rarely produced by the associated software and the formats differ between instrument vendors.
- Linear regression The absorbance-versus-time data were subject to linear regression over a suitable time range to calculate initial rates. The attached Jupyter notebook provides two tools (using interactive matplotlib graphs, and using ipywidgets), which were used to efficiently apply this analysis to a large number of datasets.
- Preprocessing data The pandas library provided functions to easily normalise the data, either to a single entry, a single row, or an entire dataframe. Further, Python functions were written to automate repetitive processing tasks in a consistent way. Examples of such normalisations included subtraction of blank readings, the conversion of absorbance values to concentrations, or the subtraction of the initial time reading from subsequent time data.
- Fitting data For fitting of initial rate data to an enzyme-kinetic rate equation (e.g., the Michaelis–Menten equation), the Python package lmfit [24] provided a high-level interface to various non-linear optimization and curve fitting routines with access to both global and local optimisation algorithms. This is further discussed in Section 3.4 below.
- Data Visualization A leading visualisation and 2D-plotting library for Python is matplotlib, which was used in this analysis because of its powerful and flexible design, its interoperability with numpy and scipy, and its excellent integration into Jupyter notebooks. Figure 3 shows the experimental data and model fit (see below) for this experiment.
3.4. Fitting Experimental Data to Obtain Kinetic Parameters
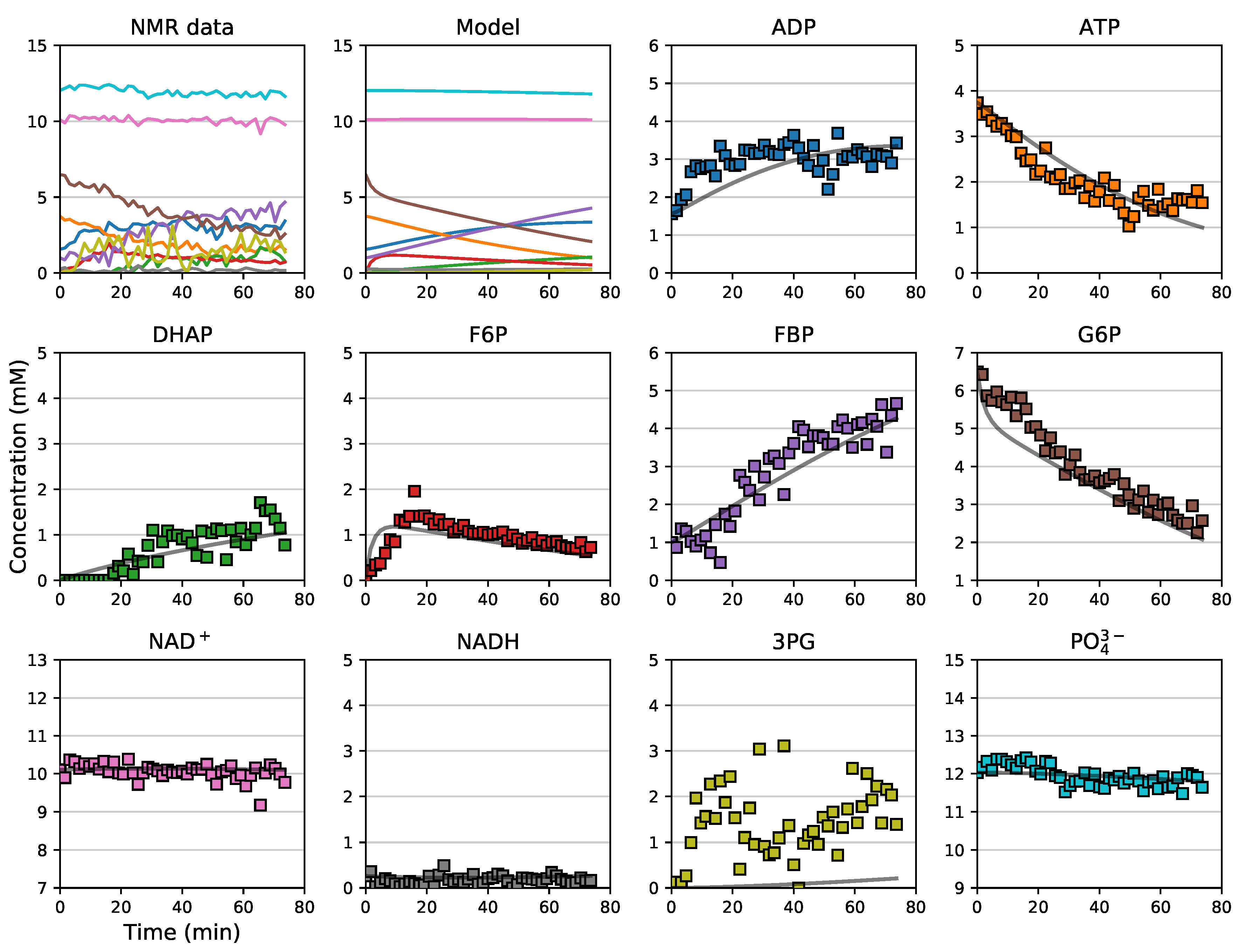
3.5. Assembly and Validation of a Larger Kinetic Model of a Pathway
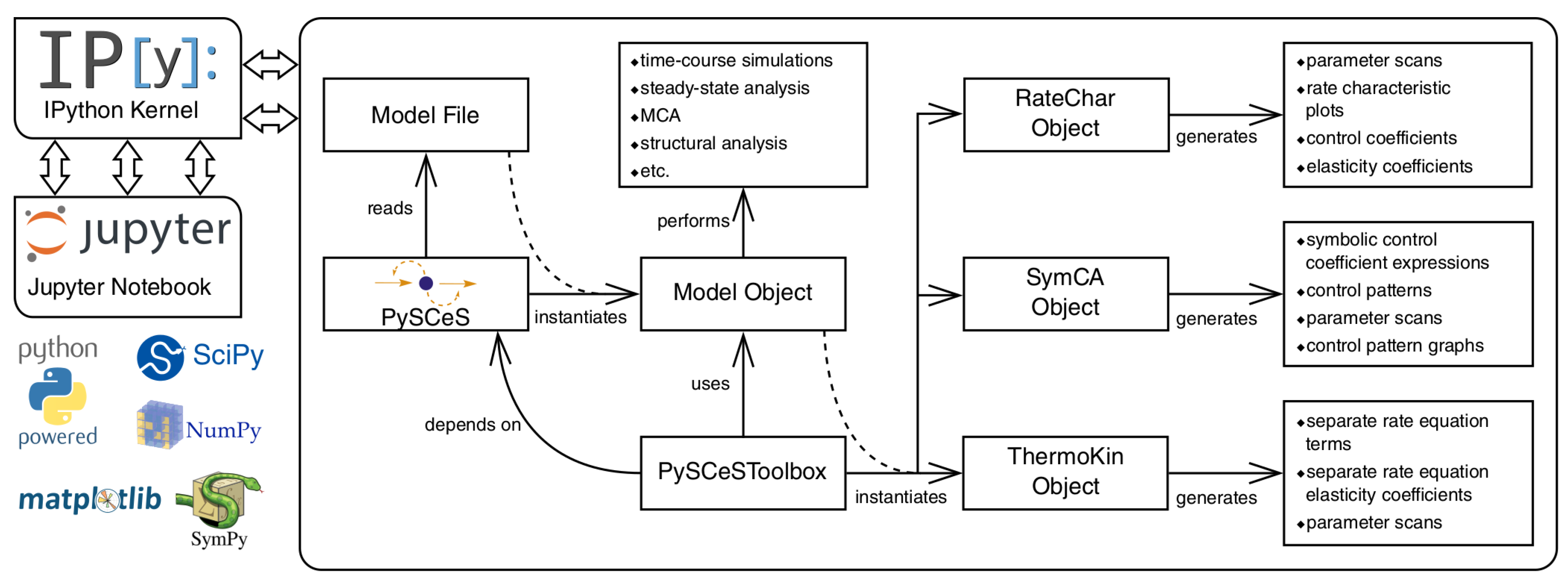
3.6. Further Model Analysis: MCA, GSDA and PyscesToolbox
- RateChar This module performs GSDA by fixing each variable metabolite in turn (thus making it a system parameter) and varying it below and above its steady-state value. This allows one to identify regulatory metabolites as well as routes of regulation in the network. This approach was computationally applied [36] to the analysis of published models of pyruvate metabolism in Lactococcus lactis [37] and aspartate-derived amino acid synthesis in Arabidopsis thaliana [38].
- SymCA This module performs symbolic metabolic control analysis by generating algebraic expressions for the control coefficients in terms of the elasticity coefficients, using the SymPy Python module for symbolic algebra [39]. These expressions are then used to evaluate and visualise so-called control patterns in the network and quantify their relative contribution to the overall value of the control coefficient. A control coefficient can thus be dissected into its most important components.
- ThermoKin This module calculates, for each reversible reaction in the model, the contribution of thermodynamics and kinetics to the enzyme regulation at a particular steady state using the formalism described in [34]. This contribution may vary as conditions change (e.g., as a result of changes in some model parameters), as the reaction operates closer to or further away from equilibrium.
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| API | Application Programming Interface |
| ENO | Enolase |
| FID | Free Induction Decay |
| GSDA | Generalised Supply-Demand Analysis |
| JSON | JavaScript Object Notation |
| MCA | Metabolic Control Analysis |
| NMR | Nuclear Magnetic Resonance |
| ODE | Ordinary Differential Equation |
| OSI | Open Source Initiative |
| PGM | Phosphoglycerate Mutase |
| SBML | Systems Biology Markup Language |
| SDA | Supply-Demand Analysis |
References
- Kitano, H. International alliances for quantitative modeling in systems biology. Mol. Syst. Biol. 2005, 1, 2005.0007. [Google Scholar] [CrossRef] [PubMed]
- Westerhoff, H.V.; Alberghina, L. Systems Biology: Did we know it all along? In Systems Biology; Alberghina, L., Westerhoff, H.V., Eds.; Springer: Berlin, Germany, 2005; pp. 3–9. [Google Scholar] [CrossRef]
- Snoep, J.L.; Bruggeman, F.; Olivier, B.G.; Westerhoff, H.V. Towards building the silicon cell: A modular approach. Biosystems 2006, 83, 207–216. [Google Scholar] [CrossRef] [PubMed]
- Bruggeman, F.J.; Westerhoff, H.V. The nature of systems biology. Trends Microbiol. 2007, 15, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Rohwer, J.M.; Hanekom, A.J.; Crous, C.; Snoep, J.L.; Hofmeyr, J.H.S. Evaluation of a simplified generic bi-substrate rate equation for computational systems biology. IEE Proc. Syst. Biol. 2006, 153, 338–341. [Google Scholar] [CrossRef]
- Rohwer, J.M.; Hanekom, A.J.; Hofmeyr, J.H.S. A universal rate equation for systems biology. Experimental Standard Conditions of Enzyme Characterizations. In Proceedings of the 2nd International Beilstein Workshop; Hicks, M.G., Kettner, C., Eds.; Beilstein-Institut zur Förderung der Chemischen Wissenschaften: Frankfurt, Germany, 2007; pp. 175–187. [Google Scholar]
- Rohwer, J.M. Kinetic modelling of plant metabolic pathways. J. Exp. Bot. 2012, 63, 2275–2292. [Google Scholar] [CrossRef]
- Ingalls, B. Mathematical Modelling in Systems Biology: An Introduction; MIT Press: Cambridge, MA, USA, 2012; p. 386. [Google Scholar]
- Jaqaman, K.; Danuser, G. Linking data to models: Data regression. Nat. Rev. Mol. Cell Biol. 2006, 7, 813–819. [Google Scholar] [CrossRef]
- John, R.A. Photometric assays. In Enzyme Assays. A Practical Approach, 2nd ed.; Eisenthal, R., Danson, M.J., Eds.; Oxford University Press: Oxford, UK, 2002; Chapter 2; pp. 49–78. [Google Scholar]
- Welling, G.W.; Scheffer, A.J.; Welling-Wester, S. Determination of enzyme activity by high-performance liquid chromatography. J. Chromatogr. B 1994, 659, 209–225. [Google Scholar] [CrossRef]
- Eicher, J.J.; Snoep, J.L.; Rohwer, J.M. Determining enzyme kinetics for systems biology with Nuclear Magnetic Resonance spectroscopy. Metabolites 2012, 2, 818–843. [Google Scholar] [CrossRef]
- Van der Walt, S.; Colbert, S.C.; Varoquaux, G. The NumPy array: A structure for efficient numerical computation. Comput. Sci. Eng. 2011, 13, 22–30. [Google Scholar] [CrossRef]
- Jones, E.; Oliphant, T.; Peterson, P. SciPy: Open Source Scientific Tools for Python. 2001. Available online: http://www.scipy.org/ (accessed on 12 July 2019).
- McKinney, W. Data Structures for Statistical Computing in Python. In Proceedings of the 9th Python in Science Conference, Austin, TX, USA, 28 June–3 July 2010; pp. 51–56. [Google Scholar]
- Hunter, J.D. Matplotlib: A 2D graphics environment. Comput. Sci. Eng. 2007, 9, 90–95. [Google Scholar] [CrossRef]
- Anaconda Software Distribution. Version 2-2.4.0. Computer Software. 2017. Available online: https://www.anaconda.com (accessed on 12 July 2019).
- Olivier, B.G.; Rohwer, J.M.; Hofmeyr, J.H.S. Modelling cellular systems with PySCeS. Bioinformatics 2005, 21, 560–561. [Google Scholar] [CrossRef] [PubMed]
- Hucka, M.; Finney, A.; Sauro, H.M.; Bolouri, H.; Doyle, J.C.; Kitano, H.; Arkin, A.P.; Bornstein, B.J.; Bray, D.; Cornish-Bowden, A.; et al. The systems biology markup language (SBML): A medium for representation and exchange of biochemical network models. Bioinformatics 2003, 19, 524–531. [Google Scholar] [CrossRef] [PubMed]
- Eicher, J.J. Understanding Glycolysis in Escherichia coli: A Systems Approach using Nuclear Magnetic Resonance Spectroscopy. Ph.D. Thesis, Stellenbosch University, Stellenbosch, South Africa, 2013. [Google Scholar]
- Pérez, F.; Granger, B.E. IPython: A system for interactive scientific computing. Comput. Sci. Eng. 2007, 9, 21–29. [Google Scholar] [CrossRef]
- Kluyver, T.; Ragan-Kelley, B.; Pérez, F.; Granger, B.E.; Bussonnier, M.; Frederic, J.; Kelley, K.; Hamrick, J.B.; Grout, J.; Corlay, S.; et al. Jupyter Notebooks—A publishing format for reproducible computational workflows. In Positioning and Power in Academic Publishing: Players, Agents and Agendas, Proceedings of the 20th International Conference on Electronic Publishing, Göttingen, Germany, June 2016; Loizides, F., Schmidt, B., Eds.; IOS Press: Amsterdam, The Netherlands, 2016; pp. 87–90. [Google Scholar] [CrossRef]
- Swanepoel, C.J. A systematic Investigation into the Quantitative Effect of pH Changes on the Upper Glycolytic Enzymes of Escherichia coli and Saccharomyces cerevisiae. Master’s Thesis, Stellenbosch University, Stellenbosch, South Africa, 2018. [Google Scholar]
- Newville, M.; Stensitzki, T.; Allen, D.B.; Ingargiola, A. LMFIT: Non-linear least-square minimization and curve-fitting for Python. Zenodo 2014. [Google Scholar] [CrossRef]
- Van Eunen, K.; Bouwman, J.; Daran-Lapujade, P.; Postmus, J.; Canelas, A.B.; Mensonides, F.I.C.; Orij, R.; Tuzun, I.; van den Brink, J.; Smits, G.J.; et al. Measuring enzyme activities under standardized in vivo-like conditions for systems biology. FEBS J. 2010, 277, 749–760. [Google Scholar] [CrossRef]
- García-Contreras, R.; Vos, P.; Westerhoff, H.V.; Boogerd, F.C. Why in vivo may not equal in vitro—New effectors revealed by measurement of enzymatic activities under the same in vivo-like assay conditions. FEBS J. 2012, 279, 4145–4159. [Google Scholar] [CrossRef] [PubMed]
- Kacser, H.; Burns, J.A. The control of flux. Symp. Soc. Exp. Biol. 1973, 27, 65–104. [Google Scholar] [CrossRef] [PubMed]
- Heinrich, R.; Rapoport, T.A. A linear steady-state treatment of enzymatic chains. General properties, control and effector strength. Eur. J. Biochem. 1974, 42, 89–95. [Google Scholar] [CrossRef]
- Hofmeyr, J.H.S.; Cornish-Bowden, A. Regulating the cellular economy of supply and demand. FEBS Lett. 2000, 476, 47–51. [Google Scholar] [CrossRef]
- Hofmeyr, J.H.S.; Rohwer, J.M. Supply-demand analysis: A framework for exploring the regulatory design of metabolism. Methods Enzymol. 2011, 500, 533–554. [Google Scholar] [CrossRef]
- Rohwer, J.M.; Hofmeyr, J.H.S. Identifying and characterising regulatory metabolites with generalised supply-demand analysis. J. Theor. Biol. 2008, 252, 546–554. [Google Scholar] [CrossRef]
- Reder, C. Metabolic control theory: A structural approach. J. Theor. Biol. 1988, 135, 175–201. [Google Scholar] [CrossRef]
- Hofmeyr, J.H.S. Metabolic control analysis in a nutshell. In Proceedings of the 2nd International Conference on Systems Biology, Pasadena, CA, USA, 5–7 November 2001; Yi, T.M., Hucka, M., Morohashi, M., Kitano, H., Eds.; Omnipress: Madison, WI, USA, 2001; pp. 291–300. [Google Scholar]
- Rohwer, J.M.; Hofmeyr, J.H.S. Kinetic and thermodynamic aspects of enzyme control and regulation. J. Phys. Chem. B 2010, 114, 16280–16289. [Google Scholar] [CrossRef] [PubMed]
- Christensen, C.D.; Hofmeyr, J.H.S.; Rohwer, J.M. PySCeSToolbox: A collection of metabolic pathway analysis tools. Bioinformatics 2018, 34, 124–125. [Google Scholar] [CrossRef] [PubMed]
- Christensen, C.D.; Hofmeyr, J.H.S.; Rohwer, J.M. Tracing regulatory routes in metabolism using generalised supply-demand analysis. BMC Syst. Biol. 2015, 9, 89. [Google Scholar] [CrossRef] [PubMed]
- Hoefnagel, M.H.N.; Starrenburg, M.J.C.; Martens, D.E.; Hugenholtz, J.; Kleerebezem, M.; Swam, I.I.V.; Bongers, R.; Westerhoff, H.V.; Snoep, J.L. Metabolic engineering of lactic acid bacteria, the combined approach: Kinetic modelling, metabolic control and experimental analysis. Microbiology 2002, 148, 1003–1013. [Google Scholar] [CrossRef] [PubMed]
- Curien, G.; Bastien, O.; Robert-Genthon, M.; Cornish-Bowden, A.; Cárdenas, M.L.; Dumas, R. Understanding the regulation of aspartate metabolism using a model based on measured kinetic parameters. Mol. Syst. Biol. 2009, 5, 271. [Google Scholar] [CrossRef]
- Meurer, A.; Smith, C.P.; Paprocki, M.; Čertík, O.; Kirpichev, S.B.; Rocklin, M.; Kumar, A.; Ivanov, S.; Moore, J.K.; Singh, S.; et al. SymPy: Symbolic computing in Python. PeerJ Comput. Sci. 2017, 3, e103. [Google Scholar] [CrossRef]
- Christensen, C.D.; Hofmeyr, J.H.S.; Rohwer, J.M. Delving deeper: Relating the behaviour of a metabolic system to the properties of its components using symbolic metabolic control analysis. PLoS ONE 2018, 13, e0207983. [Google Scholar] [CrossRef]
- Olivier, B.G.; Rohwer, J.M.; Hofmeyr, J.H.S. Modelling cellular processes with Python and SciPy. Mol. Biol. Rep. 2002, 29, 249–254. [Google Scholar] [CrossRef]
- Ashyraliyev, M.; Fomekong-Nanfack, Y.; Kaandorp, J.A.; Blom, J.G. Systems biology: Parameter estimation for biochemical models. FEBS J. 2009, 276, 886–902. [Google Scholar] [CrossRef] [PubMed]
- Cedersund, G.; Roll, J. Systems biology: Model based evaluation and comparison of potential explanations for given biological data. FEBS J. 2009, 276, 903–922. [Google Scholar] [CrossRef] [PubMed]
- Ekmekci, B.; Mcanany, C.E.; Mura, C. An Introduction to Programming for Bioscientists: A Python-Based Primer. PLoS Comput. Biol. 2016, 12, e1004867. [Google Scholar] [CrossRef] [PubMed]
- Hinsen, K. High-level scientific programming with Python. In Proceedings of the International Conference on Computational Science—Part III, Amsterdam, The Netherlands, 21–24 April 2002; Sloot, P.M., Tan, C.J.K., Dongarra, J., Hoekstra, A.G., Eds.; Springer: Berlin/Heidelberg, Germany, 2002; pp. 691–700. [Google Scholar]
- Peterson, P. F2PY: A tool for connecting Fortran and Python programs. Int. J. Comput. Sci. Eng. 2009, 4, 296. [Google Scholar] [CrossRef]
- Dalcin, L.; Bradshaw, R.; Smith, K.; Citro, C.; Behnel, S.; Seljebotn, D. Cython: The best of both worlds. Comput. Sci. Eng. 2011, 13, 31–39. [Google Scholar] [CrossRef]
- Choi, K.; Medley, J.K.; Cannistra, C.; König, M.; Smith, L.; Stocking, K.; Sauro, H.M. Tellurium: A Python based modeling and reproducibility platform for systems biology. bioRxiv 2016. Available online: https://www.biorxiv.org/content/early/2016/06/02/054601.full.pdf (accessed on 12 July 2019). [CrossRef]
- Somogyi, E.T.; Bouteiller, J.M.; Glazier, J.A.; König, M.; Medley, J.K.; Swat, M.H.; Sauro, H.M. libRoadRunner: A high performance SBML simulation and analysis library. Bioinformatics 2015, 31, 3315–3321. [Google Scholar] [CrossRef]
- Ebenhöh, O.; van Aalst, M.; Saadat, N.P.; Nies, T.; Matuszyńska, A. Building mathematical models of biological systems with modelbase. J. Open Res. Softw. 2018, 6. [Google Scholar] [CrossRef]
- Ebrahim, A.; Lerman, J.A.; Palsson, B.O.; Hyduke, D.R. COBRApy: COnstraints-Based Reconstruction and Analysis for Python. BMC Syst. Biol. 2013, 7, 74. [Google Scholar] [CrossRef]
- Poolman, M.G. ScrumPy: Metabolic modelling with Python. IEE Proc. Syst. Biol. 2006, 153, 375–378. [Google Scholar] [CrossRef]
- Smith, R.W.; van Rosmalen, R.P.; Martins Dos Santos, V.A.P.; Fleck, C. DMPy: A Python package for automated mathematical model construction of large-scale metabolic systems. BMC Syst. Biol. 2018, 12, 72. [Google Scholar] [CrossRef] [PubMed]
- Hoops, S.; Sahle, S.; Gauges, R.; Lee, C.; Pahle, J.; Simus, N.; Singhal, M.; Xu, L.; Mendes, P.; Kummer, U. COPASI—A COmplex PAthway SImulator. Bioinformatics 2006, 22, 3067–3074. [Google Scholar] [CrossRef] [PubMed]
- Olivier, B.G.; Snoep, J.L. Web-based kinetic modelling using JWS Online. Bioinformatics 2004, 20, 2143–2144. [Google Scholar] [CrossRef] [PubMed]
- Le Novère, N.; Bornstein, B.; Broicher, A.; Courtot, M.; Donizelli, M.; Dharuri, H.; Li, L.; Sauro, H.; Schilstra, M.; Shapiro, B.; et al. BioModels Database: A free, centralized database of curated, published, quantitative kinetic models of biochemical and cellular systems. Nucleic Acids Res. 2006, 34, D689–D691. [Google Scholar] [CrossRef] [PubMed]
- Wolstencroft, K.; Krebs, O.; Snoep, J.L.; Stanford, N.J.; Bacall, F.; Golebiewski, M.; Kuzyakiv, R.; Nguyen, Q.; Owen, S.; Soiland-Reyes, S.; et al. FAIRDOMHub: A repository and collaboration environment for sharing systems biology research. Nucleic Acids Res. 2017, 45, D404–D407. [Google Scholar] [CrossRef]
- Wolstencroft, K.; Owen, S.; Krebs, O.; Nguyen, Q.; Stanford, N.J.; Golebiewski, M.; Weidemann, A.; Bittkowski, M.; An, L.; Shockley, D.; et al. SEEK: A systems biology data and model management platform. BMC Syst. Biol. 2015, 9, 33. [Google Scholar] [CrossRef] [PubMed]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Badenhorst, M.; Barry, C.J.; Swanepoel, C.J.; van Staden, C.T.; Wissing, J.; Rohwer, J.M. Workflow for Data Analysis in Experimental and Computational Systems Biology: Using Python as ‘Glue’. Processes 2019, 7, 460. https://doi.org/10.3390/pr7070460
Badenhorst M, Barry CJ, Swanepoel CJ, van Staden CT, Wissing J, Rohwer JM. Workflow for Data Analysis in Experimental and Computational Systems Biology: Using Python as ‘Glue’. Processes. 2019; 7(7):460. https://doi.org/10.3390/pr7070460
Chicago/Turabian StyleBadenhorst, Melinda, Christopher J. Barry, Christiaan J. Swanepoel, Charles Theo van Staden, Julian Wissing, and Johann M. Rohwer. 2019. "Workflow for Data Analysis in Experimental and Computational Systems Biology: Using Python as ‘Glue’" Processes 7, no. 7: 460. https://doi.org/10.3390/pr7070460
APA StyleBadenhorst, M., Barry, C. J., Swanepoel, C. J., van Staden, C. T., Wissing, J., & Rohwer, J. M. (2019). Workflow for Data Analysis in Experimental and Computational Systems Biology: Using Python as ‘Glue’. Processes, 7(7), 460. https://doi.org/10.3390/pr7070460