Spatial Variations of Bacterial Communities of an Anaerobic Lagoon-Type Biodigester Fed with Dairy Manure


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
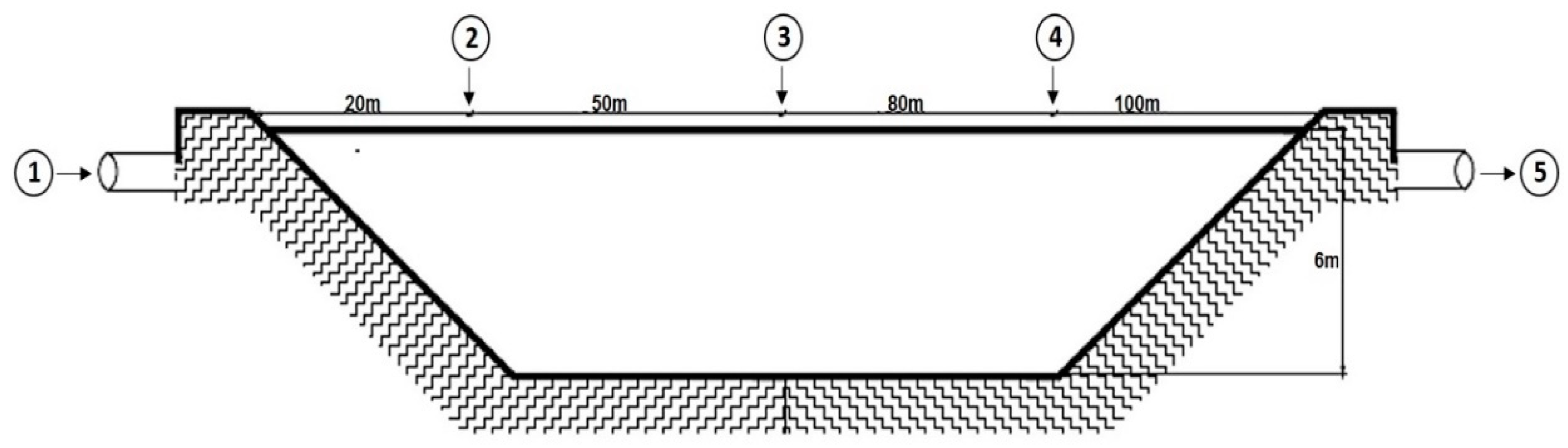
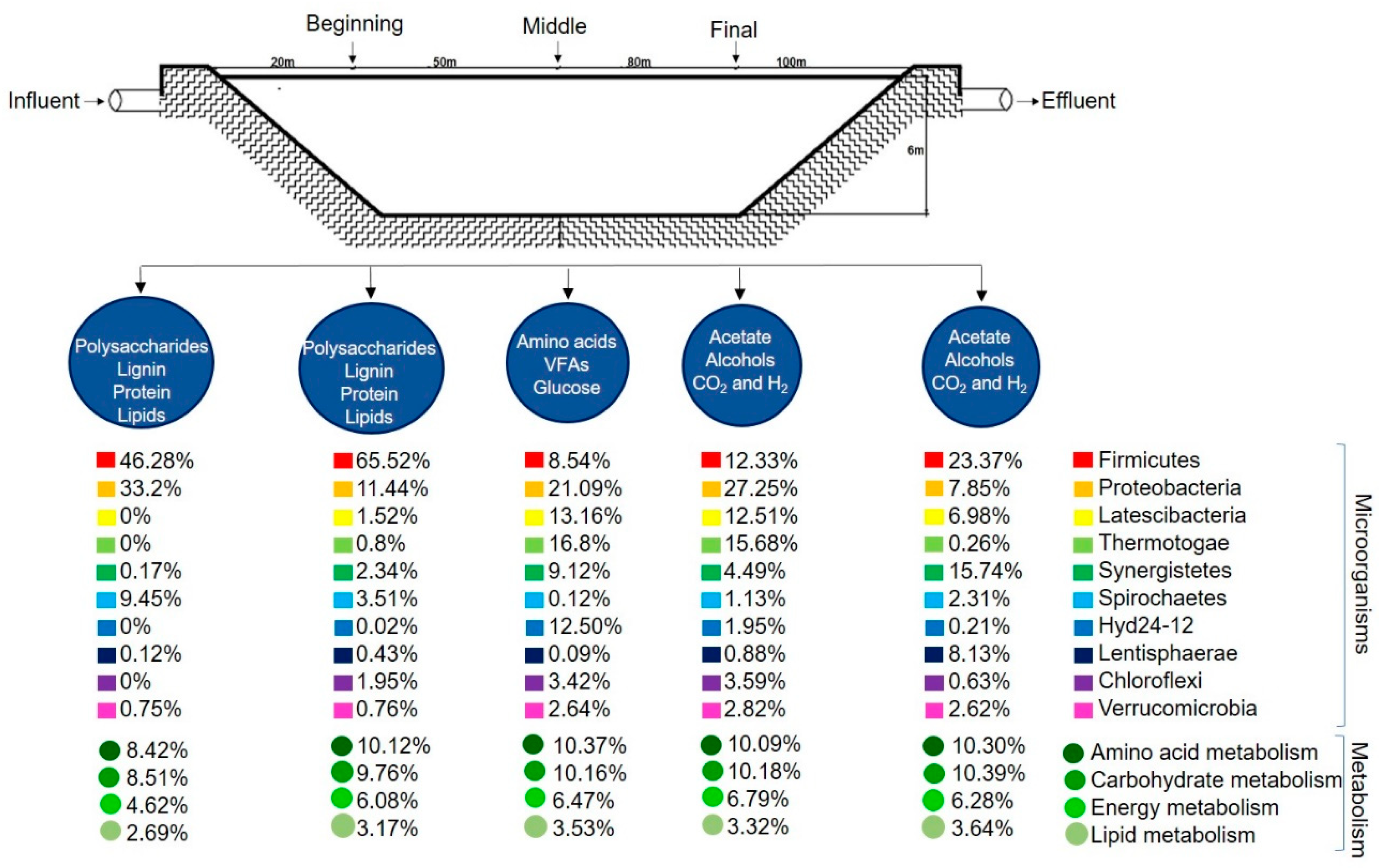
2.1. Sample Collection
2.2. Microbial Community Composition by 454 Pyrosequencing
2.3. Diversity Analysis
2.4. Functional Analyses
2.5. Nucleotide Sequences Accession Numbers
3. Results
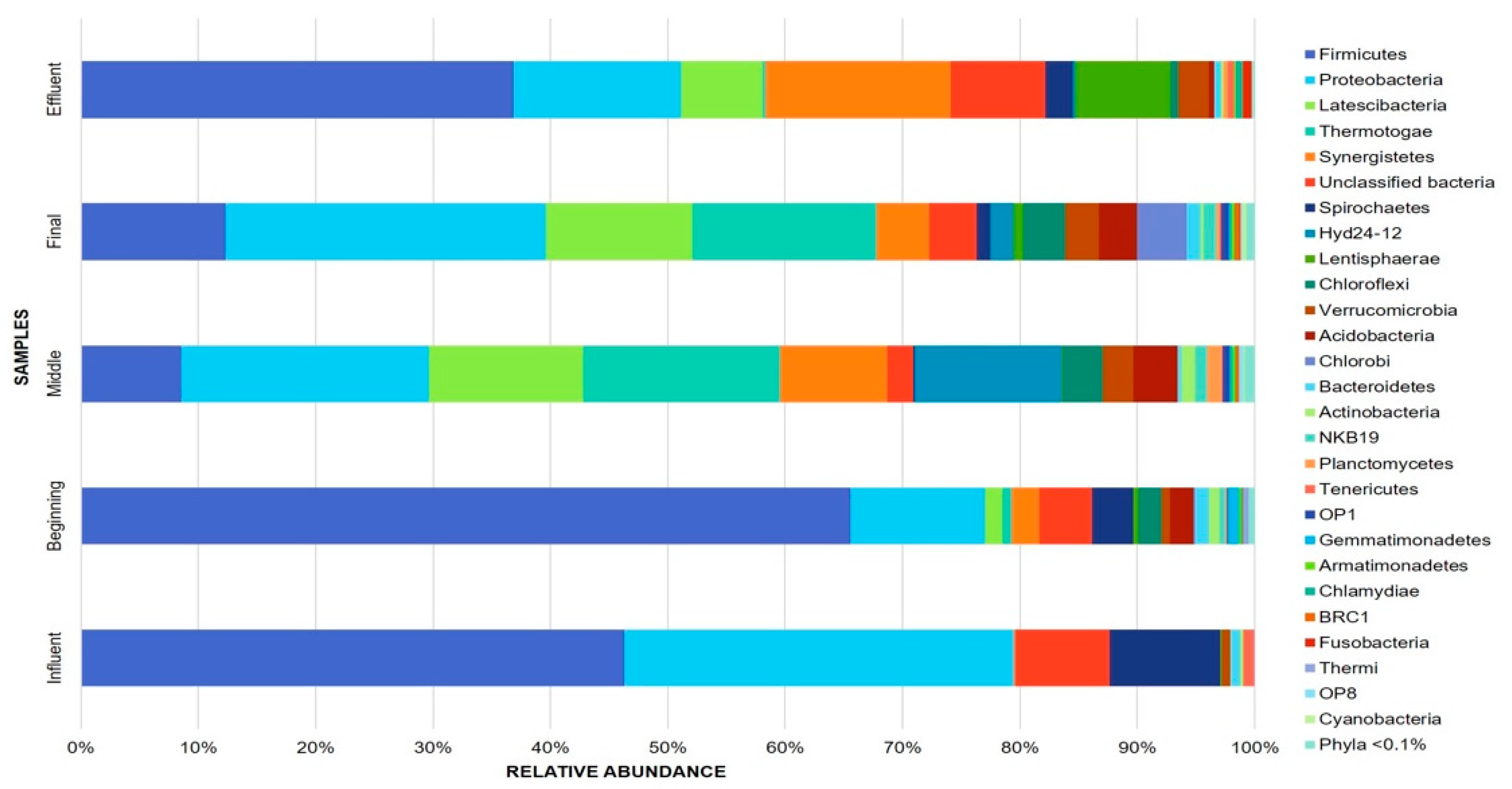
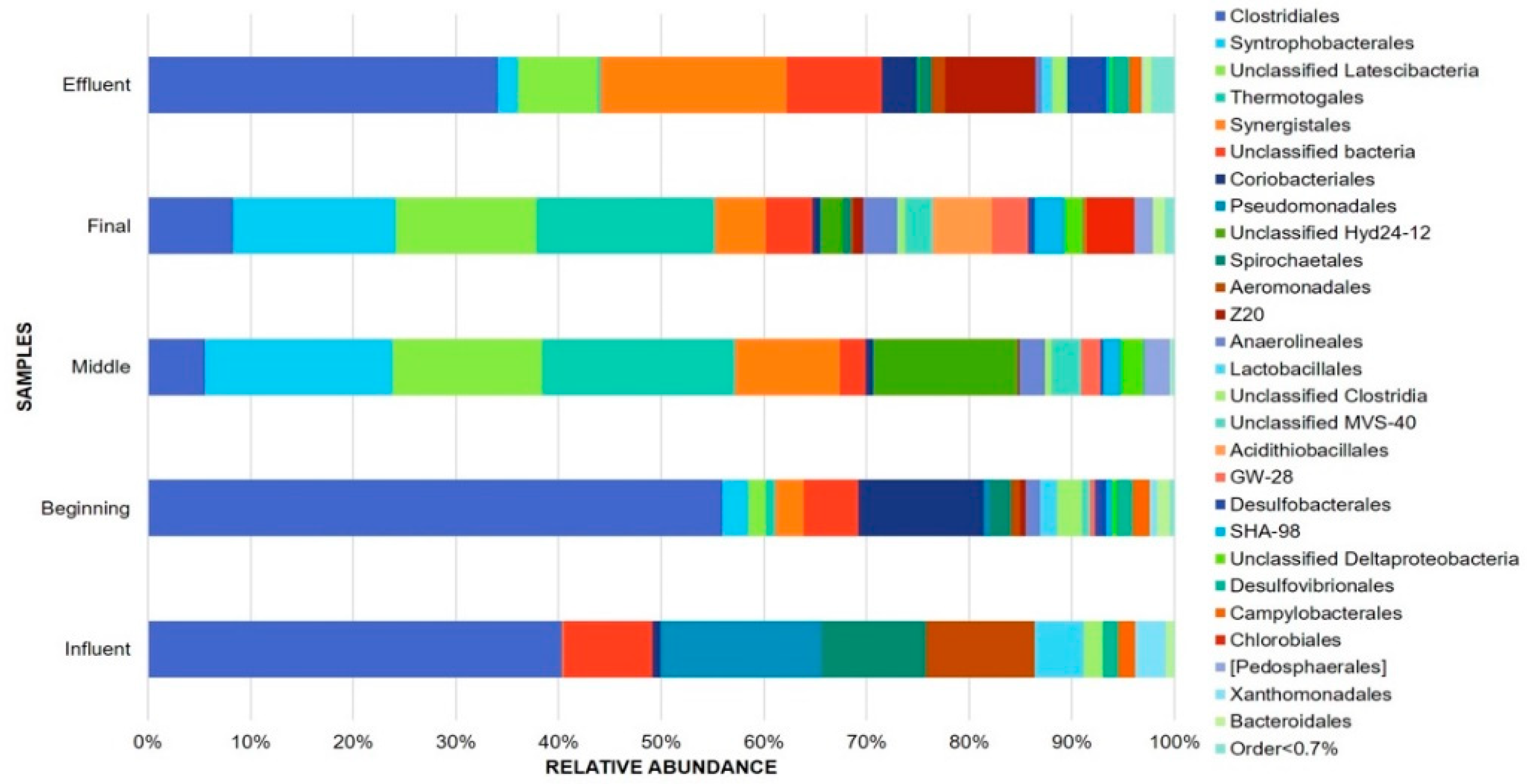
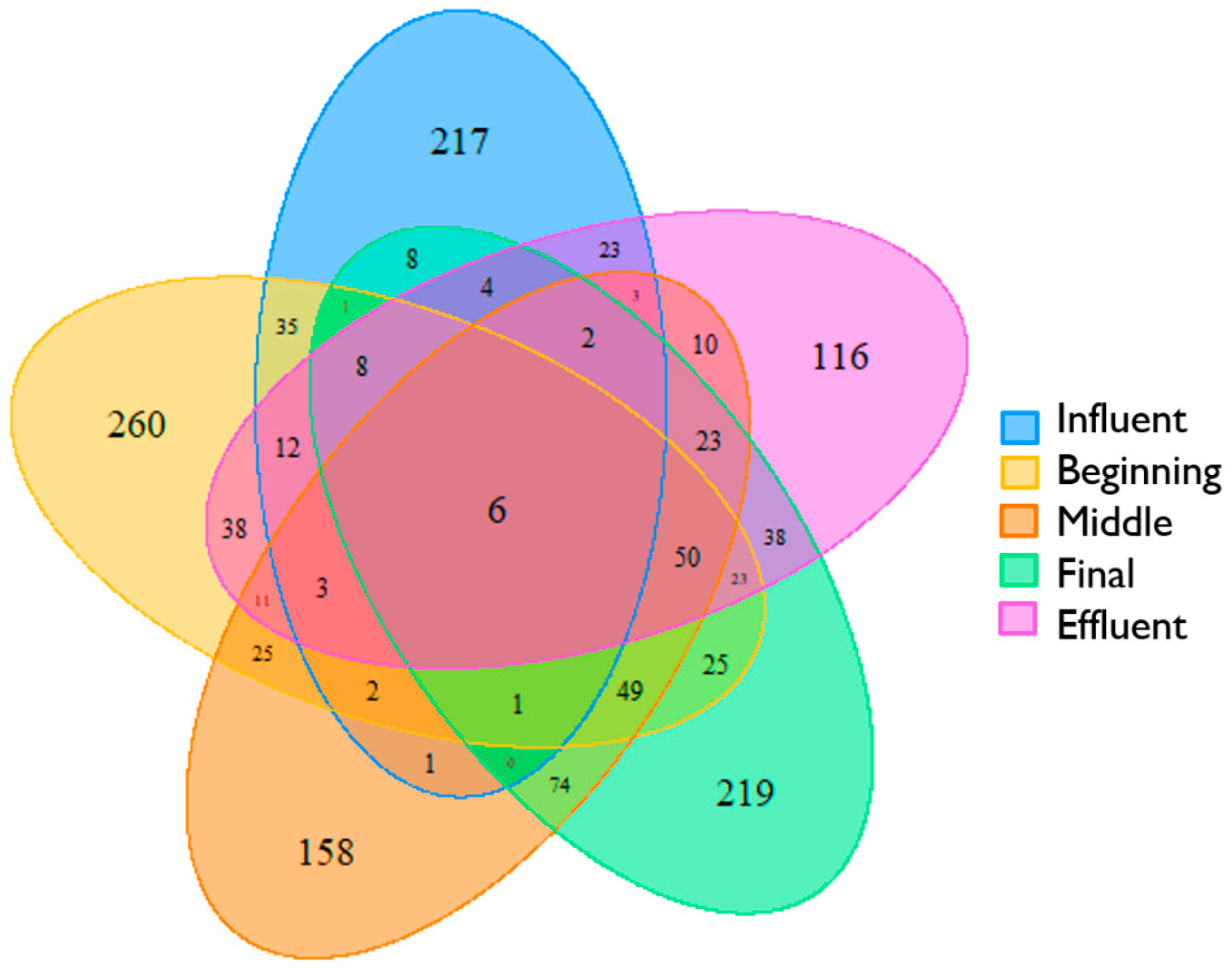
3.1. Bacterial Community Composition
3.2. Alpha Diversity
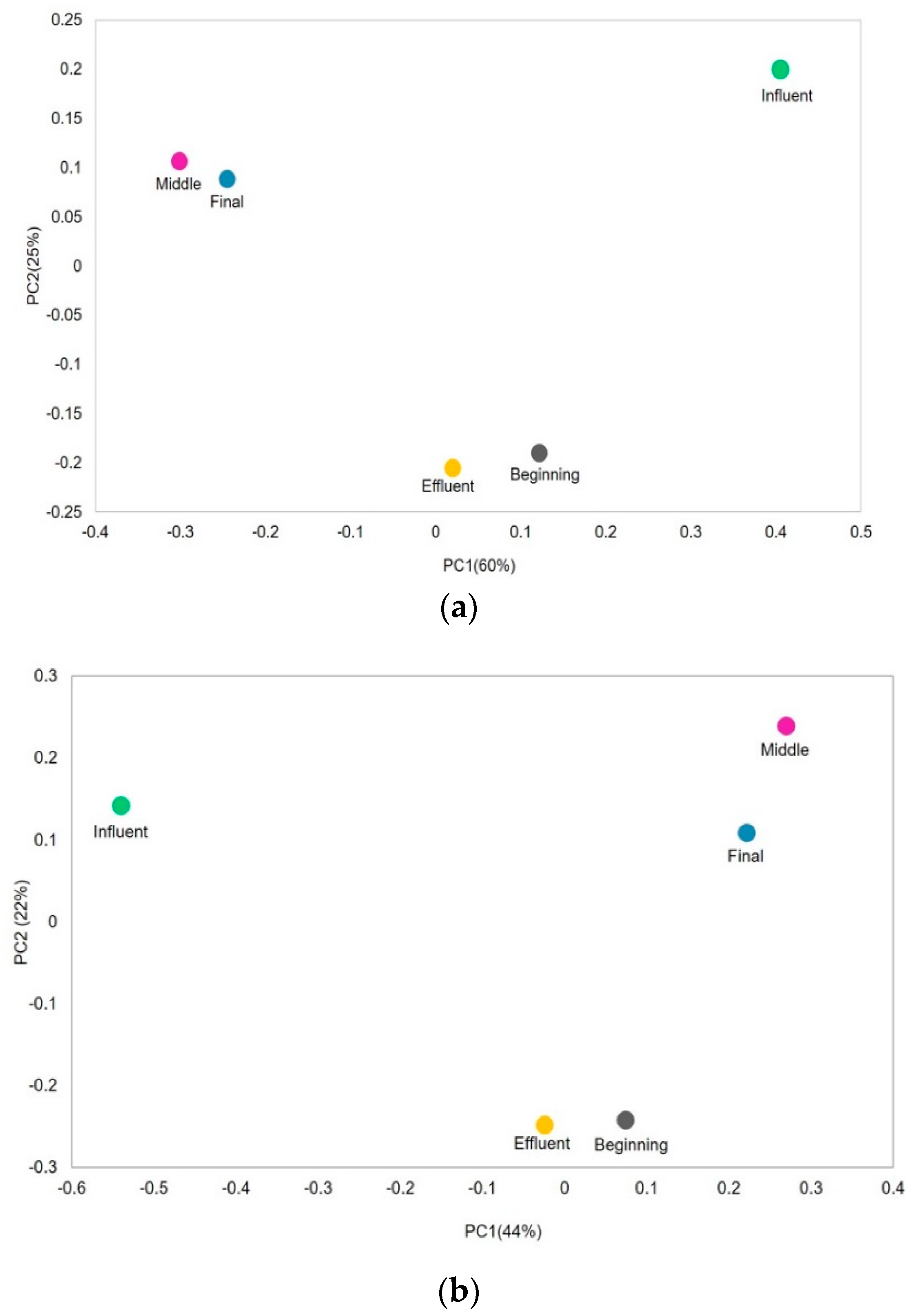
3.3. Beta Diversity
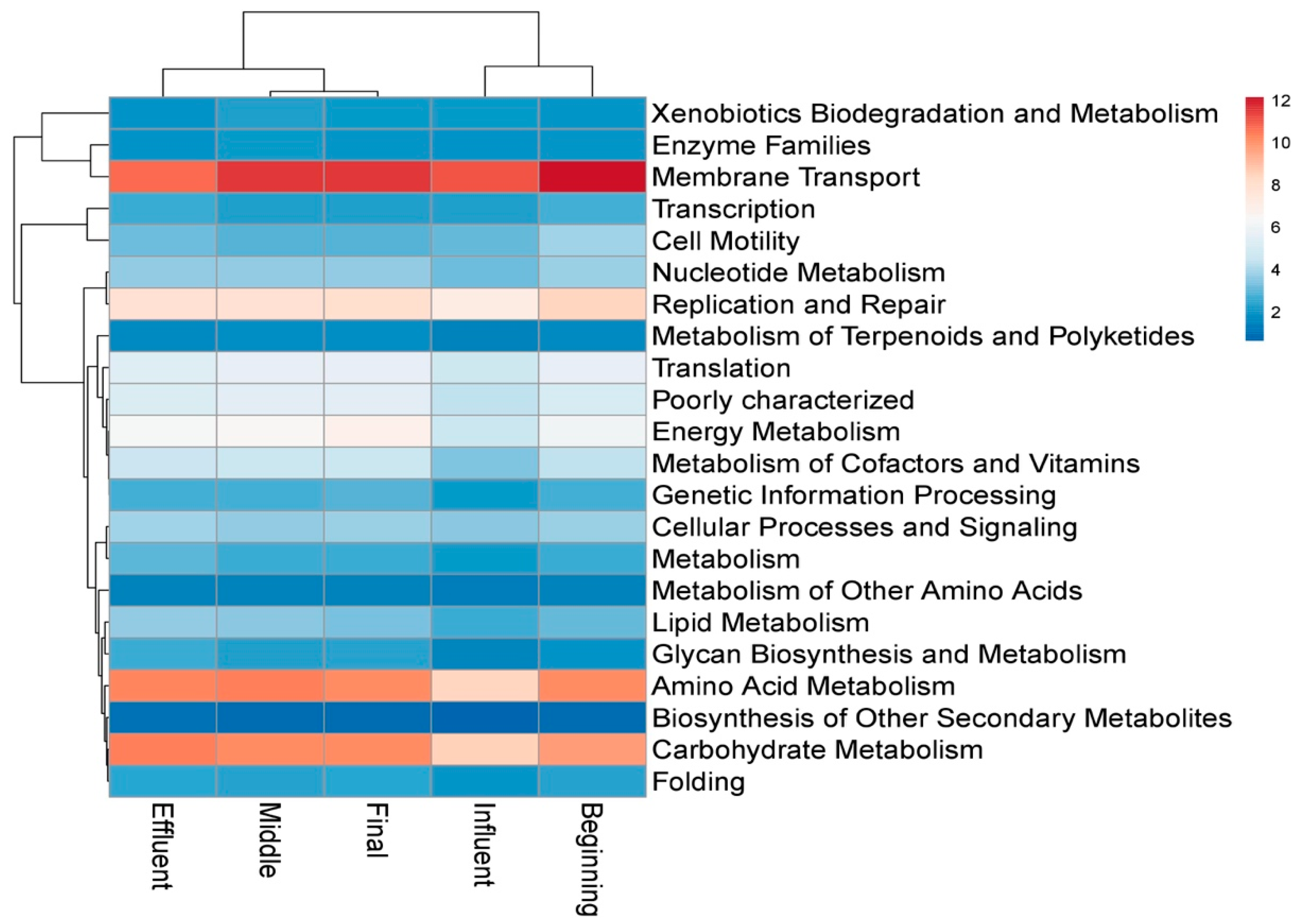
3.4. Functional Profile of Microbial Communities
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Smith, P.; Martino, D.; Cai, Z.; O’Mara, F.; Rice, C.; Scholes, B.; Howden, M.; McAllister, T.; Pan, G.; Romanenkov, V.; et al. Agriculture. In Contribution of Working Group III to the Fourth Assessment Report of the Intergovernmental Panel on Climate Change; Cambridge University Press: Cambridge, UK, 2007; pp. 498–540. [Google Scholar]
- Reisinger, A.; Clark, H. How much do direct livestock emissions actually contribute to global warming? Glob. Chang. Biol. 2018, 24, 1749–1761. [Google Scholar] [CrossRef] [PubMed]
- Bond, T.; Templeton, M.R. History and future of domestic biogas plants in the developing world. Energy Sustain. Dev. 2011, 15, 347–354. [Google Scholar] [CrossRef]
- Hernandez-De Lira, I.O.; Huber, D.H.; Espinosa-Solares, T.; Balagurusamy, N. Methane emission and bioenergy potential from livestock manures in Mexico. J. Renew. Sustain. Energy 2015, 7, 053117. [Google Scholar] [CrossRef]
- Weber, B.; Rojas-Oropeza, M.; Torres-Bernal, M.; Pampillón-González, L. Produccion de Biogas en México, Estadp Actual y Perspectivas, 1st ed.; Red Mexicana de Bioenergia: Mexico city, Mexico, 2012; pp. 1–48. [Google Scholar]
- Lee, J.; Han, G.; Shin, S.G.; Koo, T.; Cho, K.; Kim, W.; Hwang, S. Seasonal monitoring of bacteria and archaea in a full-scale thermophilic anaerobic digester treating food waste-recycling wastewater: Correlations between microbial community characteristics and process variables. Chem. Eng. J. 2016, 300, 291–299. [Google Scholar] [CrossRef]
- Sun, W.; Yu, G.; Louie, T.; Liu, T.; Zhu, C.; Xue, G.; Gao, P. From mesophilic to thermophilic digestion: The transitions of anaerobic bacterial, archaeal, and fungal community structures in sludge and manure samples. Appl. Microbiol. Biotechnol. 2015, 99, 10271–10282. [Google Scholar] [CrossRef]
- Sundberg, C.; Al-Soud, W.A.; Larsson, M.; Alm, E.; Yekta, S.S.; Svensson, B.H.; Sørensen, S.J.; Karlsson, A. 454 pyrosequencing analyses of bacterial and archaeal richness in 21 full-scale biogas digesters. FEMS Microbiol. Ecol. 2013, 85, 612–626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sul, W.J.; Cole, J.R.; Jesus, E.D.C.; Wang, Q.; Farris, R.J.; Fish, J.A.; Tiedje, J.M. Bacterial community comparisons by taxonomy-supervised analysis independent of sequence alignment and clustering. Proc. Natl. Acad. Sci. USA 2011, 108, 14637–14642. [Google Scholar] [CrossRef] [Green Version]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Peña, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Garrity, G.M.; Tiedje, J.M.; Cole, J.R. Naive bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 2007, 73, 5261–5267. [Google Scholar] [CrossRef]
- McMurdie, P.J.; Holmes, S. Phyloseq: An R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef]
- iNEXT-Package: Interpolation and Extrapolation for Species Diversity in iNEXT: Interpolation and Extrapolation for Species Diversity. Available online: https://rdrr.io/cran/iNEXT/man/iNEXT-package.html (accessed on 9 April 2019).
- Langille, M.G.I.; Zaneveld, J.; Caporaso, J.G.; McDonald, D.; Knights, D.; Reyes, J.A.; Clemente, J.C.; Burkepile, D.E.; Vega Thurber, R.L.; Knight, R.; et al. Predictive functional profiling of microbial communities using 16s rRNA marker gene sequences. Nat. Biotechnol. 2013, 31, 814–821. [Google Scholar] [CrossRef] [PubMed]
- Lozupone, C.; Knight, R. UniFrac: A new phylogenetic method for comparing microbial communities. Appl. Environ. Microbiol. 2005, 71, 8228–8235. [Google Scholar] [CrossRef] [PubMed]
- Ye, L.; Zhang, T. Bacterial Communities in different sections of a municipal wastewater treatment plant revealed by 16S rDNA 454 Pyrosequencing. Appl. Microbiol. Biotechnol. 2013, 97, 2681–2690. [Google Scholar] [CrossRef]
- Li, Y.-F.; Chen, P.-H.; Yu, Z. Spatial and temporal variations of microbial community in a mixed plug-flow loop reactor fed with dairy manure. Microb. Biotechnol. 2014, 7, 332–346. [Google Scholar] [CrossRef] [PubMed]
- Ziganshin, A.M.; Liebetrau, J.; Pröter, J.; Kleinsteuber, S. Microbial community structure and dynamics during anaerobic digestion of various agricultural waste materials. Appl. Microbiol. Biotechnol. 2013, 97, 5161–5174. [Google Scholar] [CrossRef]
- Lee, S.H.; Kang, H.J.; Lee, Y.H.; Lee, T.J.; Han, K.; Choi, Y.; Park, H.D. Monitoring bacterial community structure and variability in time scale in full-scale anaerobic digesters. J. Environ. Monit. JEM 2012, 14, 1893–1905. [Google Scholar] [CrossRef] [PubMed]
- Cardinali-Rezende, J.; Rojas-Ojeda, P.; Nascimento, A.M.A.; Sanz, J.L. Proteolytic bacterial dominance in a full-scale municipal solid waste anaerobic reactor assessed by 454 pyrosequencing technology. Chemosphere 2016, 146, 519–525. [Google Scholar] [CrossRef] [PubMed]
- Luo, G.; Angelidaki, I. Analysis of bacterial communities and bacterial pathogens in a biogas plant by the combination of ethidium monoazide, PCR and Ion torrent sequencing. Water Res. 2014, 60, 156–163. [Google Scholar] [CrossRef]
- Shin, S.G.; Koo, T.; Lee, J.; Han, G.; Cho, K.; Kim, W.; Hwang, S. Correlations between bacterial populations and process parameters in four full-scale anaerobic digesters treating sewage sludge. Bioresour. Technol. 2016, 214, 711–721. [Google Scholar] [CrossRef]
- Gao, J.; Liu, G.; Li, H.; Xu, L.; Du, L.; Yang, B. Predictive functional profiling using marker gene sequences and community diversity analyses of microbes in full-scale anaerobic sludge digesters. Bioprocess Biosyst. Eng. 2016, 7, 1115–1127. [Google Scholar] [CrossRef]
- St-Pierre, B.; Wright, A.D.G. Comparative metagenomic analysis of bacterial populations in three full-scale mesophilic anaerobic manure digesters. Appl. Microbiol. Biotechnol. 2014, 98, 2709–2717. [Google Scholar] [CrossRef] [PubMed]
- Nelson, M.C.; Morrison, M.; Yu, Z. A meta-analysis of the microbial diversity observed in anaerobic digesters. Bioresour. Technol. 2011, 102, 3730–3739. [Google Scholar] [CrossRef] [PubMed]
- Møller, H.B.; Moset, V.; Brask, M.; Weisbjerg, M.R.; Lund, P. Feces composition and manure derived methane yield from dairy cows: Influence of diet with focus on fat supplement and roughage type. Atmos. Environ. 2014, 94, 36–43. [Google Scholar] [CrossRef]
- Naushad, H.S.; Gupta, R.S. Phylogenomics and molecular signatures for species from the plant pathogen-containing order Xanthomonadales. PLoS ONE 2013, 8, e55216. [Google Scholar] [CrossRef] [PubMed]
- Martin-Carnahan, A.; Joseph, S.W. Aeromonadalesord. Nov. In Bergey’s Manual® of Systematic Bacteriology: Volume Two the Proteobacteria Part B The Gammaproteobacteria; Brenner, D.J., Krieg, N.R., Staley, J.T., Garrity, G.M., Boone, D.R., De Vos, P., Goodfellow, M., Rainey, F.A., Schleifer, K.H., Eds.; Springer: Boston, MA, USA, 2005; pp. 556–587. [Google Scholar]
- Fu, S.F.; He, S.; Shi, X.S.; Katukuri, N.R.; Dai, M.; Guo, R.B. The chemical properties and microbial community characterization of the thermophilic microaerobic pretreatment process. Bioresour. Technol. 2015, 198, 497–502. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Xu, X.J.; Xie, P.; Yuan, Y.; Zhou, X.; Wang, A.J.; Lee, D.J.; Ren, N.Q. Pyrosequencing reveals microbial community dynamics in integrated simultaneous desulfurization and denitrification process at different influent nitrate concentrations. Chemosphere 2017, 171, 294–301. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Ren, N.; Wang, A.; Yu, Z.; Lee, D.J. Microbial community of granules in expanded granular sludge bed reactor for simultaneous biological removal of sulfate, nitrate and lactate. Appl. Microbiol. Biotechnol. 2008, 79, 1071–1077. [Google Scholar] [CrossRef]
- Youssef, N.H.; Farag, I.F.; Rinke, C.; Hallam, S.J.; Woyke, T.; Elshahed, M.S. In silico analysis of the metabolic potential and niche specialization of candidate phylum “Latescibacteria” (WS3). PLoS ONE 2015, 10, e0127499. [Google Scholar] [CrossRef]
- Zhou, J.; Yang, J.; Yu, Q.; Yong, X.; Xie, X.; Zhang, L.; Wei, P.; Jia, H. Different organic loading rates on the biogas production during the anaerobic digestion of rice straw: A pilot study. Bioresour. Technol. 2017, 244, 865–871. [Google Scholar] [CrossRef]
- Li, A.; Chu, Y.; Wang, X.; Ren, L.; Yu, J.; Liu, X.; Yan, J.; Zhang, L.; Wu, S.; Li, S. A pyrosequencing-based metagenomic study of methane-producing microbial community in solid-state biogas reactor. Biotechnol. Biofuels 2013, 6, 3. [Google Scholar] [CrossRef]
- Schlüter, A.; Bekel, T.; Diaz, N.N.; Dondrup, M.; Eichenlaub, R.; Gartemann, K.-H.; Krahn, I.; Krause, L.; Krömeke, H.; Kruse, O.; et al. The metagenome of a biogas-producing microbial community of a production-scale biogas plant fermenter analysed by the 454-pyrosequencing technology. J. Biotechnol. 2008, 136, 77–90. [Google Scholar] [CrossRef] [PubMed]
- Levén, L.; Eriksson, A.R.B.; Schnürer, A. Effect of process temperature on bacterial and archaeal communities in two methanogenic bioreactors treating organic household waste. FEMS Microbiol. Ecol. 2007, 59, 683–693. [Google Scholar] [CrossRef] [PubMed]
- Pap, B.; Györkei, Á.; Boboescu, I.Z.; Nagy, I.K.; Bíró, T.; Kondorosi, É.; Maróti, G. Temperature-dependent transformation of biogas-producing microbial communities points to the increased importance of hydrogenotrophic methanogenesis under thermophilic operation. Bioresour. Technol. 2015, 177, 375–380. [Google Scholar] [CrossRef] [PubMed]
- Pervin, H.M.; Dennis, P.G.; Lim, H.J.; Tyson, G.W.; Batstone, D.J.; Bond, P.L. Drivers of microbial community composition in mesophilic and thermophilic temperature-phased anaerobic digestion pre-treatment reactors. Water Res. 2013, 47, 7098–7108. [Google Scholar] [CrossRef] [PubMed]
- Müller, B.; Sun, L.; Westerholm, M.; Schnürer, A. Bacterial community composition and fhs profiles of low- and high-ammonia biogas digesters reveal novel syntrophic acetate-oxidising bacteria. Biotechnol. Biofuels 2016, 9, 48. [Google Scholar] [CrossRef] [PubMed]
- Regueiro, L.; Carballa, M.; Lema, J.M. Outlining microbial community dynamics during temperature drop and subsequent recovery period in anaerobic co-digestion systems. J. Biotechnol. 2014, 192, 179–186. [Google Scholar] [CrossRef]
- Allison, S.D.; Martiny, J.B. Resistance, resilience, and redundancy in microbial communities. Proc. Natl. Acad. Sci. USA 2008, 105, 11512–11519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loreau, M.; de Mazancourt, C. Biodiversity and ecosystem stability: A synthesis of underlying mechanisms. Ecol. Lett. 2013, 16, 106–115. [Google Scholar] [CrossRef]
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
García-Lozano, M.; Hernández-De Lira, I.O.; Huber, D.H.; Balagurusamy, N. Spatial Variations of Bacterial Communities of an Anaerobic Lagoon-Type Biodigester Fed with Dairy Manure. Processes 2019, 7, 408. https://doi.org/10.3390/pr7070408
García-Lozano M, Hernández-De Lira IO, Huber DH, Balagurusamy N. Spatial Variations of Bacterial Communities of an Anaerobic Lagoon-Type Biodigester Fed with Dairy Manure. Processes. 2019; 7(7):408. https://doi.org/10.3390/pr7070408
Chicago/Turabian StyleGarcía-Lozano, Marleny, Inty Omar Hernández-De Lira, David H. Huber, and Nagamani Balagurusamy. 2019. "Spatial Variations of Bacterial Communities of an Anaerobic Lagoon-Type Biodigester Fed with Dairy Manure" Processes 7, no. 7: 408. https://doi.org/10.3390/pr7070408
APA StyleGarcía-Lozano, M., Hernández-De Lira, I. O., Huber, D. H., & Balagurusamy, N. (2019). Spatial Variations of Bacterial Communities of an Anaerobic Lagoon-Type Biodigester Fed with Dairy Manure. Processes, 7(7), 408. https://doi.org/10.3390/pr7070408