Theoretical Study of the Adsorption Process of Antimalarial Drugs into Acrylamide-Base Hydrogel Model Using DFT Methods: The First Approach to the Rational Design of a Controlled Drug Delivery System

 ,
,  , ,
, , 

Abstract
1. Introduction
2. Materials and Methods
Computational Methods
3. Results
3.1. Geometrical Structures and Binding Energy
3.2. Natural Bond Orbital (NBO) Analysis
3.3. Water Environment Behavior
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Organisation Mondiale de la Santé. World Malaria Report 2012 WHO Global Malaria Programme; World Health Organization: Geneva, Swizerland, 2012; ISBN 978-92-4-156453-3. [Google Scholar]
- Al Qaraghuli, M.M.; Obeid, M.A.; Aldulaimi, O.; Ferro, V.A. Control of malaria by bio-therapeutics and drug delivery systems. J. Med. Microbiol. Diagn. 2017, 6, 260. [Google Scholar] [CrossRef]
- Liu, J.; Xiao, Y.; Allen, C. Polymer–drug compatibility: A guide to the development of delivery systems for the anticancer agent, ellipticine. J. Pharm. Sci. 2004, 93, 132–143. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.; Zhao, W.; Yu, J.; Li, Y.; Zhao, C. Recent Development of pH-Responsive Polymers for Cancer Nanomedicine. Molecules 2019, 24, 4. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Sun, H.; Liu, Y.; Hou, W.; Yang, Y.; Cai, R.; Cui, C.; Zhang, P.; Pan, X.; Li, X.; et al. Self-Assembled Aptamer-Grafted Hyperbranched Polymer Nanocarrier for Targeted and Photoresponsive Drug Delivery. Angew. Chem. Int. Ed. 2018, 57, 17048–17052. [Google Scholar] [CrossRef] [PubMed]
- Externally Triggered Heat and Drug Release from Magnetically Controlled Nanocarriers|ACS Applied Polymer Materials. Available online: https://pubs.acs.org/doi/pdf/10.1021/acsapm.8b00100 (accessed on 27 May 2019).
- Murambiwa, P.; Masola, B.; Govender, T.; Mukaratirwa, S.; Musabayane, C.T. Anti-malarial drug formulations and novel delivery systems: A review. Acta Trop. 2011, 118, 71–79. [Google Scholar] [CrossRef] [PubMed]
- Ganji, F.; Vasheghani-Farahani, S.; Vasheghani-Farahani, E. Theoretical description of hydrogel swelling: A review. Iran. Polym. J. 2010, 19, 375–398. [Google Scholar]
- Huang, Y.; Jin, X.; Liu, H.; Hu, Y. A molecular thermodynamic model for the swelling of thermo-sensitive hydrogels. Fluid Phase Equilibria 2008, 263, 96–101. [Google Scholar] [CrossRef]
- Cai, S.; Suo, Z. Mechanics and chemical thermodynamics of phase transition in temperature-sensitive hydrogels. J. Mech. Phys. Solids 2011, 59, 2259–2278. [Google Scholar] [CrossRef]
- Du, X.; Zhou, J.; Shi, J.; Xu, B. Supramolecular Hydrogelators and Hydrogels: From Soft Matter to Molecular Biomaterials. Chem. Rev. 2015, 115, 13165–13307. [Google Scholar] [CrossRef]
- Huynh, L.; Neale, C.; Pomès, R.; Allen, C. Computational approaches to the rational design of nanoemulsions, polymeric micelles, and dendrimers for drug delivery. Nanomed.: Nanotechnol. Biol. Med. 2012, 8, 20–36. [Google Scholar] [CrossRef]
- Shen, E.; Kipper, M.J.; Dziadul, B.; Lim, M.-K.; Narasimhan, B. Mechanistic relationships between polymer microstructure and drug release kinetics in bioerodible polyanhydrides. J. Control. Release 2002, 82, 115–125. [Google Scholar] [CrossRef]
- Park, J.; Ye, M.; Park, K. Biodegradable polymers for microencapsulation of drugs. Molecules 2005, 10, 146–161. [Google Scholar] [CrossRef] [PubMed]
- Weiss, R.G.; Terech, P. (Eds.) Molecular Gels: Materials with Self-Assembled Fibrillar Networks; Springer: Dordrecht, The Netherlands, 2006; ISBN 978-1-4020-3352-0. [Google Scholar]
- Zweep, N.; Hopkinson, A.; Meetsma, A.; Browne, W.R.; Feringa, B.L.; van Esch, J.H. balancing hydrogen bonding and van der waals interactions in cyclohexane-based bisamide and bisurea organogelators. Langmuir 2009, 25, 8802–8809. [Google Scholar] [CrossRef] [PubMed]
- Hoy, R.S.; Fredrickson, G.H. Thermoreversible associating polymer networks. I. Interplay of thermodynamics, chemical kinetics, and polymer physics. J. Chem. Phys. 2009, 131, 224902. [Google Scholar] [CrossRef] [PubMed]
- Maniruzzaman, M.; Pang, J.; Morgan, D.J.; Douroumis, D. Molecular Modeling as a Predictive Tool for the Development of Solid Dispersions. Available online: https://pubs.acs.org/doi/abs/10.1021/mp500510m (accessed on 3 November 2018).
- Maniruzzaman, M.; Morgan, D.J.; Mendham, A.P.; Pang, J.; Snowden, M.J.; Douroumis, D. Drug–polymer intermolecular interactions in hot-melt extruded solid dispersions. Int. J. Pharm. 2013, 443, 199–208. [Google Scholar] [CrossRef] [PubMed]
- Schmaljohann, D. Thermo- and pH-responsive polymers in drug delivery. Adv. Drug Deliv. Rev. 2006, 58, 1655–1670. [Google Scholar] [CrossRef] [PubMed]
- Seiffert, S.; Sprakel, J. Physical chemistry of supramolecular polymer networks. Chem. Soc. Rev. 2012, 41, 909–930. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, F. Theoretical Study of Molecular Association and Thermoreversible Gelation in Polymers. Polym. J. 2002, 34, 479–509. [Google Scholar] [CrossRef]
- Peppas, N.A.; Huang, Y.; Torres-Lugo, M.; Ward, J.H.; Zhang, J. Physicochemical Foundations and Structural Design of Hydrogels in Medicine and Biology. Annu. Rev. Biomed. Eng. 2000, 2, 9–29. [Google Scholar] [CrossRef]
- Chun, B.J.; Lu, J.; Weck, M.; Jang, S.S. Characterization of molecular association of poly(2-oxazoline)s-based micelles with various epoxides and diols via the Flory–Huggins theory: A molecular dynamics simulation approach. Phys. Chem. Chem. Phys. 2015, 17, 29161–29170. [Google Scholar] [CrossRef]
- Bahar, I.; Erbil, H.Y.; Baysal, B.M.; Erman, B. Determination of polymer-solvent interaction parameter from swelling of networks: The system poly(2-hydroxyethyl methacrylate)-diethylene glycol. Macromolecules 1987, 20, 1353–1356. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Marquez, E.; Domínguez, R.M.; Mora, J.R.; Córdova, T.; Chuchani, G. Experimental and theoretical studies of the homogeneous, unimolecular gas-phase elimination kinetics of trimethyl orthovalerate and trimethyl orthochloroacetate. J. Phys. Chem. A 2010, 114, 4203–4209. [Google Scholar] [CrossRef] [PubMed]
- Exner, K.S.; Over, H. Kinetics of electrocatalytic reactions from first-principles: A critical comparison with the ab initio thermodynamics approach. Acc. Chem. Res. 2017, 50, 1240–1247. [Google Scholar] [CrossRef] [PubMed]
- Reed, A.E.; Curtiss, L.A.; Weinhold, F. Intermolecular interactions from a natural bond orbital, donor-acceptor viewpoint. Chem. Rev. 1988, 88, 899–926. [Google Scholar] [CrossRef]
- Shirota, H.; Ushiyama, H. Hydrogen-bonding dynamics in aqueous solutions of amides and acids: Monomer, dimer, trimer, and polymer. J. Phys. Chem. B 2008, 112, 13542–13551. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Chen, J.; Shen, Q.; Fu, W.; Wu, W. Molecular insights on the cyclic peptide nanotube-mediated transportation of antitumor drug 5-fluorouracil. Mol. Pharm. 2010, 7, 1985–1994. [Google Scholar] [CrossRef]
- Albertorio, F.; Hughes, M.E.; Golovchenko, J.A.; Branton, D. Base dependent DNA–carbon nanotube interactions: Activation enthalpies and assembly–disassembly control. Nanotechnology 2009, 20, 395101. [Google Scholar] [CrossRef]
- Sparks, T.C.; Lorsbach, B.A. Agrochemical Discovery—Building the Next Generation of Insect Control Agents. In ACS Symposium Series; Gross, A.D., Ozoe, Y., Coats, J.R., Eds.; American Chemical Society: Washington, DC, USA, 2017; Volume 1264, pp. 1–17. ISBN 978-0-8412-3257-0. [Google Scholar]
- Baldi, A. Computational approaches for drug design and discovery: An overview. Syst. Rev. Pharm. 2010, 1, 99. [Google Scholar] [CrossRef]
- Gallo, M.; Favila, A.; Glossman-Mitnik, D. DFT studies of functionalized carbon nanotubes and fullerenes as nanovectors for drug delivery of antitubercular compounds. Chem. Phys. Lett. 2007, 447, 105–109. [Google Scholar] [CrossRef]
- Karataş, D.; Tekin, A.; Bahadori, F.; Çelik, M.S. Interaction of curcumin in a drug delivery system including a composite with poly(lactic-co-glycolic acid) and montmorillonite: A density functional theory and molecular dynamics study. J. Mater. Chem. B 2017, 5, 8070–8082. [Google Scholar] [CrossRef]
- Kaur, J.; Singla, P.; Goel, N. Adsorption of oxazole and isoxazole on BNNT surface: A DFT study. Appl. Surf. Sci. 2015, 328, 632–640. [Google Scholar] [CrossRef]
- Liu, Z.; Fan, A.C.; Rakhra, K.; Sherlock, S.; Goodwin, A.; Chen, X.; Yang, Q.; Felsher, D.W.; Dai, H. Supramolecular stacking of doxorubicin on carbon nanotubes for in vivo cancer therapy. Angew. Chem. Int. Ed. Engl. 2009, 48, 7668–7672. [Google Scholar] [CrossRef] [PubMed]
- Qin, W.; Li, X.; Bian, W.-W.; Fan, X.-J.; Qi, J.-Y. Density functional theory calculations and molecular dynamics simulations of the adsorption of biomolecules on graphene surfaces. Biomaterials 2010, 31, 1007–1016. [Google Scholar] [CrossRef] [PubMed]
- Geerlings, P.; De Proft, F.; Langenaeker, W. Conceptual density functional theory. Chem. Rev. 2003, 103, 1793–1874. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian16 Revision B.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Sun, H.; Kabb, C.P.; Dai, Y.; Hill, M.R.; Ghiviriga, I.; Bapat, A.P.; Sumerlin, B.S. Macromolecular metamorphosis via stimulus-induced transformations of polymer architecture. Nat. Chem. 2017, 9, 817–823. [Google Scholar] [CrossRef] [PubMed]
- Souza, B.S.; Mora, J.R.; Wanderlind, E.H.; Clementin, R.M.; Gesser, J.C.; Fiedler, H.D.; Nome, F.; Menger, F.M. Transforming a Stable Amide into a Highly Reactive One: Capturing the Essence of Enzymatic Catalysis. Angew. Chem. Int. Ed. 2017, 56, 5345–5348. [Google Scholar] [CrossRef] [PubMed]
- Mora, J.R.; Cervantes, C.; Marquez, E. New Insight into the Chloroacetanilide Herbicide Degradation Mechanism through a Nucleophilic Attack of Hydrogen Sulfide. Int. J. Mol. Sci. 2018, 19, 2864. [Google Scholar] [CrossRef] [PubMed]
- McQuarrie, D.A. Statistical Mechanics; University Science Books: Sausalito, CA, USA, 2000; ISBN 978-1-891389-15-3. [Google Scholar]
- Tan, T.T.M.; Rode, B.M. Molecular modelling of polymers, 3. Prediction of glass transition temperatures of poly(acrylic acid), poly(methacrylic acid) and polyacrylamide derivatives. Macromol. Theory Simul. 1996, 5, 467–475. [Google Scholar] [CrossRef]
- Scaranto, J.; Mallia, G.; Harrison, N.M. An efficient method for computing the binding energy of an adsorbed molecule within a periodic approach. The application to vinyl fluoride at rutile TiO2(110) surface. Comput. Mater. Sci. 2011, 50, 2080–2086. [Google Scholar] [CrossRef]
- Chunsrivirot, S.; Trout, B.L. Free Energy of Binding of a Small Molecule to an Amorphous Polymer in a Solvent. Langmuir 2011, 27, 6910–6919. [Google Scholar] [CrossRef] [PubMed]
- Gutowski, M.; Van Lenthe, J.H.; Verbeek, J.; Van Duijneveldt, F.B.; Chałasinski, G. The basis set superposition error in correlated electronic structure calculations. Chem. Phys. Lett. 1986, 124, 370–375. [Google Scholar] [CrossRef]
- Kruse, H.; Grimme, S. A geometrical correction for the inter- and intra-molecular basis set superposition error in Hartree-Fock and density functional theory calculations for large systems. J. Chem. Phys. 2012, 136, 154101. [Google Scholar] [CrossRef] [PubMed]
- Ghose, A.K.; Crippen, G.M. Atomic Physicochemical Parameters for Three-Dimensional Structure-Directed Quantitative Structure-Activity Relationships I. Partition Coefficients as a Measure of Hydrophobicity. J. Comput. Chem. 1986, 7, 565–577. [Google Scholar] [CrossRef]
- Zheng, Y.-Z.; Zhou, Y.; Liang, Q.; Chen, D.-F.; Guo, R.; Lai, R.-C. Hydrogen-bonding Interactions between Apigenin and Ethanol/Water: A Theoretical Study. Sci. Rep. 2016, 6, 34647. [Google Scholar] [CrossRef] [PubMed]
- Magdaline, J.D.; Chithambarathanu, T. Natural bond orbital analysis and vibrational spectroscopic studies of 2-furoic acid using density functional theory. Appl. Phys. 2012, 50, 7. [Google Scholar]
- Niklasson, A.M.N.; Challacombe, M. Density Matrix Perturbation Theory. Phys. Rev. Lett. 2004, 92. [Google Scholar] [CrossRef] [PubMed]




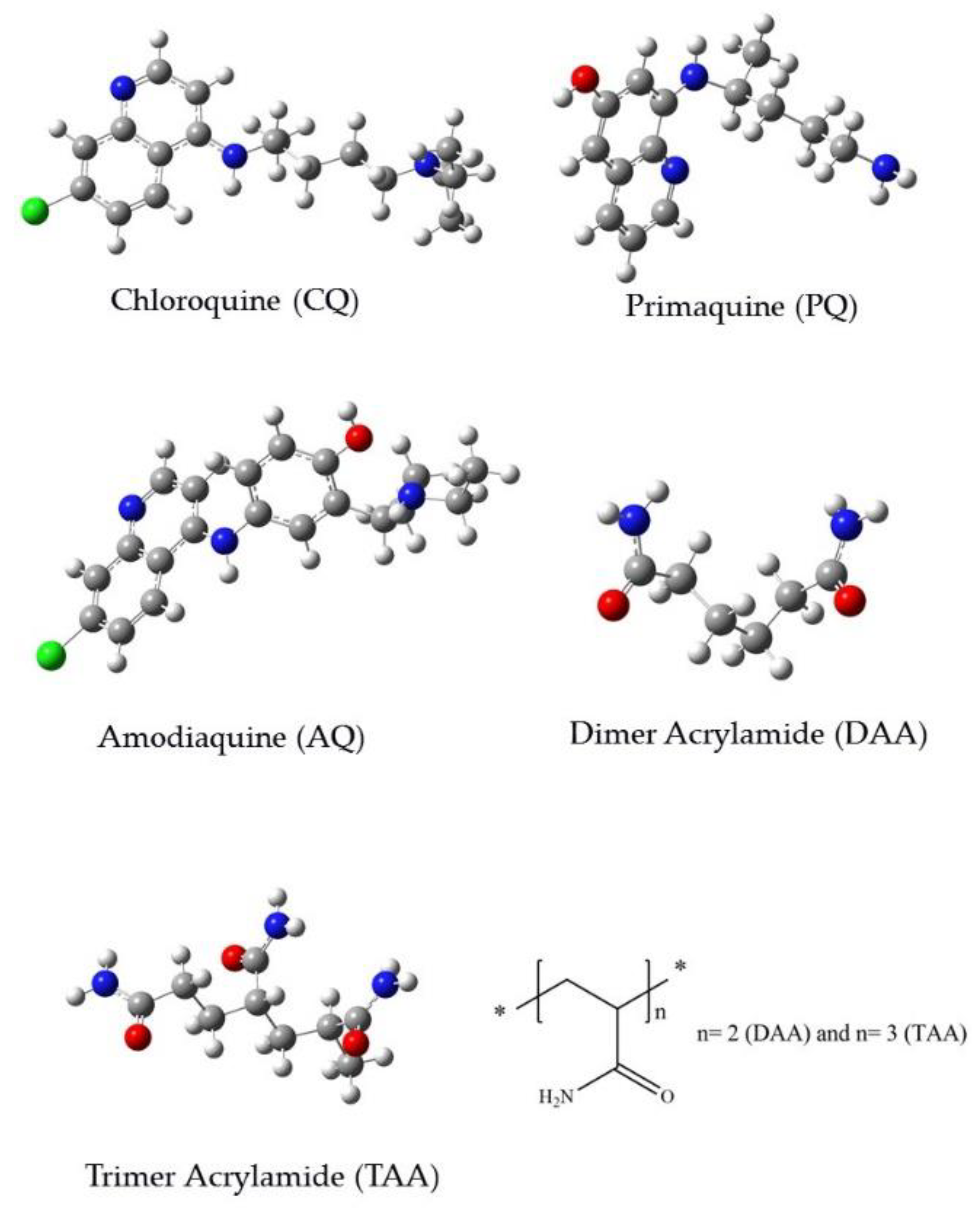{kind=link}
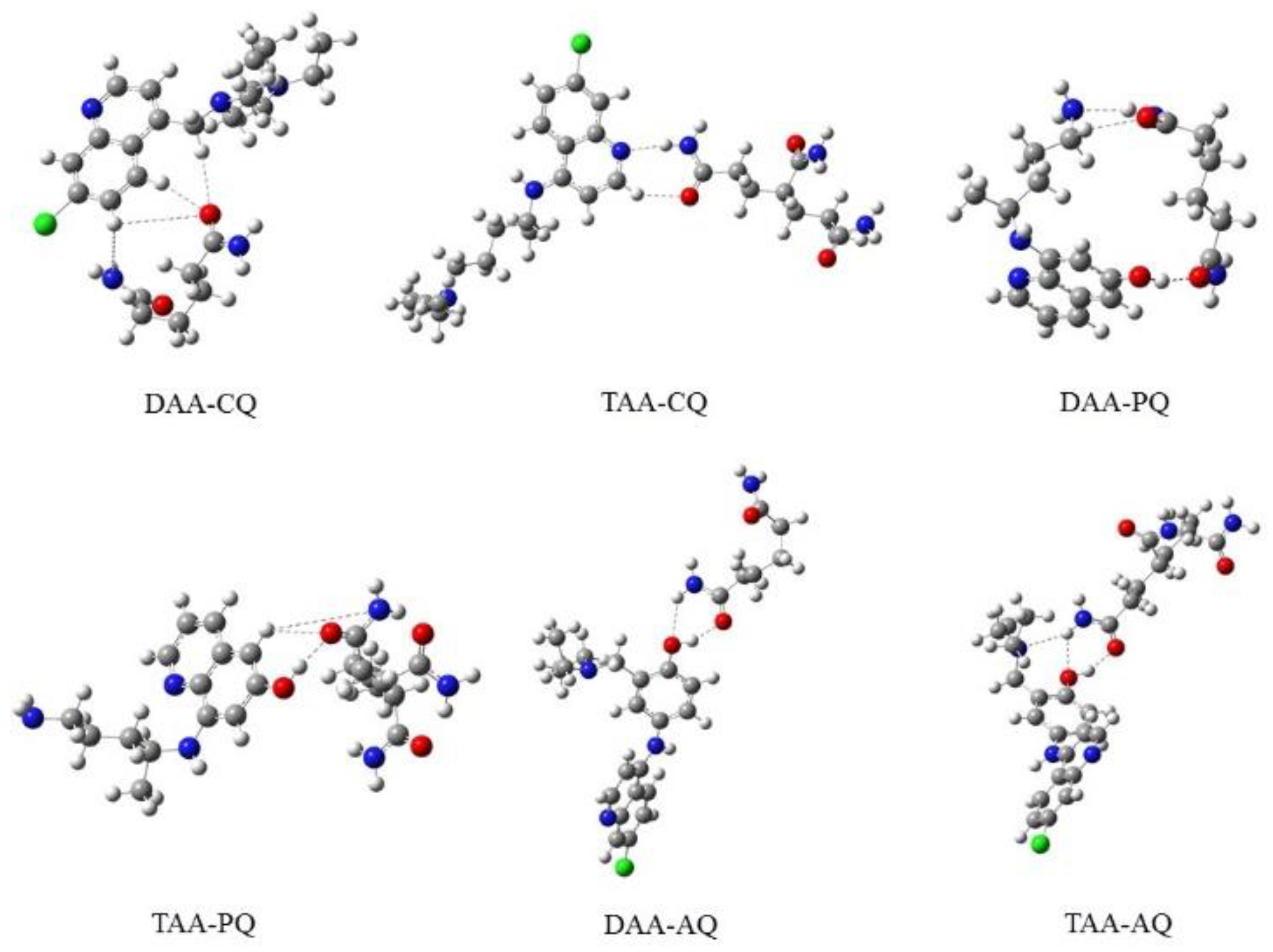{kind=link}
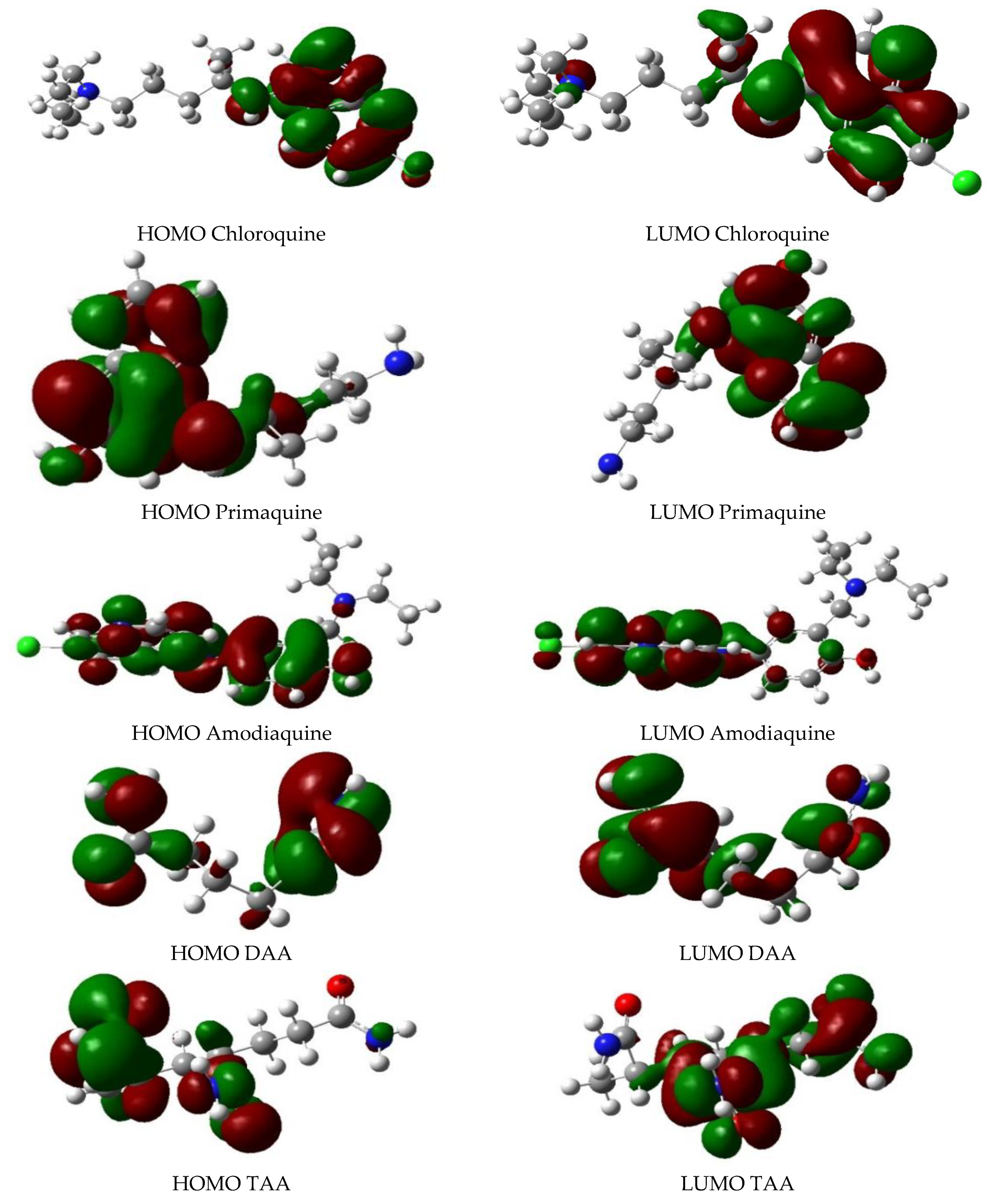{kind=link}
| Complex | Bond Length Å | |||||
|---|---|---|---|---|---|---|
| DAA-CQ | 53O-20H | 53O-29H | 53O-14H | 53O-2C | 49N-14H | 49N-50H |
| 2.450 | 3.181 | 3.364 | 1.230 | 3.542 | 1.89 | |
| DAA-PQ | 5O-59H | 5O-31H | 5O=4C | 58O-59H | 19N-20H | 55N-20H |
| 2.877 | 2.853 | 2.233 | 0.977 | 1.025 | 2.104 | |
| DAA-AQ | 5O-50H | 5O-48H | 49O-50H | 49O-2H | 1N-2H | 1N-50H |
| 2.738 | 2.996 | 0.990 | 2.292 | 1.014 | 2.679 | |
| TAA-CQ | 76O-13H | 76O-75C | 17N-78H | 77N-78H | 77N-13H | 77N-75C |
| 2.339 | 2.233 | 2.004 | 1.027 | 3.149 | 1.356 | |
| TAA-PQ | 65O-37H | 65O-9H | 650-64C | 36O-62H | 36O-37H | 36O-6C |
| 2.738 | 2.752 | 1.242 | 2.682 | 0.989 | 1.354 | |
| TAA-AQ | 75O-29H | 75O-27H | 28O-29H | 28O-77H | 76N-29H | 76N-77H |
| 2.692 | 3.177 | 0.994 | 2.297 | 2.687 | 1.021 | |
| Complex | 10−5 EF (kcal/mol) | 10−5 EH (kJ/mol) | 10−5 EC (kJ/mol) | ΔEb (kJ/mol) |
|---|---|---|---|---|
| DAA-CQ | −5.21 | −3.11 | −11.43 | −03.15 |
| DAA-PQ | −4.92 | −3.11 | −8.030 | −06.51 |
| DAA-AQ | −9.25 | −3.11 | −1.230 | −09.78 |
| TAA-CQ | −8.31 | −4.66 | −12.98 | −05.96 |
| TAA-PQ | −4.92 | −4.66 | −22.78 | −13.20 |
| TAA-AQ | −9.25 | −4.66 | −13.91 | −11.51 |
| Descriptors | DAA-CQ | TAA-CQ | DAA-PQ | TAA-PQ | DAA-AQ | TAA-AQ |
|---|---|---|---|---|---|---|
| HOMO (kJ/mol) | −125.330 | −135.991 | −120.711 | −116.332 | −124.690 | −129.202 |
| LUMO (kJ/mol) | −40.420 | −36.712 | −29.243 | −26.882 | −33.200 | −35.202 |
| -(HOMO-LUMO) (kJ/mol) | 84.901 | 99.281 | 91.472 | 89.453 | 91.491 | 94.002 |
| Global Hardness(n) (kJ/mol) | 42.451 | 49.645 | 45.731 | 44.721 | 45.750 | 47.001 |
| Electronic potential (µ) | −82.881 | −86.353 | −74.971 | −71.612 | −78.941 | −82.201 |
| ΔN (Hydrogel-Drugs) (kJ/mol) | −0.0403 | −0.036 | −0.111 | −0.099 | −0.040 | −0.032 |
| Complex | Donor (i) | Acceptor (j) | E2ij (kJ/mol) |
|---|---|---|---|
| TAA-CQ | LP(1) 76N | BD* 77N-H78 | 14.39 |
| LP(1) Cl | BD* 11C-H13 | 4.140 | |
| LP(2) Cl | BD* 11C-H13 | 8.870 | |
| TAA-PQ | LP(1) 65O | BD* 36O-H37 | 29.12 |
| LP(2) 65O | BD* 36O-H37 | 80.50 | |
| LP(2) 36O | BD* 61C-H62 | 2.170 | |
| TAA-AQ | LP(1) 65O | BD* 36O-H37 | 29.46 |
| LP(2) 65O | BD* 36O-H37 | 97.74 | |
| LP(2) 36O | BD* 61C-H62 | 2.180 |
| Descriptor | TAA-AQ | TAA-CQ | TAA-PQ |
|---|---|---|---|
| LogP | −7.550 | −10.00 | −13.48 |
| Gibbs free energy in water (kJ/mol) | −146.54 | −135.75 | −125.23 |
| Dipole momentum, µ, (Debye) | 6.9400 | 6.5190 | 7.4930 |
| Stabilization energy (EH2O-Egas) kJ/mol | −24.96 | −31.32 | −22.27 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cortes, E.; Márquez, E.; Mora, J.R.; Puello, E.; Rangel, N.; De Moya, A.; Trilleras, J. Theoretical Study of the Adsorption Process of Antimalarial Drugs into Acrylamide-Base Hydrogel Model Using DFT Methods: The First Approach to the Rational Design of a Controlled Drug Delivery System. Processes 2019, 7, 396. https://doi.org/10.3390/pr7070396
Cortes E, Márquez E, Mora JR, Puello E, Rangel N, De Moya A, Trilleras J. Theoretical Study of the Adsorption Process of Antimalarial Drugs into Acrylamide-Base Hydrogel Model Using DFT Methods: The First Approach to the Rational Design of a Controlled Drug Delivery System. Processes. 2019; 7(7):396. https://doi.org/10.3390/pr7070396
Chicago/Turabian StyleCortes, Eliceo, Edgar Márquez, José R. Mora, Esneyder Puello, Norma Rangel, Aldemar De Moya, and Jorge Trilleras. 2019. "Theoretical Study of the Adsorption Process of Antimalarial Drugs into Acrylamide-Base Hydrogel Model Using DFT Methods: The First Approach to the Rational Design of a Controlled Drug Delivery System" Processes 7, no. 7: 396. https://doi.org/10.3390/pr7070396
APA StyleCortes, E., Márquez, E., Mora, J. R., Puello, E., Rangel, N., De Moya, A., & Trilleras, J. (2019). Theoretical Study of the Adsorption Process of Antimalarial Drugs into Acrylamide-Base Hydrogel Model Using DFT Methods: The First Approach to the Rational Design of a Controlled Drug Delivery System. Processes, 7(7), 396. https://doi.org/10.3390/pr7070396