Separating Electronic from Steric Effects in Ethene/α-Olefin Copolymerization: A Case Study on Octahedral [ONNO] Zr-Catalysts

 ,
,  ,
,  ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Polymerization Experiments
2.2. DFT Calculations
3. Results and Discussion
3.1. Ethene/1-Butene Copolymerization
3.2. Computational Modeling
3.2.1. Connection between Electronic/Steric Properties of R1R2 and E/B Copolymerization Performance
3.2.2. Quantitative Kinetic Modeling of Reactivity Ratios for R1R2 in E/P Copolymerization
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Stürzel, M.; Mihan, S.; Mülhaupt, R. From Multisite Polymerization Catalysis to Sustainable Materials and All-Polyolefin Composites. Chem. Rev. 2016, 116, 1398–1433. [Google Scholar] [CrossRef] [PubMed]
- Sauter, D.; Taoufik, M.; Boisson, C. Polyolefins, a Success Story. Polymers 2017, 9, 185. [Google Scholar] [CrossRef] [PubMed]
- Simpson, D.M.; Vaugham, G.A. Encyclopedia of Polymer Science and Technology, 4th ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2014; Volume 5. [Google Scholar]
- Bensason, S.; Minick, J.; Moet, A.; Chum, S.; Hiltner, A.; Baer, E. Classification of homogeneous ethylene-octene copolymers based on comonomer content. J. Polym. Sci. Part B Polym. Phys. 1996, 34, 1301–1315. [Google Scholar] [CrossRef]
- Xu, X.; Xu, J.; Feng, L.; Chen, W. Effect of short chain-branching distribution on crystallinity and modulus of metallocene-based ethylene–butene copolymers. J. Appl. Polym. Sci. 2000, 77, 1709–1715. [Google Scholar] [CrossRef]
- Burfield, D.R. Correlation between crystallinity and ethylene content in LLDPE and related ethylene copolymers: Demonstration of the applicability of a simple empirical relationship. Macromolecules 1987, 20, 3020–3023. [Google Scholar] [CrossRef]
- Baier, M.C.; Zuideveld, M.A.; Mecking, S. Post-Metallocenes in the Industrial Production of Polyolefins. Angew. Chem. Int. Ed. 2014, 53, 9722–9744. [Google Scholar] [CrossRef] [PubMed]
- Klosin, J.; Fontaine, P.P.; Figueroa, R. Development of Group IV Molecular Catalysts for High Temperature Ethylene-α-Olefin Copolymerization Reactions. Acc. Chem. Res. 2015, 48, 2004–2016. [Google Scholar] [CrossRef] [PubMed]
- Kaminsky, W. The discovery of metallocene catalysts and their present state of the art. J. Polym. Sci. Part A Polym. Chem. 2004, 42, 3911–3921. [Google Scholar] [CrossRef]
- Ehm, C.; Zaccaria, F.; Cipullo, R. From Mechanistic Investigation to Quantitative Prediction. In Computational Quantum Chemistry; Soroush, M., Ed.; Elsevier: Amsterdam, The Netherlands, 2019; pp. 287–326. [Google Scholar]
- Hooper, M.S.; Michalak, A. Computational Modeling of Polymerization Catalysts. In Handbook of Transition Metal Polymerization Catalysts; Wiley-VCH: Weinheim, Germany, 2018. [Google Scholar]
- Rappé, A.K.; Skiff, W.M.; Casewit, C.J. Modeling Metal-Catalyzed Olefin Polymerization. Chem. Rev. 2000, 100, 1435–1456. [Google Scholar] [CrossRef]
- Corradini, P.; Guerra, G.; Cavallo, L. Do New Century Catalysts Unravel the Mechanism of Stereocontrol of Old Ziegler−Natta Catalysts? Acc. Chem. Res. 2004, 37, 231–241. [Google Scholar] [CrossRef]
- Coates, G.W. Precise Control of Polyolefin Stereochemistry Using Single-Site Metal Catalysts. Chem. Rev. 2000, 100, 1223–1252. [Google Scholar] [CrossRef] [PubMed]
- Resconi, L.; Cavallo, L.; Fait, A.; Piemontesi, F. Selectivity in Propene Polymerization with Metallocene Catalysts. Chem. Rev. 2000, 100, 1253–1346. [Google Scholar] [CrossRef]
- Busico, V.; Cipullo, R. Microstructure of polypropylene. Prog. Polym. Sci. 2001, 26, 443–533. [Google Scholar] [CrossRef]
- Talarico, G.; Budzelaar, P.H.M. Analysis of Stereochemistry Control in Homogeneous Olefin Polymerization Catalysis. Organometallics 2014, 33, 5974–5982. [Google Scholar] [CrossRef]
- Castro, L.; Therukauff, G.; Vantomme, A.; Welle, A.; Haspeslagh, L.; Brusson, J.M.; Maron, L.; Carpentier, J.F.; Kirillov, E. A Theoretical Outlook on the Stereoselectivity Origins of Isoselective Zirconocene Propylene Polymerization Catalysts. Chem. Eur. J. 2018, 24, 10784–10792. [Google Scholar] [CrossRef] [PubMed]
- Ehm, C.; Vittoria, A.; Goryunov, G.P.; Kulyabin, P.S.; Budzelaar, P.H.M.; Voskoboynikov, A.Z.; Busico, V.; Uborsky, D.V.; Cipullo, R. Connection of Stereoselectivity, Regioselectivity, and Molecular Weight Capability in rac-R′2Si(2-Me-4-R-indenyl)2ZrCl2 Type Catalysts. Macromolecules 2018, 51, 8073–8083. [Google Scholar] [CrossRef]
- Talarico, G.; Budzelaar, P.H.M. Ligand Coordination Driven by Monomer and Polymer Chain: The Intriguing Case of Salalen–Ti Catalyst for Propene Polymerization. Macromolecules 2017, 50, 5332–5336. [Google Scholar] [CrossRef]
- Domski, G.J.; Eagan, J.M.; De Rosa, C.; Di Girolamo, R.; LaPointe, A.M.; Lobkovsky, E.B.; Talarico, G.; Coates, G.W. Combined Experimental and Theoretical Approach for Living and Isoselective Propylene Polymerization. ACS Catal. 2017, 7, 6930–6937. [Google Scholar] [CrossRef]
- Milano, G.; Cavallo, L.; Guerra, G. Site Chirality as a Messenger in Chain-End Stereocontrolled Propene Polymerization. J. Am. Chem. Soc. 2002, 124, 13368–13369. [Google Scholar] [CrossRef]
- Brintzinger, H.H.; Fischer, D.; Mülhaupt, R.; Rieger, B.; Waymouth, R.M. Stereospecific Olefin Polymerization with Chiral Metallocene Catalysts. Angew. Chem. Int. Ed. Eng. 1995, 34, 1143–1170. [Google Scholar] [CrossRef]
- Kakugo, M.; Naito, Y.; Mizunuma, K.; Miyatake, T. Carbon-13 NMR determination of monomer sequence distribution in ethylene-propylene copolymers prepared with δ-titanium trichloride-diethylaluminum chloride. Macromolecules 1982, 15, 1150–1152. [Google Scholar] [CrossRef]
- Randall, J.C. A review of high resolution liquind 13carbon nuclear magnetic resonance characterizations of ethylene-based polymers. J. Macromol. Sci. Part C 1989, 29, 201–317. [Google Scholar] [CrossRef]
- Busico, V.; Cipullo, R.; Segre, A.L. Advances in the 13C NMR characterization of ethene/propene copolymers, 1. Macromol. Chem. Phys. 2002, 203, 1403–1412. [Google Scholar] [CrossRef]
- Dankova, M.; Waymouth, R.M. High Comonomer Selectivity in Ethylene/Hexene Copolymerization by Unbridged Indenyl Metallocenes. Macromolecules 2003, 36, 3815–3820. [Google Scholar] [CrossRef]
- Reybuck, S.E.; Meyer, A.; Waymouth, R.M. Copolymerization Behavior of Unbridged Indenyl Metallocenes: Substituent Effects on the Degree of Comonomer Incorporation. Macromolecules 2002, 35, 637–643. [Google Scholar] [CrossRef]
- Kunz, K.; Erker, G.; Kehr, G.; Fröhlich, R.; Jacobsen, H.; Berke, H.; Blacque, O. Formation of Cyclodimeric (sp2-C1)-Bridged Cp/-Oxido (“CpC1O”MIVX2) Group 4 Metal Ziegler−Natta Catalyst SystemsHow Important Is the “Constrained Geometry” Effect? J. Am. Chem. Soc. 2002, 124, 3316–3326. [Google Scholar] [CrossRef] [PubMed]
- Möhring, P.C.; Coville, N.J. The influence of cyclopentadienyl ring substituent steric and electronic effects on the ethylene-α-olefin copolymerisation behaviour of (CpR)2ZrCl2ethylalumoxane catalysts. J. Mol. Catal. A Chem. 1995, 96, 181–195. [Google Scholar] [CrossRef]
- Thornberry, M.P.; Reynolds, N.T.; Deck, P.A.; Fronczek, F.R.; Rheingold, A.L.; Liable-Sands, L.M. Synthesis, Structure, and Olefin Polymerization Catalytic Behavior of Aryl-Substituted Zirconocene Dichlorides. Organometallics 2004, 23, 1333–1339. [Google Scholar] [CrossRef]
- Lehmus, P.; Kokko, E.; Härkki, O.; Leino, R.; Luttikhedde, H.J.G.; Näsman, J.H.; Seppälä, J.V. Homo- and Copolymerization of Ethylene and α-Olefins over 1- and 2-Siloxy-Substituted Ethylenebis(indenyl)zirconium and Ethylenebis(tetrahydroindenyl)zirconium Dichlorides. Macromolecules 1999, 32, 3547–3552. [Google Scholar] [CrossRef]
- Zaccaria, F.; Ehm, C.; Budzelaar, P.H.M.; Busico, V. Accurate Prediction of Copolymerization Statistics in Molecular Olefin Polymerization Catalysis: The Role of Entropic, Electronic, and Steric Effects in Catalyst Comonomer Affinity. ACS Catal. 2017, 7, 1512–1519. [Google Scholar] [CrossRef]
- Yano, A.; Hasegawa, S.; Kaneko, T.; Sone, M.; Sato, M.; Akimoto, A. Ethylene/1-hexene copolymerization with Ph2C(Cp)(Flu)ZrCl2 derivatives: Correlation between ligand structure and copolymerization behavior at high temperature. Macromol. Chem. Phys. 1999, 200, 1542–1553. [Google Scholar] [CrossRef]
- Gassman, P.G.; Mickelson, J.W.; Sowa, J.R. 1,2,3,4-Tetramethyl-5-(trifluoromethyl)cyclopentadienide: A unique ligand with the steric properties of pentamethylcyclopentadienide and the electronic properties of cyclopentadienide. J. Am. Chem. Soc. 1992, 114, 6942–6944. [Google Scholar] [CrossRef]
- Nsiri, H.; Belaid, I.; Larini, P.; Thuilliez, J.; Boisson, C.; Perrin, L. Ethylene–Butadiene Copolymerization by Neodymocene Complexes: A Ligand Structure/Activity/Polymer Microstructure Relationship Based on DFT Calculations. ACS Catal. 2016, 6, 1028–1036. [Google Scholar] [CrossRef]
- Friederichs, N.; Wang, B.; Budzelaar, P.H.M.; Coussens, B.B. A combined experimental—molecular modeling approach for ethene–propene copolymerization with C2-symmetric metallocenes. J. Mol. Catal. A Chem. 2005, 242, 91–104. [Google Scholar] [CrossRef]
- Laine, A.; Coussens, B.B.; Hirvi, J.T.; Berthoud, A.; Friederichs, N.; Severn, J.R.; Linnolahti, M. Effect of Ligand Structure on Olefin Polymerization by a Metallocene/Borate Catalyst: A Computational Study. Organometallics 2015, 34, 2415–2421. [Google Scholar] [CrossRef]
- Zaccaria, F.; Cipullo, R.; Budzelaar, P.H.M.; Busico, V.; Ehm, C. Backbone rearrangement during olefin capture as the rate limiting step in molecular olefin polymerization catalysis and its effect on comonomer affinity. J. Polym. Sci. Part A Polym. Chem. 2017, 55, 2807–2814. [Google Scholar] [CrossRef]
- Tshuva, E.Y.; Goldberg, I.; Kol, M. Isospecific Living Polymerization of 1-Hexene by a Readily Available Nonmetallocene C2-Symmetrical Zirconium Catalyst. J. Am. Chem. Soc. 2000, 122, 10706–10707. [Google Scholar] [CrossRef]
- Busico, V.; Cipullo, R.; Pellecchia, R.; Ronca, S.; Roviello, G.; Talarico, G. Design of stereoselective Ziegler–Natta propene polymerization catalysts. Proc. Natl. Acad. Sci. USA 2006, 103, 15321–15326. [Google Scholar] [CrossRef]
- Ciancaleoni, G.; Fraldi, N.; Cipullo, R.; Busico, V.; Macchioni, A.; Budzelaar, P.H.M. Structure/Properties Relationship for Bis(phenoxyamine)Zr(IV)-Based Olefin Polymerization Catalysts: A Simple DFT Model To Predict Catalytic Activity. Macromolecules 2012, 45, 4046–4053. [Google Scholar] [CrossRef]
- Ciancaleoni, G.; Fraldi, N.; Budzelaar, P.H.M.; Busico, V.; Cipullo, R.; Macchioni, A. Structure−Activity Relationship in Olefin Polymerization Catalysis: Is Entropy the Key? J. Am. Chem. Soc. 2010, 132, 13651–13653. [Google Scholar] [CrossRef]
- Busico, V.; Cipullo, R.; Friederichs, N.; Ronca, S.; Talarico, G.; Togrou, M.; Wang, B. Block Copolymers of Highly Isotactic Polypropylene via Controlled Ziegler−Natta Polymerization. Macromolecules 2004, 37, 8201–8203. [Google Scholar] [CrossRef]
- Busico, V.; Cipullo, R.; Ronca, S.; Budzelaar, P.H.M. Mimicking Ziegler-Natta Catalysts in Homogeneous Phase, 1. C2-Symmetric Octahedral Zr(IV) Complexes with Tetradentate [ONNO]-Type Ligands. Macromol. Rapid Commun. 2001, 22, 1405–1410. [Google Scholar] [CrossRef]
- Segal, S.; Goldberg, I.; Kol, M. Zirconium and Titanium Diamine Bis(phenolate) Catalysts for α-Olefin Polymerization: From Atactic Oligo(1-hexene) to Ultrahigh-Molecular-Weight Isotactic Poly(1-hexene). Organometallics 2005, 24, 200–202. [Google Scholar] [CrossRef]
- Cohen, A.; Kopilov, J.; Goldberg, I.; Kol, M. C1-Symmetric Zirconium Complexes of [ONNO′]-Type Salan Ligands: Accurate Control of Catalyst Activity, Isospecificity, and Molecular Weight in 1-Hexene Polymerization. Organometallics 2009, 28, 1391–1405. [Google Scholar] [CrossRef]
- Busico, V.; Cipullo, R.; Romanelli, V.; Ronca, S.; Togrou, M. Reactivity of Secondary Metal−Alkyls in Catalytic Propene Polymerization: How Dormant Are “Dormant Chains”? J. Am. Chem. Soc. 2005, 127, 1608–1609. [Google Scholar] [CrossRef]
- Busico, V.; Cipullo, R.; Friederichs, N.; Ronca, S.; Togrou, M. The First Molecularly Characterized Isotactic Polypropylene-block-polyethylene Obtained via “Quasi-Living” Insertion Polymerization. Macromolecules 2003, 36, 3806–3808. [Google Scholar] [CrossRef]
- Preston, A.Z.; Kim, J.; Medvedev, G.A.; Delgass, W.N.; Caruthers, J.M.; Abu-Omar, M.M. Steric and Solvation Effects on Polymerization Kinetics, Dormancy, and Tacticity of Zr-Salan Catalysts. Organometallics 2017, 36, 2237–2244. [Google Scholar] [CrossRef]
- Ciancaleoni, G.; Fraldi, N.; Budzelaar, P.H.M.; Busico, V.; Macchioni, A. Activation of a bis(phenoxy-amine) precatalyst for olefin polymerisation: First evidence for an outer sphere ion pair with the methylborate counterion. Dalton Trans. 2009, 8824–8827. [Google Scholar] [CrossRef]
- Busico, V.; Cipullo, R.; Cutillo, F.; Friederichs, N.; Ronca, S.; Wang, B. Improving the Performance of Methylalumoxane: A Facile and Efficient Method to Trap “Free” Trimethylaluminum. J. Am. Chem. Soc. 2003, 125, 12402–12403. [Google Scholar] [CrossRef]
- Zaccaria, F.; Zuccaccia, C.; Cipullo, R.; Budzelaar, P.H.M.; Macchioni, A.; Busico, V.; Ehm, C. BHT-Modified MAO: Cage Size Estimation, Chemical Counting of Strongly Acidic Al Sites, and Activation of a Ti-Phosphinimide Precatalyst. ACS Catal. 2019, 9, 2996–3010. [Google Scholar] [CrossRef]
- Kissin, Y. Isospecific Polymerization of Olefins: With Heterogeneous Ziegler-Natta Catalysts; Springer: New York, NY, USA, 1985. [Google Scholar]
- Hamm, G.E. In “Copolymerization”; Hamm, G.E., Ed.; Wiley-Interscience: New York, NY, USA, 1964; Chapter I. [Google Scholar]
- Ehm, C.; Budzelaar, P.H.M.; Busico, V. Calculating accurate barriers for olefin insertion and related reactions. J. Organomet. Chem. 2015, 775, 39–49. [Google Scholar] [CrossRef]
- Gaussian 09, Revision B.1; Gaussian, Inc.: Wallingford, CT, USA, 2009; for the full citation see the Supporting Information.
- Baker, J. PQS, version 2.4; Parallel Quantum Solutions: Fayetteville, AR, USA, 2001. [Google Scholar]
- Baker, J. An algorithm for the location of transition states. J. Comput. Chem. 1986, 7, 385–395. [Google Scholar] [CrossRef]
- Budzelaar, P.H.M. Geometry optimization using generalized, chemically meaningful constraints. J. Comput. Chem. 2007, 28, 2226–2236. [Google Scholar] [CrossRef]
- Tao, J.; Perdew, J.P.; Staroverov, V.N.; Scuseria, G.E. Climbing the Density Functional Ladder: Nonempirical Meta\char21{}Generalized Gradient Approximation Designed for Molecules and Solids. Phys. Rev. Lett. 2003, 91, 146401. [Google Scholar] [CrossRef] [PubMed]
- Peterson, K.A.; Figgen, D.; Dolg, M.; Stoll, H. Energy-consistent relativistic pseudopotentials and correlation consistent basis sets for the 4d elements Y–Pd. J. Chem. Phys. 2007, 126, 142101. [Google Scholar] [CrossRef] [PubMed]
- Schuchardt, K.L.; Didier, B.T.; Elsethagen, T.; Sun, L.; Gurumoorthi, V.; Chase, J.; Li, J.; Windus, T.L. Basis Set Exchange: A Community Database for Computational Sciences. J. Chem. Inf. Model. 2007, 47, 1045–1052. [Google Scholar] [CrossRef] [PubMed]
- Whitten, J.L. Coulombic potential energy integrals and approximations. J. Chem. Phys. 1973, 58, 4496–4501. [Google Scholar] [CrossRef]
- Baerends, E.J.; Ellis, D.E.; Ros, P. Self-consistent molecular Hartree—Fock—Slater calculations I. The computational procedure. Chem. Phys. 1973, 2, 41–51. [Google Scholar] [CrossRef]
- Feyereisen, M.; Fitzgerald, G.; Komornicki, A. Use of approximate integrals in ab initio theory. An application in MP2 energy calculations. Chem. Phys. Lett. 1993, 208, 359–363. [Google Scholar] [CrossRef]
- Vahtras, O.; Almlöf, J.; Feyereisen, M.W. Integral approximations for LCAO-SCF calculations. Chem. Phys. Lett. 1993, 213, 514–518. [Google Scholar] [CrossRef]
- Flisak, Z.; Ziegler, T. “Dormant” secondary metal-alkyl complexes are not omnipresent. Proc. Natl. Acad. Sci. USA 2006, 103, 15338–15342. [Google Scholar] [CrossRef] [PubMed]
- Foster, J.P.; Weinhold, F. Natural hybrid orbitals. J. Am. Chem. Soc. 1980, 102, 7211–7218. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other function. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar] [CrossRef]
- Tobisch, S.; Ziegler, T. Catalytic Oligomerization of Ethylene to Higher Linear α-Olefins Promoted by the Cationic Group 4 [(η5-Cp-(CMe2-bridge)-Ph)MII(ethylene)2]+ (M = Ti, Zr, Hf) Active Catalysts: A Density Functional Investigation of the Influence of the Metal on the Catalyt. J. Am. Chem. Soc. 2004, 126, 9059–9071. [Google Scholar] [CrossRef] [PubMed]
- Crabtree, R.H. Deactivation in Homogeneous Transition Metal Catalysis: Causes, Avoidance, and Cure. Chem. Rev. 2015, 115, 127–150. [Google Scholar] [CrossRef] [PubMed]
- Cipullo, R.; Melone, P.; Yu, Y.; Iannone, D.; Busico, V. Olefin polymerisation catalysts: When perfection is not enough. Dalton Trans. 2015, 44, 12304–12311. [Google Scholar] [CrossRef] [PubMed]
- Zaccaria, F.; Ehm, C.; Budzelaar, P.H.M.; Busico, V.; Cipullo, R. Catalyst Mileage in Olefin Polymerization: The Peculiar Role of Toluene. Organometallics 2018, 37, 2872–2879. [Google Scholar] [CrossRef]
- Desert, X.; Carpentier, J.-F.; Kirillov, E. Quantification of active sites in single-site group 4 metal olefin polymerization catalysis. Coord. Chem. Rev. 2019, 386, 50–68. [Google Scholar] [CrossRef]
- Alameddin, N.G.; Ryan, M.F.; Eyler, J.R.; Siedle, A.R.; Richardson, D.E. Intrinsic Ancillary Ligand Effects in Cationic Zirconium Polymerization Catalysts: Reactions of [L2ZrCH3]+ Cations with H2 and C2H4. Organometallics 1995, 14, 5005–5007. [Google Scholar] [CrossRef]
- Richardson, D.E.; Alameddin, N.G.; Ryan, M.F.; Hayes, T.; Eyler, J.R.; Siedle, A.R. Intrinsic Ancillary Ligand Effects in Cationic Zirconium Polymerization Catalysts: Gas-Phase Reactions of [L2ZrCH3]+ Cations with Alkenes. J. Am. Chem. Soc. 1996, 118, 11244–11253. [Google Scholar] [CrossRef]
- Macchioni, A. Ion Pairing in Transition-Metal Organometallic Chemistry. Chem. Rev. 2005, 105, 2039–2074. [Google Scholar] [CrossRef] [PubMed]
- Resconi, L.; Camurati, I.; Sudmeijer, O. Chain transfer reactions in propylene polymerization with zirconocene catalysts. Top. Catal. 1999, 7, 145–163. [Google Scholar] [CrossRef]
- Ehm, C.; Antinucci, G.; Budzelaar, P.H.M.; Busico, V. Catalyst activation and the dimerization energy of alkylaluminium compounds. J. Organomet. Chem. 2014, 772–773, 161–171. [Google Scholar] [CrossRef]
- Ehm, C.; Budzelaar, P.H.M.; Busico, V. Metal–carbon bond strengths under polymerization conditions: 2,1-insertion as a catalyst stress test. J. Catal. 2017, 351, 146–152. [Google Scholar] [CrossRef]
- Ehm, C.; Budzelaar, P.H.M.; Busico, V. Tuning the Relative Energies of Propagation and Chain Termination Barriers in Polyolefin Catalysis through Electronic and Steric Effects. Eur. J. Inorg. Chem. 2017, 3343–3349. [Google Scholar] [CrossRef]
- Ishii, A.; Ikuma, K.; Nakata, N.; Nakamura, K.; Kuribayashi, H.; Takaoki, K. Zirconium and Hafnium Complexes with Cycloheptane- or Cyclononane-Fused [OSSO]-Type Bis(phenolato) Ligands: Synthesis, Structure, and Highly Active 1-Hexene Polymerization and Ring-Size Effects of Fused Cycloalkanes on the Activity. Organometallics 2017, 36, 3954–3966. [Google Scholar] [CrossRef]
- Liptak, M.D.; Gross, K.C.; Seybold, P.G.; Feldgus, S.; Shields, G.C. Absolute pKa Determinations for Substituted Phenols. J. Am. Chem. Soc. 2002, 124, 6421–6427. [Google Scholar] [CrossRef]
- Falivene, L.; Credendino, R.; Poater, A.; Petta, A.; Serra, L.; Oliva, R.; Scarano, V.; Cavallo, L. SambVca 2. A Web Tool for Analyzing Catalytic Pockets with Topographic Steric Maps. Organometallics 2016, 35, 2286–2293. [Google Scholar] [CrossRef]
- Clavier, H.; Correa, A.; Cavallo, L.; Escudero-Adán, E.C.; Benet-Buchholz, J.; Slawin, A.M.Z.; Nolan, S.P. [Pd(NHC)(allyl)Cl] Complexes: Synthesis and Determination of the NHC Percent Buried Volume (%Vbur) Steric Parameter. Eur. J. Inorg. Chem. 2009, 2009, 1767–1773. [Google Scholar] [CrossRef]
- Poater, A.; Cosenza, B.; Correa, A.; Giudice, S.; Ragone, F.; Scarano, V.; Cavallo, L. SambVca: A Web Application for the Calculation of the Buried Volume of N-Heterocyclic Carbene Ligands. Eur. J. Inorg. Chem. 2009, 2009, 1759–1766. [Google Scholar] [CrossRef]
- Galimberti, M.; Mascellani, N.; Piemontesi, F.; Camurati, I. Random ethene/propene copolymerization from a catalyst system based on a “constrained geometry” half-sandwich complex. Macromol. Rapid Commun. 1999, 20, 214–218. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
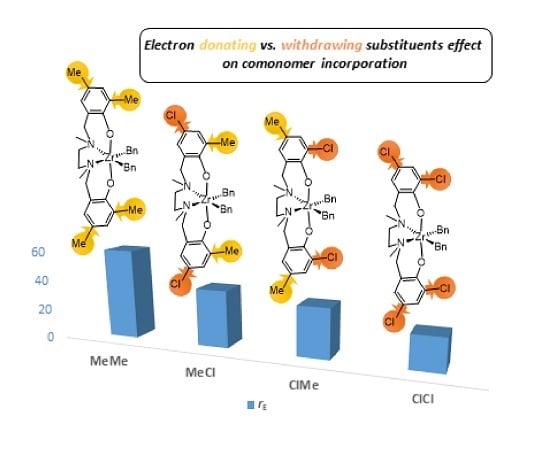
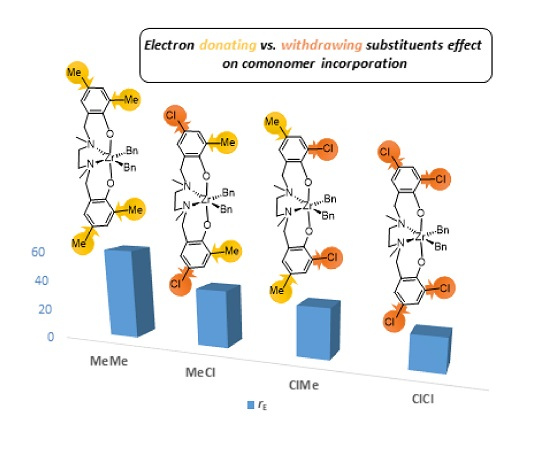
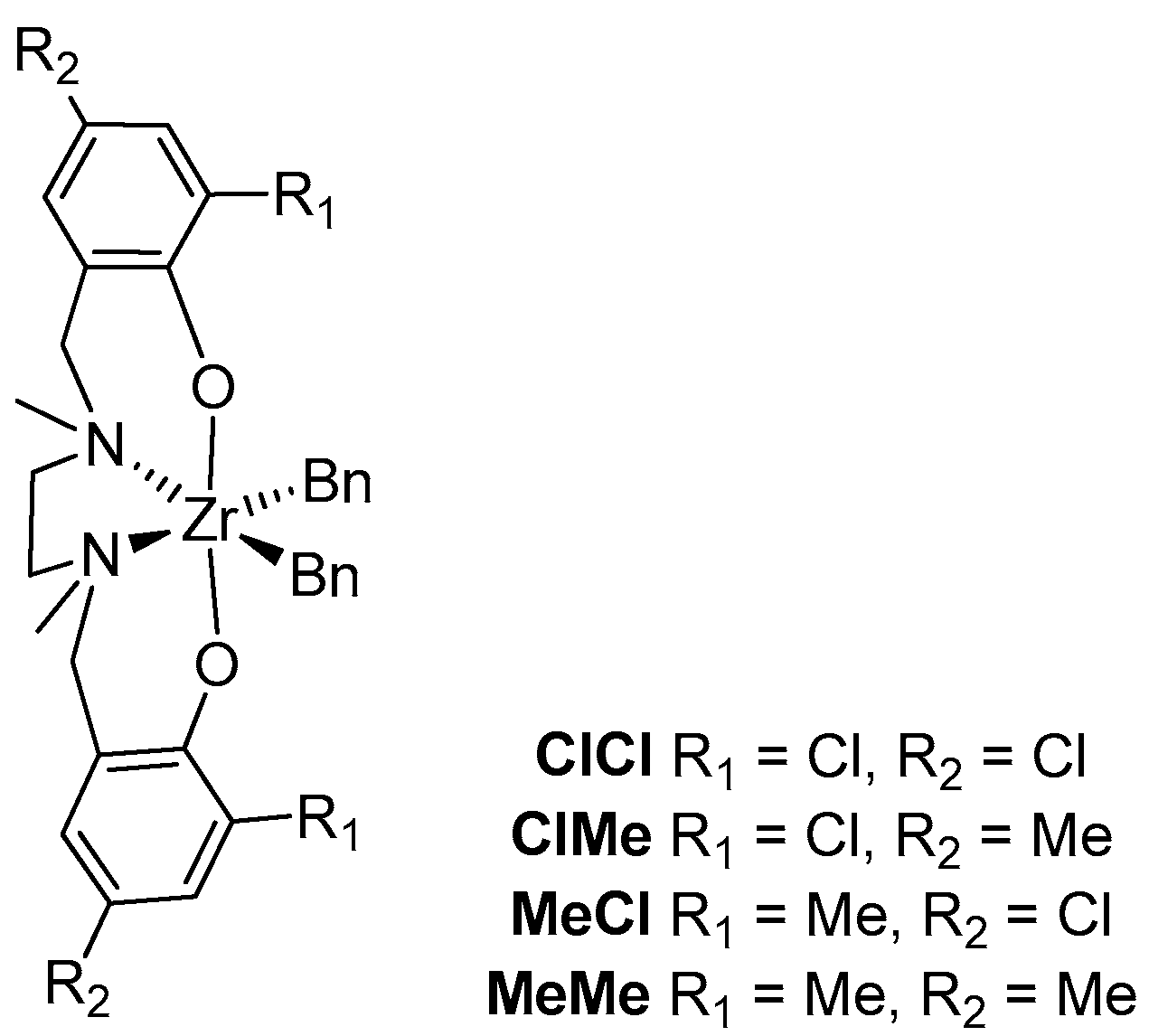
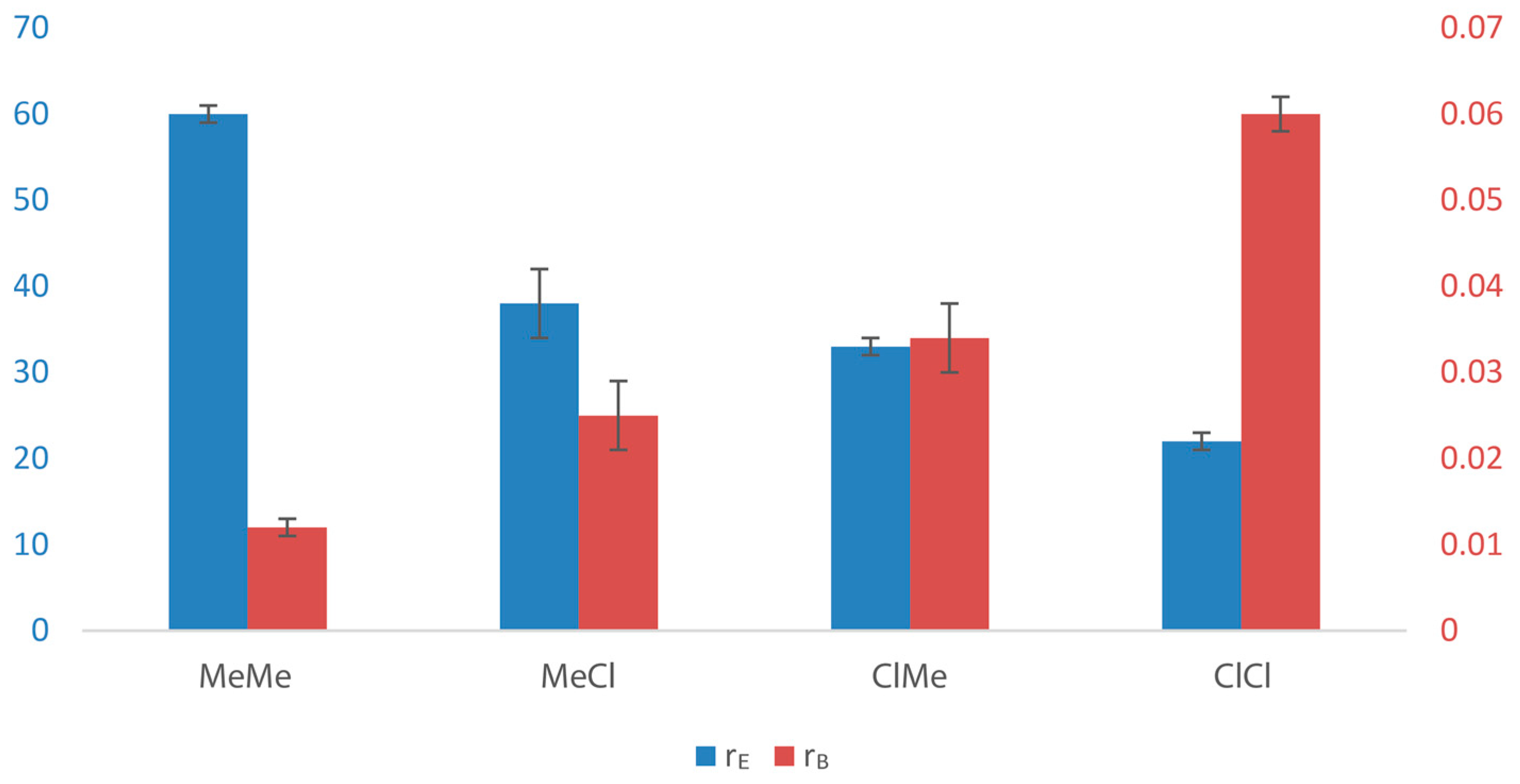
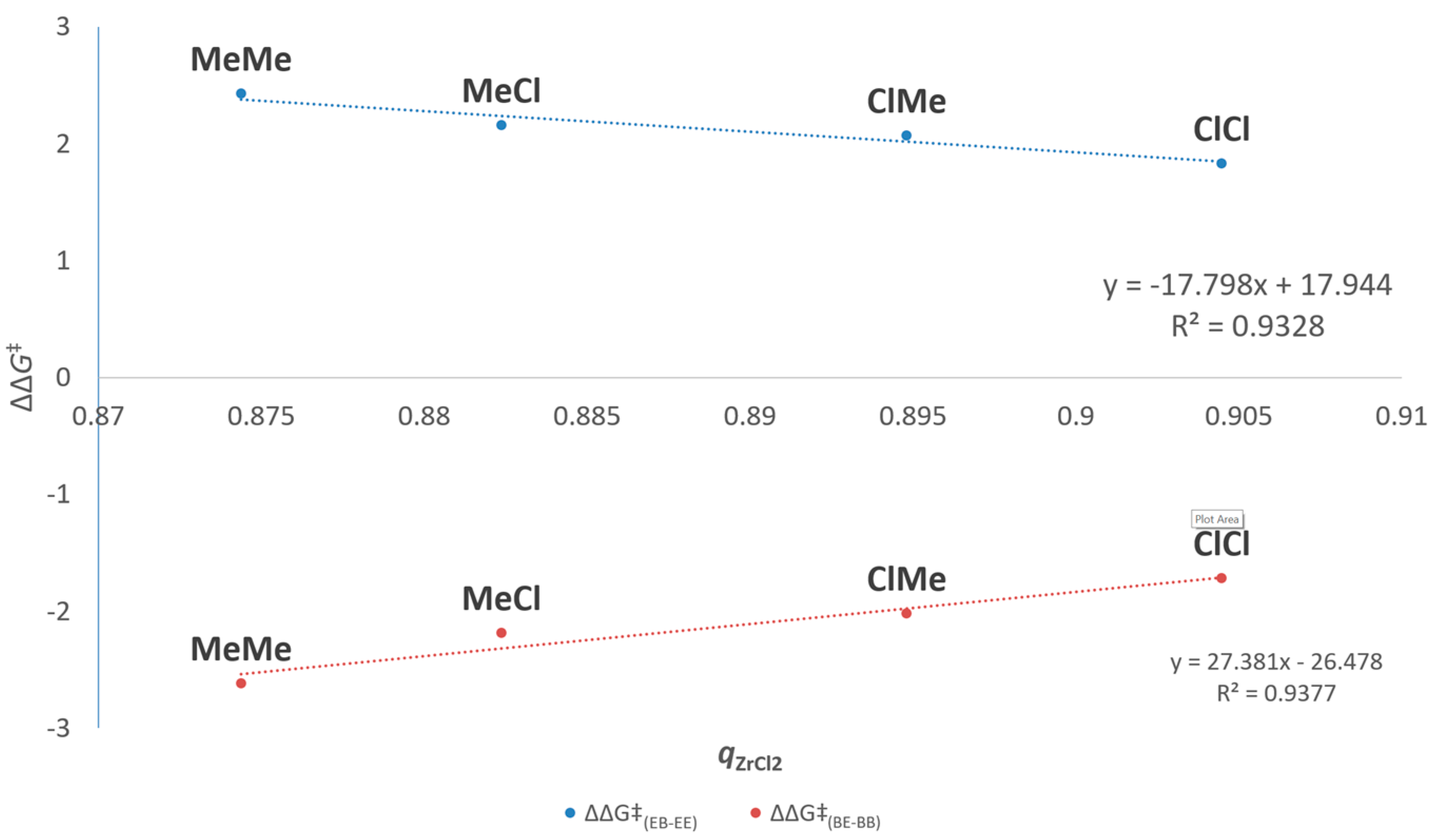
| Entry | Catalyst | Rp(a) | [B]cop (b) | Pn(c) ×10−2 | rE(d) | ΔΔG‡(EB-EE) (e) | rB(d) | ΔΔG‡(BE-BB) (e) | rErB |
|---|---|---|---|---|---|---|---|---|---|
| mol% | |||||||||
| 1 | MeMe | 1 | 24 | 1.00 | 60(1) | 2.4 | 0.012(1) | −2.6 | 0.7 |
| 2 | MeCl | 7 | 32 | 0.53 | 38(4) | 2.2 | 0.025(4) | −2.2 | 1.0 |
| 3 | ClMe | 31 | 37 | 0.24 | 33(1) | 2.1 | 0.034(4) | −2.0 | 1.1 |
| 4 | ClCl | 360 | 43 | 0.22 | 22(1) | 1.8 | 0.06(2) | −1.7 | 1.3 |
| Entry. | Catalyst | Total %Vbur | %Vbur per Quadrant | |||
|---|---|---|---|---|---|---|
| SW | NW | NE | SE | |||
| 1 | MeMe | 49.4 | 48.4 | 50.5 | 48.5 | 50.3 |
| 2 | MeCl | 49.5 | 48.5 | 50.3 | 48.5 | 50.6 |
| 3 | ClMe | 49.1 | 47.7 | 50.4 | 47.7 | 50.5 |
| 4 | ClCl | 49.1 | 47.7 | 50.5 | 47.7 | 50.5 |
| 5 | AdMe | 57.8 | 61.3 | 53.8 | 61.7 | 54.2 |
| Entry | Catalyst | rE | ΔΔG‡(EP-EE) | rP | ΔΔG‡(PE-PP) | rErP |
|---|---|---|---|---|---|---|
| Experimental | ||||||
| 1 | MeMe | 41 | 2.2 | 0.03 | −2.1 | 1.2 |
| 2 | ClCl | 8.7 | 1.3 | 0.19 | −1.0 | 1.7 |
| DFT–INS only | ||||||
| 3 | MeMe | - | 1.5 [0.7] | - | −1.6 [−0.5] | |
| 4 | ClCl | - | 1.3 [0.0] | - | −1.2 [0.2] | |
| DFT–INS and BBRA | ||||||
| 5 | MeMe | - | 1.5 [0.7] | - | −1.6 [−0.5] | |
| 6 | ClCl | - | 0.6 [0.7] | - | −1.2 [0.2] | |
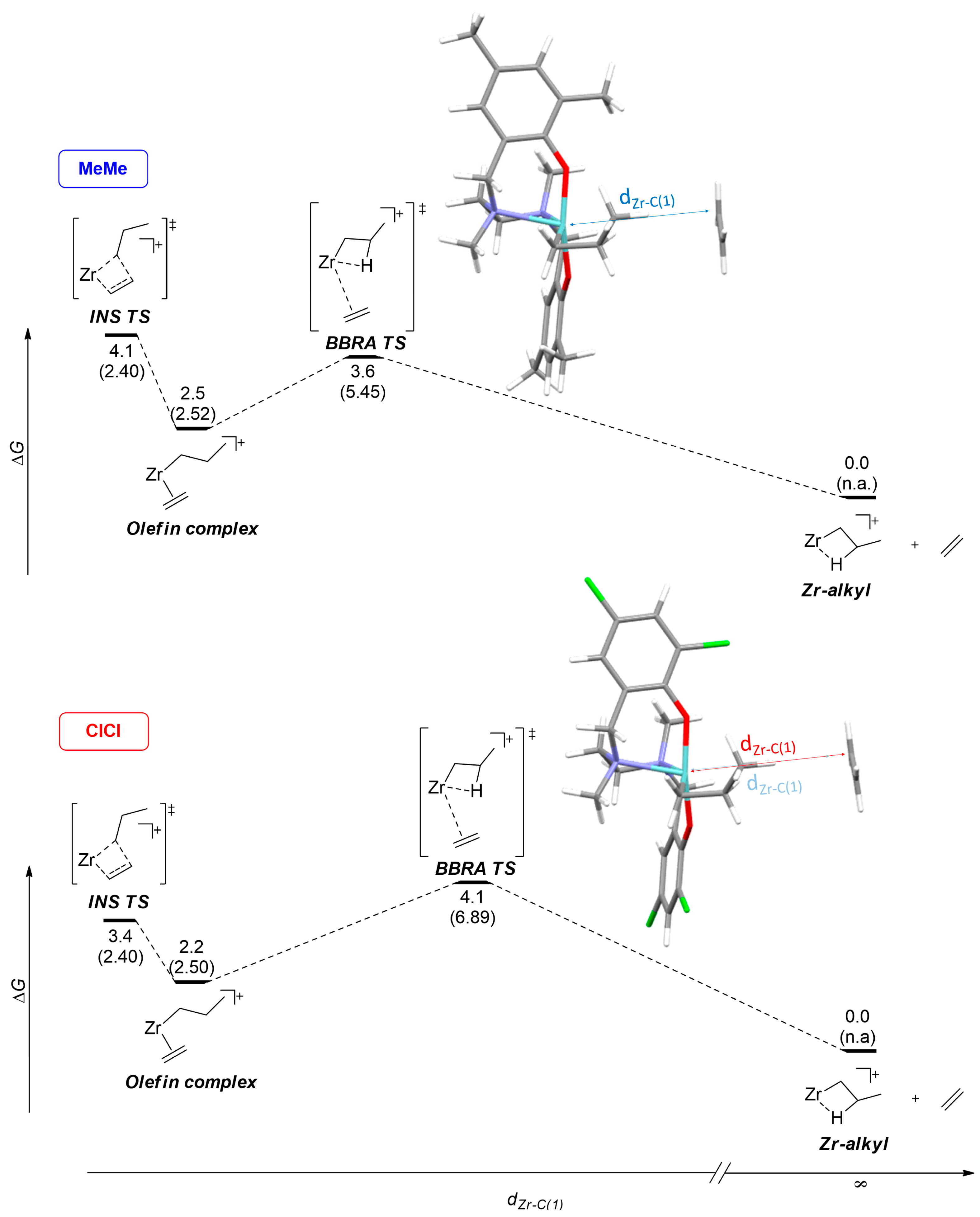
| Entry | Catalyst | Process | ΔG‡ | ΔH‡ | ΔS‡ |
|---|---|---|---|---|---|
| 1 | MeMe | INS | 4.1 | −3.7 | 26 |
| 2 | BBRA | 3.6 | −0.7 | 14 | |
| 3 | ClCl | INS | 3.4 | −4.6 | 27 |
| 4 | BBRA | 4.1 | −0.4 | 15 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zaccaria, F.; Cipullo, R.; Correa, A.; Budzelaar, P.H.M.; Busico, V.; Ehm, C. Separating Electronic from Steric Effects in Ethene/α-Olefin Copolymerization: A Case Study on Octahedral [ONNO] Zr-Catalysts. Processes 2019, 7, 384. https://doi.org/10.3390/pr7060384
Zaccaria F, Cipullo R, Correa A, Budzelaar PHM, Busico V, Ehm C. Separating Electronic from Steric Effects in Ethene/α-Olefin Copolymerization: A Case Study on Octahedral [ONNO] Zr-Catalysts. Processes. 2019; 7(6):384. https://doi.org/10.3390/pr7060384
Chicago/Turabian StyleZaccaria, Francesco, Roberta Cipullo, Andrea Correa, Peter H. M. Budzelaar, Vincenzo Busico, and Christian Ehm. 2019. "Separating Electronic from Steric Effects in Ethene/α-Olefin Copolymerization: A Case Study on Octahedral [ONNO] Zr-Catalysts" Processes 7, no. 6: 384. https://doi.org/10.3390/pr7060384
APA StyleZaccaria, F., Cipullo, R., Correa, A., Budzelaar, P. H. M., Busico, V., & Ehm, C. (2019). Separating Electronic from Steric Effects in Ethene/α-Olefin Copolymerization: A Case Study on Octahedral [ONNO] Zr-Catalysts. Processes, 7(6), 384. https://doi.org/10.3390/pr7060384