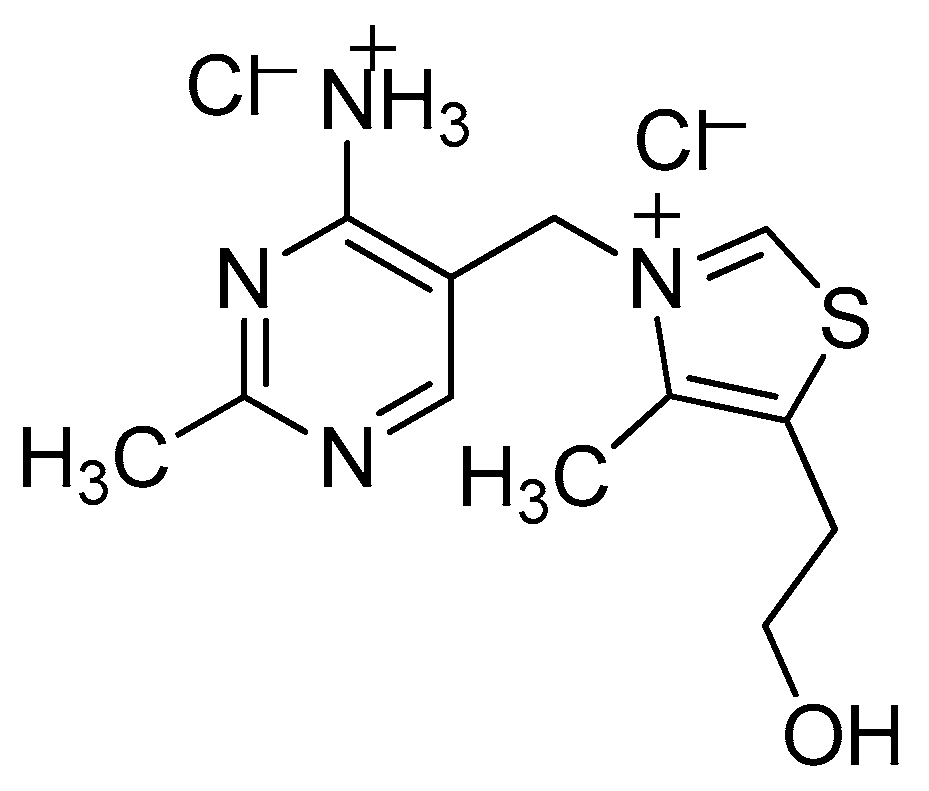
VB1 Promoted Green Synthesis of Chalcones and Its Neuroprotection Potency Evaluation

Abstract
: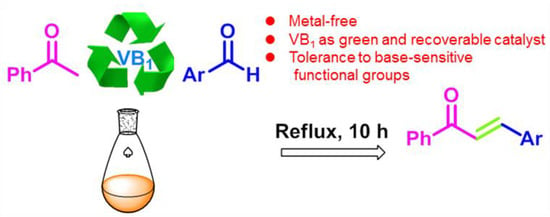
1. Introduction
2. Results and Discussion
2.1. Optimization of the Reaction Conditions
2.2. The Scope of the Substrates
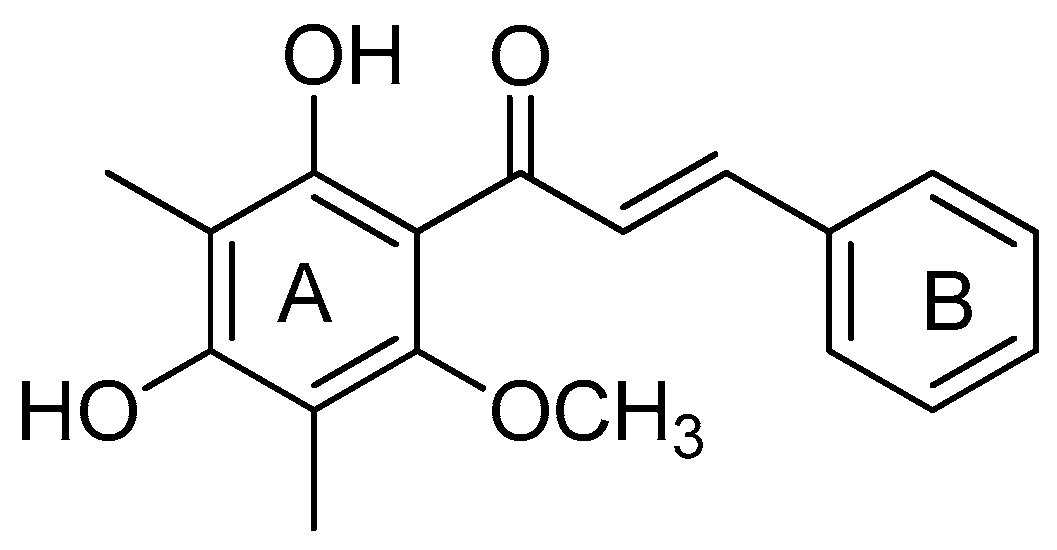
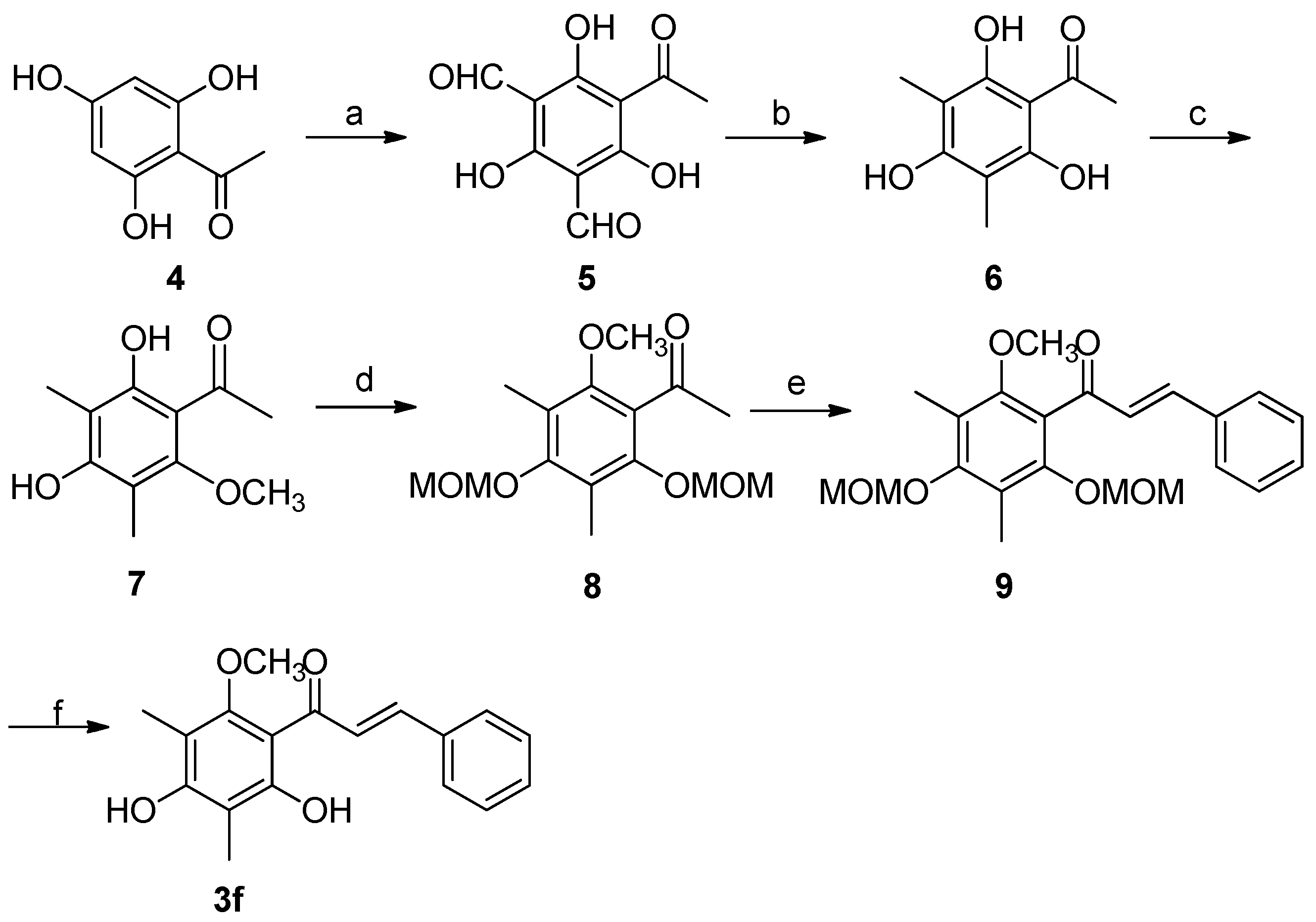
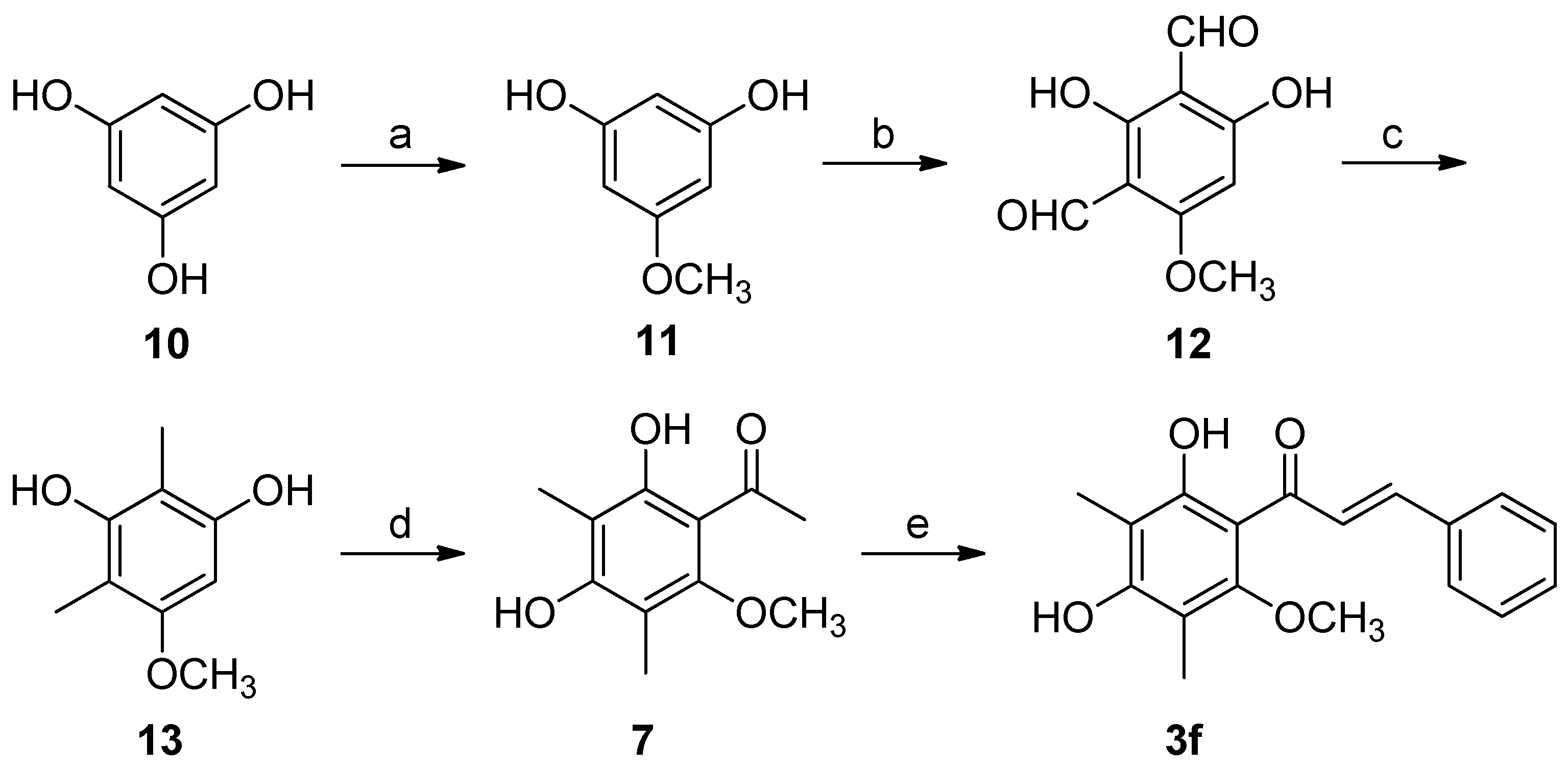
2.3. Synthesis of Natural Product 3f
2.4. Biological Activity
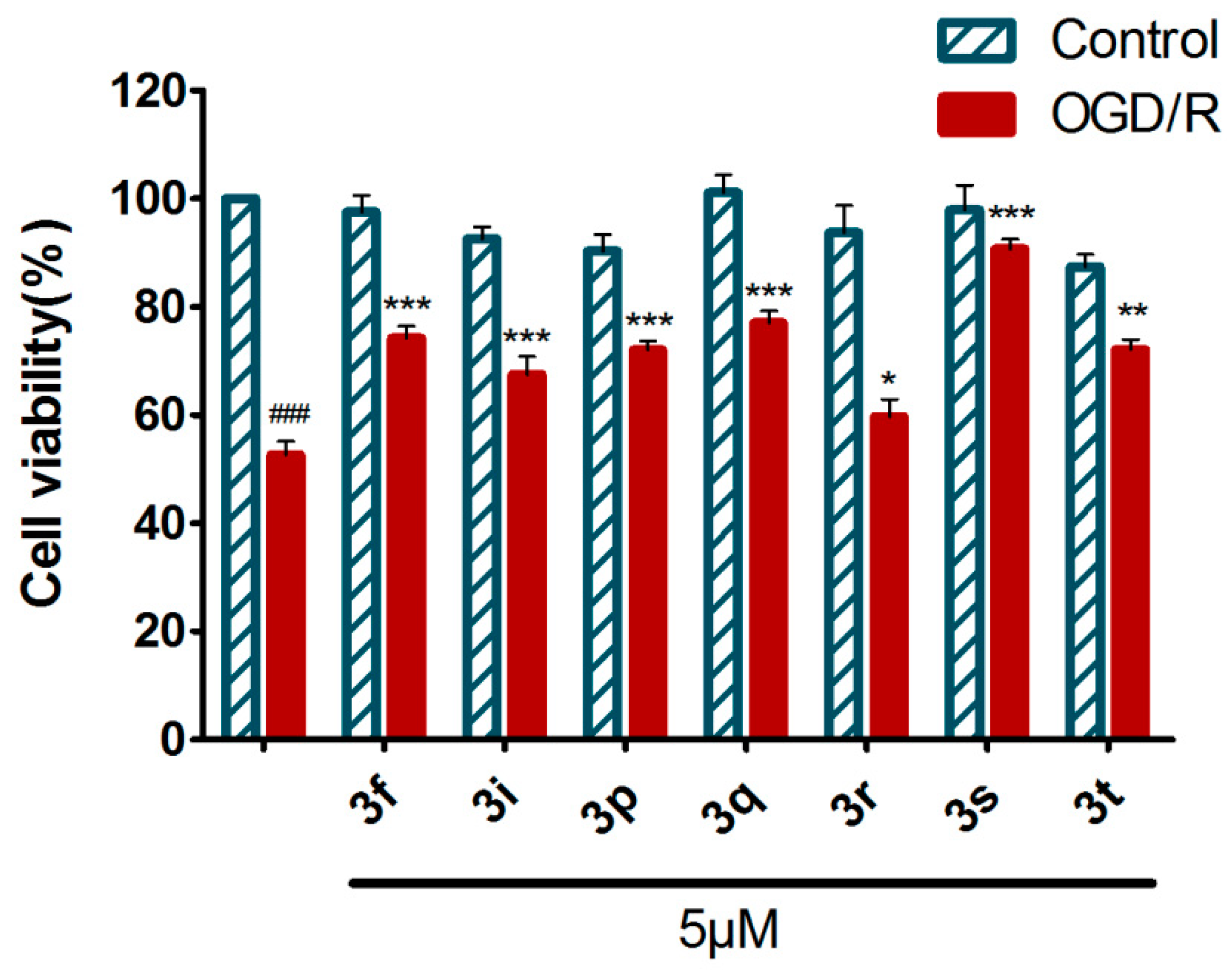
2.4.1. Neuroprotection Against Oxygen-Glucose Deprivation/Reperfusion Injury of 3f and Derivatives 3g–t
3. Experimental
3.1. Biological Assays
3.2. Chemistry
3.2.1. General Procedure for the Synthesis of Compounds 3a–e, 3g–r
3.2.2. General Procedure for the Synthesis of Compound 3f
3.2.3. General Procedure for the Synthesis of Compounds 3s–t
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Supporting Information
Conflicts of Interest
References
- Zhuang, C.L.; Zhang, W.; Sheng, C.Q.; Zhang, W.N.; Xing, C.G.; Miao, Z.Y. Chalcone: A Privileged Structure in Medicinal Chemistry. Chem. Rev. 2017, 117, 7762–7810. [Google Scholar] [CrossRef] [PubMed]
- Kakati, D.; Barua, N.C. Total synthesis and assignment of the absolute stereochemistry of xanthoangelol. Development of a highly efficient method for Claisen–Schmidt condensation. Tetrahedron 2014, 70, 637–642. [Google Scholar] [CrossRef]
- Dung, N.T.; Kim, J.M.; Kang, S.C. Chemical composition, antimicrobial and antioxidant activities of the essential oil and the ethanol extract of Cleistocalyx operculatus (Roxb.) Merr and Perry buds. Food Chem. Toxicol. 2008, 46, 3632–3639. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.G.; Qian, J.; Lu, Y.H. Hepatoprotective Effects of 2′,4′-Dihydroxy-6′-methoxy-3′,5′-dimethylchalcone on CCl4-Induced Acute Liver Injury in Mice. J. Agric. Food Chem. 2011, 59, 12821–12829. [Google Scholar] [CrossRef] [PubMed]
- Ye, C.L.; Lu, Y.H.; Wei, D.Z. Flavonoids from Cleistocalyx operculatus. Phytochemistry. 2004, 65, 445–447. [Google Scholar] [CrossRef]
- Huang, H.; Niu, J.; Lu, Y. Multidrug resistance reversal effect of DMC derived from buds of Cleistocalyx operculatus in human hepatocellular tumor xenograft model. J. Sci. Food Agric. 2012, 92, 135–140. [Google Scholar] [CrossRef]
- Qian, F.; Ye, C.L.; Wei, D.Z.; Lu, Y.H.; Yang, S.L. In Vitro and In Vivo Reversal of Cancer Cell Multidrug Resistance by 2′,4′-Dihydroxy-6′-methoxy-3′,5′-dimethylchalcone. J. Chemother. 2005, 17, 309–314. [Google Scholar] [CrossRef]
- Min, B.S.; Thu, C.; van Dat, N.T.; Dang, N.H.; Jang, H.S.; Hung, T.M. Antioxidative Flavonoids from Cleistocalyx operculatus Buds. Chem. Pharm. Bull. 2008, 56, 1725–1728. [Google Scholar] [CrossRef]
- Martínez, A.; Conde, E.; Moure, A. Protective effect against oxygen reactive species and skin fibroblast stimulation of Couroupita guianensis leaf extracts. Nat. Prod. Res. 2012, 26, 37–41. [Google Scholar] [CrossRef] [PubMed]
- Su, M.Y.; Huang, H.Y.; Li, L.; Lu, Y.H. Protective Effects of 2′,4′-Dihydroxy-6′-methoxy-3′,5′-dimethylchalcone to PC12 Cells against Cytotoxicity Induced by Hydrogen Peroxide. J. Agric. Food Chem. 2011, 59, 521–527. [Google Scholar] [CrossRef]
- Ko, H.; Kim, Y.J.; Amor, E.C.; Lee, J.W.; Kim, H.C.; Kim, H.J.; Yang, H.O. Induction of autophagy by dimethyl cardamonin is associated with proliferative arrest in human colorectal carcinoma HCT116 and LOVO cells. J. Cell Biochem. 2011, 112, 2471–2479. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.F.; Xie, B.F.; Zhou, J.M.; Feng, G.K.; Liu, Z.C.; Wei, X.Y.; Zhang, F.X.; Liu, M.F.; Zeng, Y.X. Blockade of Vascular Endothelial Growth Factor Receptor Signal Pathway and Antitumor Activity of ON-III (2′,4′-Dihydroxy-6′-methoxy-3′,5′-dimethylchalcone), a Component from Chinese Herbal Medicine. Mol. Pharmacol. 2005, 67, 1444–1450. [Google Scholar] [CrossRef] [PubMed]
- Ye, C.L.; Liu, J.W.; Wei, D.Z.; Lu, Y.H.; Qian, F. In vitro anti-tumor activity of 2′,4′-dihydroxy-6′-methoxy-3′,5′-dimethylchalcone against six established human cancer cell lines. Pharmacol. Res. 2004, 50, 505–510. [Google Scholar] [CrossRef] [PubMed]
- Dao, T.T.; Tung, B.T.; Nguyen, P.H.; Thuong, P.T.; Yoo, S.S.; Kim, E.H.; Kim, S.K.; Oh, W.K. C-Methylated Flavonoids from Cleistocalyx operculatus and Their Inhibitory Effects on Novel Influenza A (H1N1) Neuraminidase. J. Nat. Prod. 2010, 3423, 1636–1642. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.X.; Liu, M.F.; Lu, R.R. Studies on the chemical constituents from the bud of Cleistocalyx operculatus. Operculatus. Acta Bot. Sin. 1990, 32, 469–472. [Google Scholar]
- Hossain, M.A. Synthesis of 2, 4-Dihydroxy-d-Methoxy-3, 5-Dimethylchalcone. Bangladesh J. Sci. Ind. Res. 2001, 36, 50–54. [Google Scholar]
- Khang, P.V.; Hu, L.H.; Ma, L. One-Pot Synthesis of Isoindolin-1-ones with Thiamine Hydrochloride (VB1) as a Catalyst and Their Inhibitory Activity Against Cancer Cell Lines. Polycycl. Aromat. Compd. 2017. [Google Scholar] [CrossRef]
- Liu, J.H.; Ma, L.; Hu, L.H. Thiamine hydrochloride (VB1): An efficient promoter for the one-pot synthesis of benzo[4,5]imidazo[1,2-a]pyrimidine and [1,2,4]triazolo[1,5-a]pyrimidine derivatives in water medium. Green Chem. 2012, 14, 840–846. [Google Scholar] [CrossRef]
- Shao, Z.K.; Wang, L.J.; Liu, S.; Wang, X.F. Tetramethylpyrazine Protects Neurons from Oxygen-Glucose Deprivation-Induced Death. Death. Med. Sci. Monit. 2017, 23, 5277–5282. [Google Scholar] [CrossRef]
- Parka, S.Y.; Choib, Y.W.; Park, G. Nrf2-mediated neuroprotection against oxygen-glucose deprivation/reperfusion injury by emodin via AMPK-dependent inhibition of GSK-3β. J. Pharm. Pharmacol. 2018, 70, 525–535. [Google Scholar] [CrossRef]
- Kim, S.J.; Bae, S.W.; Lee, J.S.; Park, J.W. Recyclable gold nanoparticle catalyst for the aerobic alcohol oxidation and C–C bond forming reaction between primary alcohols and ketones under ambient conditions. Tetrahedron 2009, 65, 1461–1466. [Google Scholar] [CrossRef]
- Abid, M.; Bhat, A.R.; Athar, F.; Azam, A. Synthesis and antiamoebic activity of new 1-N-substituted thiocarbamoyl-3,5-diphenyl-2-pyrazoline derivatives and their Pd(II) complexes. Eur. J. Med. Chem. 2009, 44, 417–425. [Google Scholar] [CrossRef]
- Sarda, S.R.; Jadhav, W.N.; Tekale, S.U.; Jadhav, G.V.; Patil, B.R.; Suryawanshi, G.S.; Pawar, R.P. Phosphonium Ionic Liquid Catalyzed an Efficient Synthesis of Chalcones. Lett. Org. Chem. 2009, 6, 481–484. [Google Scholar] [CrossRef]
- Pavan, F.R.; Leite, C.Q.F.; Coelho, R.G.; Coutinho, I.D.; Honda, N.K.; Cardoso, C.A.L.; Vilegas, W.; Leite, S.R.A.; Sato, D.N. Evaluation of anti-Mycobacterium tuberculosis activity of Campomanesia adamantium (Myrtaceae). Quím. Nova 2009, 32, 1222–1226. [Google Scholar] [CrossRef]
- Krishna, B.M.; Chaganty, R.B. Cardamonin and alpinetin from the seeds of Alpinia speciosa. Phytochemistry 1973, 12, 238. [Google Scholar] [CrossRef]
- Aponte, J.C.; Castillo, D.; Estevez, Y.; Gonzalez, G.; Arevalo, J.; Hammond, G.B.; Sauvain, M. In vitro and in vivo anti-Leishmania activity of polysubstituted synthetic chalcones. Bioorg. Med. Chem. Lett. 2010, 20, 100–103. [Google Scholar] [CrossRef]
- Zhang, B.; Duan, D.; Ge, C.; Yao, J.; Liu, Y.; Li, X.; Fang, J. Synthesis of Xanthohumol Analogues and Discovery of Potent Thioredoxin Reductase Inhibitor as Potential Anticancer Agent. J. Med. Chem. 2015, 58, 1795–1805. [Google Scholar] [CrossRef]
- Stevens, J.M.; Taylor, A.M.; Nickerson, G.B.; Ivancic, M.; Henning, J.; Haunold, A.; Deinzer, M.L. Prenylflavonoid variation in Humulus lupulus: Distribution and taxonomic significance of xanthogalenol and 4′-O-methylxanthohumol. Phytochemistry 2000, 53, 759–775. [Google Scholar] [CrossRef]
- Vogel, S.; Heilmann, J. Synthesis, Cytotoxicity, and Antioxidative Activity of Minor Prenylated Chalcones from Humulus lupulus. J. Nat. Prod. 2008, 71, 1237–1241. [Google Scholar] [CrossRef]
- Navarini, A.L.; Chiaradia, L.D.; Mascarello, A.; Fritzen, M.; Nunes, R.J.; Yunes, R.A.; Creczynski-Pasa, T.B. Hydroxychalcones induce apoptosis in B16-F10 melanoma cells via GSH and ATP depletion. Eur. J. Med. Chem. 2009, 44, 1630–1637. [Google Scholar] [CrossRef]
- Sim, H.M.; Loh, K.Y.; Yeo, W.K.; Lee, C.Y.; Go, M.L. Aurones as Modulators of ABCG2 and ABCB1: Synthesis and Structure-Activity Relationships. Chem. Med. Chem. 2011, 6, 713–724. [Google Scholar] [CrossRef] [PubMed]
- Ducki, S.; Rennison, D.; Woo, M.; Kendall, A.; Chabert, J.F.; McGown, A.T.; Lawrence, N.J. Combretastatin-like chalcones as inhibitors of microtubule polymerization. Part 1: Synthesis and biological evaluation of antivascular activity. Bioorg. Med. Chem. 2009, 17, 7698–7710. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | ||||
|---|---|---|---|---|
| Entry | Solvent | Temperature (oC) | Time (h) | Yield of 3a b (%) |
| 1 | 1,4-Dioxane | 80 | 24 | 0 |
| 2 | THF | Reflux | 24 | 0 |
| 3 | toluene | 80 | 24 | 0 |
| 4 | DMF | 80 | 24 | 0 |
| 5 | DMSO | 80 | 24 | 0 |
| 6 | EtOH | Reflux | 24 | 64 |
| 7 | EtOH–H2O = 2:3 | Reflux | 10 | 77 |
| 8 c | EtOH–H2O = 1:1 | Reflux | 10 | 84, 84, 83,78 |
| 9 | EtOH–H2O = 2:1 | Reflux | 10 | 80 |
| 10 | EtOH–H2O = 4:1 | Reflux | 10 | 79 |
| 11 | EtOH–H2O = 8:1 | Reflux | 10 | 72 |
| Entry | VB1 (mol%) | Time (h) | Yield of 3ab (%) |
|---|---|---|---|
| 1 | 30 | 10 | 84 |
| 2 | 20 | 10 | 83 |
| 3 | 15 | 15 | 76 |
 | |||||
|---|---|---|---|---|---|
| Entry | Product | R1 | R2 | Time (h) | Yield b (%) |
| 1 | 3a | H | C6H5 | 10 | 82 |
| 2 | 3b | 4-Cl | C6H5 | 10 | 81 |
| 3 | 3c | 2-OH-4,5-(C4H8) | 2-Furyl | 14 | 71 |
| 4 c | 3d | 2-OH-4,5-(C4H8) | 2-Thienyl | 14 | 73 d |
| 5 | 3e | 2-OH-4,5-(C4H8) | 3-Pyridyl | 10 | 79 |
| 6 | 3f | 2,4-(OH)2-6-OCH3 -3,5-(CH3)2 | C6H5 | 10 | 82 |
| 7 | 3g | 2,4-(OH)2-6-OCH3 | C6H5 | 10 | 80 |
| 8 | 3h | 2,4-(OH)2-6-OCH3 | 3-OCH3C6H4 | 10 | 83 |
| 9 | 3i | 2,4-(OH)2-6-OCH3 | 4-OCH3C6H4 | 10 | 85 |
| 10 | 3j | 2,4-(OH)2-6-OCH3 | 3-OHC6H4 | 10 | 82 |
| 11 | 3k | 2,4-(OH)2-6-OCH3 | 4-OHC6H4 | 10 | 82 |
| 12 | 3l | 2,4-(OH)2-6-OCH3 | 3-ClC6H4 | 13 | 76 |
| 13 | 3m | 2,4-(OH)2-6-OCH3 | 4-ClC6H4 | 13 | 77 |
| 14 | 3n | 2-OH-4,6-(OCH3)2 | 4-OHC6H4 | 10 | 83 |
| 15 | 3o | 2-OH-4,6-(OCH3)2 | 3-NO2C6H4 | 13 | 79 |
| 16 | 3p | 2-OH-4,6-(OCH3)2 | 4-OCH3C6H4 | 10 | 83 |
| 17 | 3q | 2-OH-3,4,5-(OCH3)3 | 3,4-(OCH3)2C6H3 | 10 | 82 |
| 18 | 3r | 3,4,5-(OCH3)3 | 3,4-(OCH3)2C6H3 | 10 | 87 |
| 19 | 3s | 2-OH-4,6-(OCH3)2-3-CH2CHC(CH3)2 | 3,4-(Methylenedioxy)C6H3 | 10 | 85 |
| 20 | 3t | 2-OH- 4,6-(OCH3)2-3-CH2CHC(CH3)2 | 3,4,5-(OCH3)3C6H2 | 10 | 83 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yin, H.; Shi, X.; Wang, H.; Liu, G.; Ma, L. VB1 Promoted Green Synthesis of Chalcones and Its Neuroprotection Potency Evaluation. Processes 2019, 7, 236. https://doi.org/10.3390/pr7040236
Yin H, Shi X, Wang H, Liu G, Ma L. VB1 Promoted Green Synthesis of Chalcones and Its Neuroprotection Potency Evaluation. Processes. 2019; 7(4):236. https://doi.org/10.3390/pr7040236
Chicago/Turabian StyleYin, Huanhuan, Ximeng Shi, Hao Wang, Guixia Liu, and Lei Ma. 2019. "VB1 Promoted Green Synthesis of Chalcones and Its Neuroprotection Potency Evaluation" Processes 7, no. 4: 236. https://doi.org/10.3390/pr7040236
APA StyleYin, H., Shi, X., Wang, H., Liu, G., & Ma, L. (2019). VB1 Promoted Green Synthesis of Chalcones and Its Neuroprotection Potency Evaluation. Processes, 7(4), 236. https://doi.org/10.3390/pr7040236