A Machine Learning-based Pipeline for the Classification of CTX-M in Metagenomics Samples


 ,
,  ,
,  , and
, and
Abstract
:1. Introduction
1.1. Metagenomics
1.2. Machine Learning
2. Materials and Methods
2.1. Selection of the CTX-M and Metagenome Baseline Reference Database for the Study
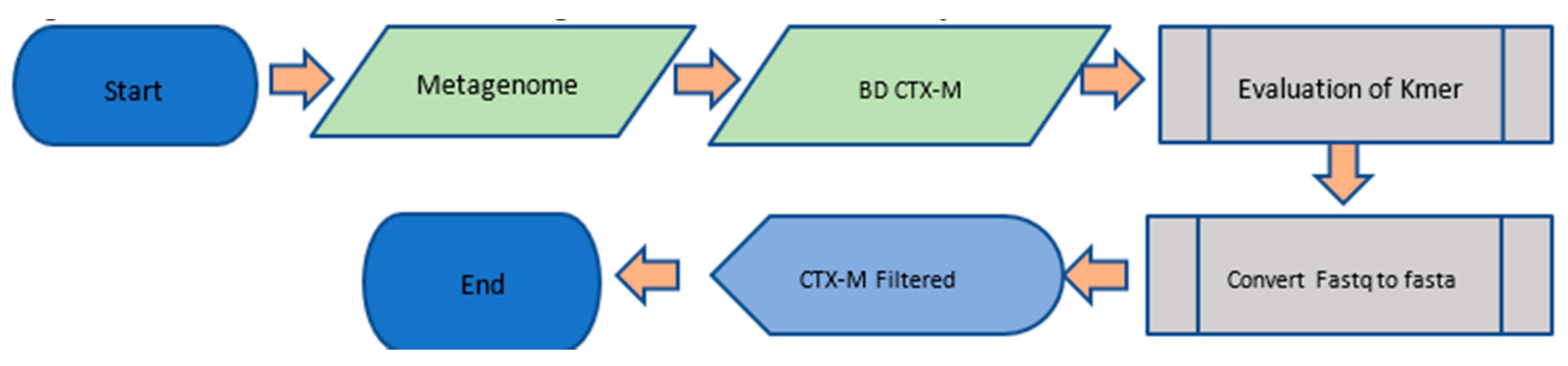
| Algorithm 1. Bioinformatic pipeline for filtering and formatting input data. |
| Parameterize the initial mapping with Duk using odd K-mers. Execute tests using different K-mers. Name: Pre-filter CTX-M Start For k-mer values between 17 and 65 Do Execute duk with each k-mer against the reference database Save results in a single file “duk_results” Finish do Best_K-mer < 0 Best p-value < 0 For each line in “duk_results” file Do Find p-value of each k-mer If (P-value found is larger than Best p-value) Best p-value < p-value found Best_K-mer < k-mer found End if Convert output file of best k-mer to FASTA format Format the FASTA file for the ANN (X, y) For each end of CTX-M sequence Do Separate CTX-M group from each sequence. Finish do End |
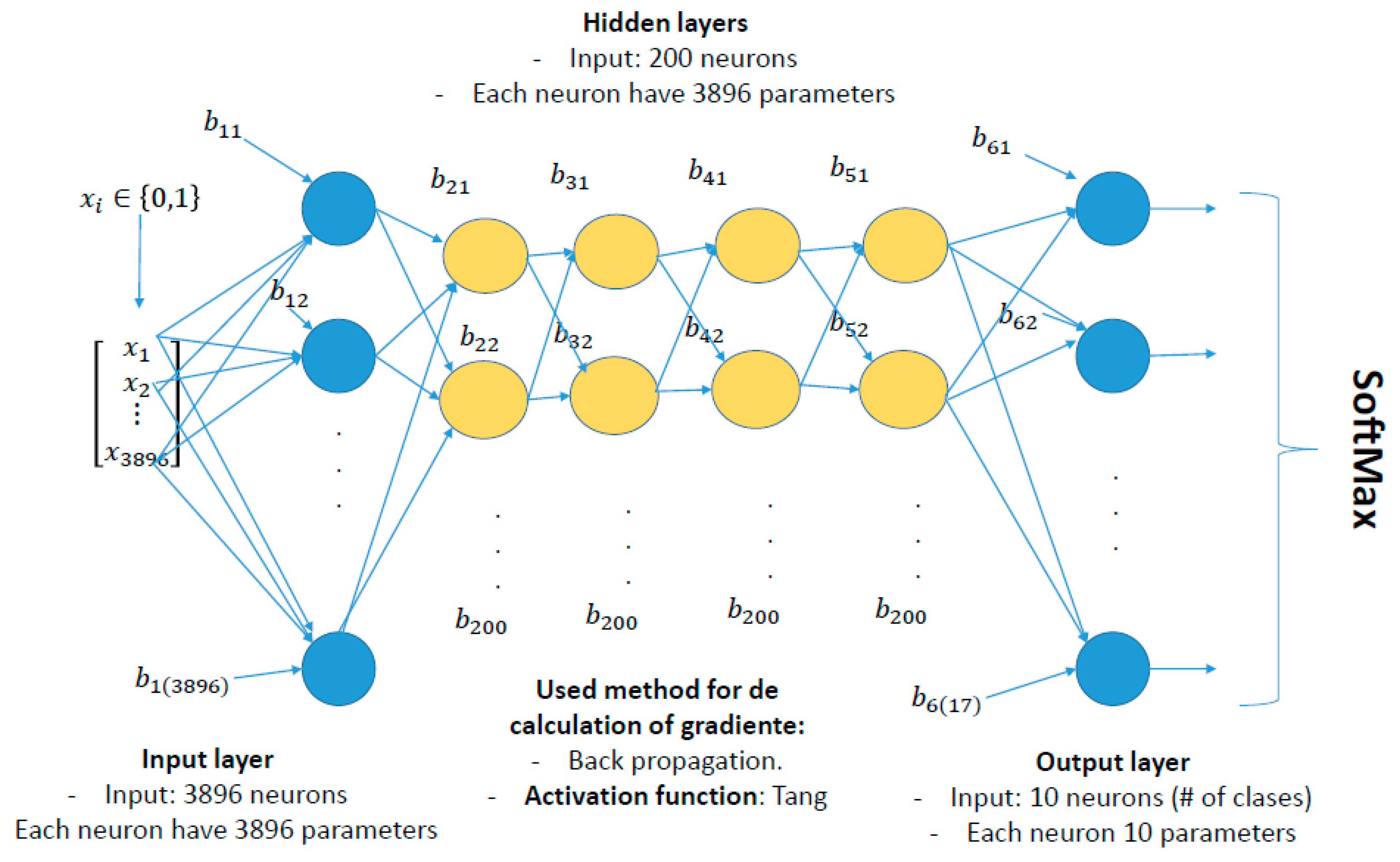
2.2. Defining an Optimal Neural Network Architecture
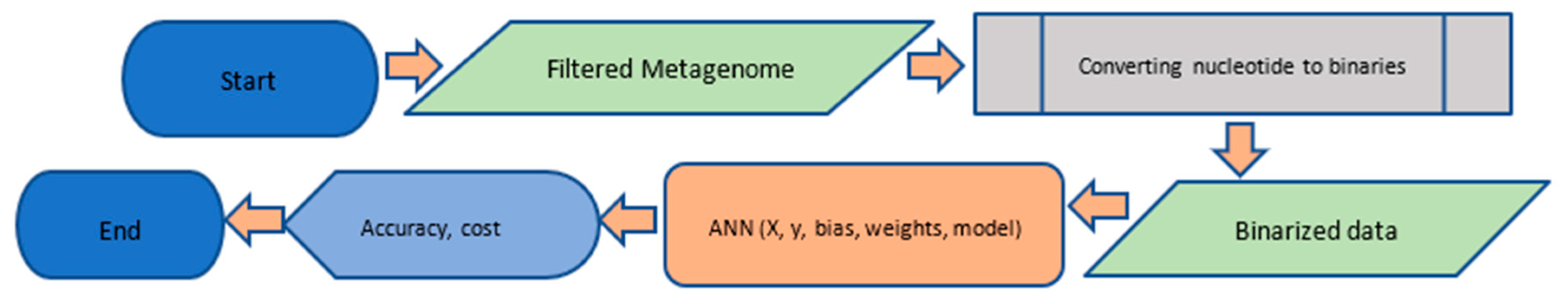
2.3. Data Standardization for the Neural Network
3. Analysis of Results
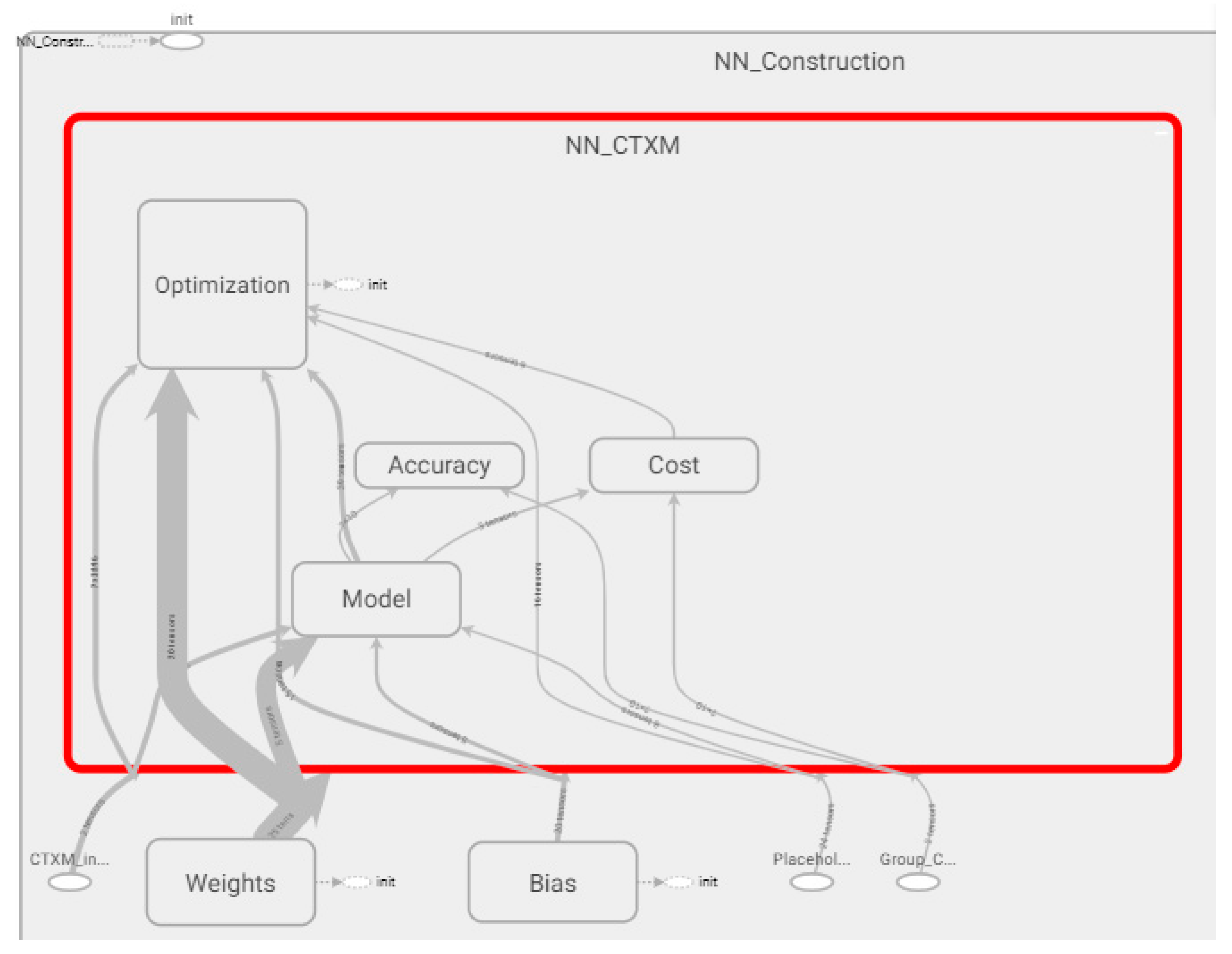
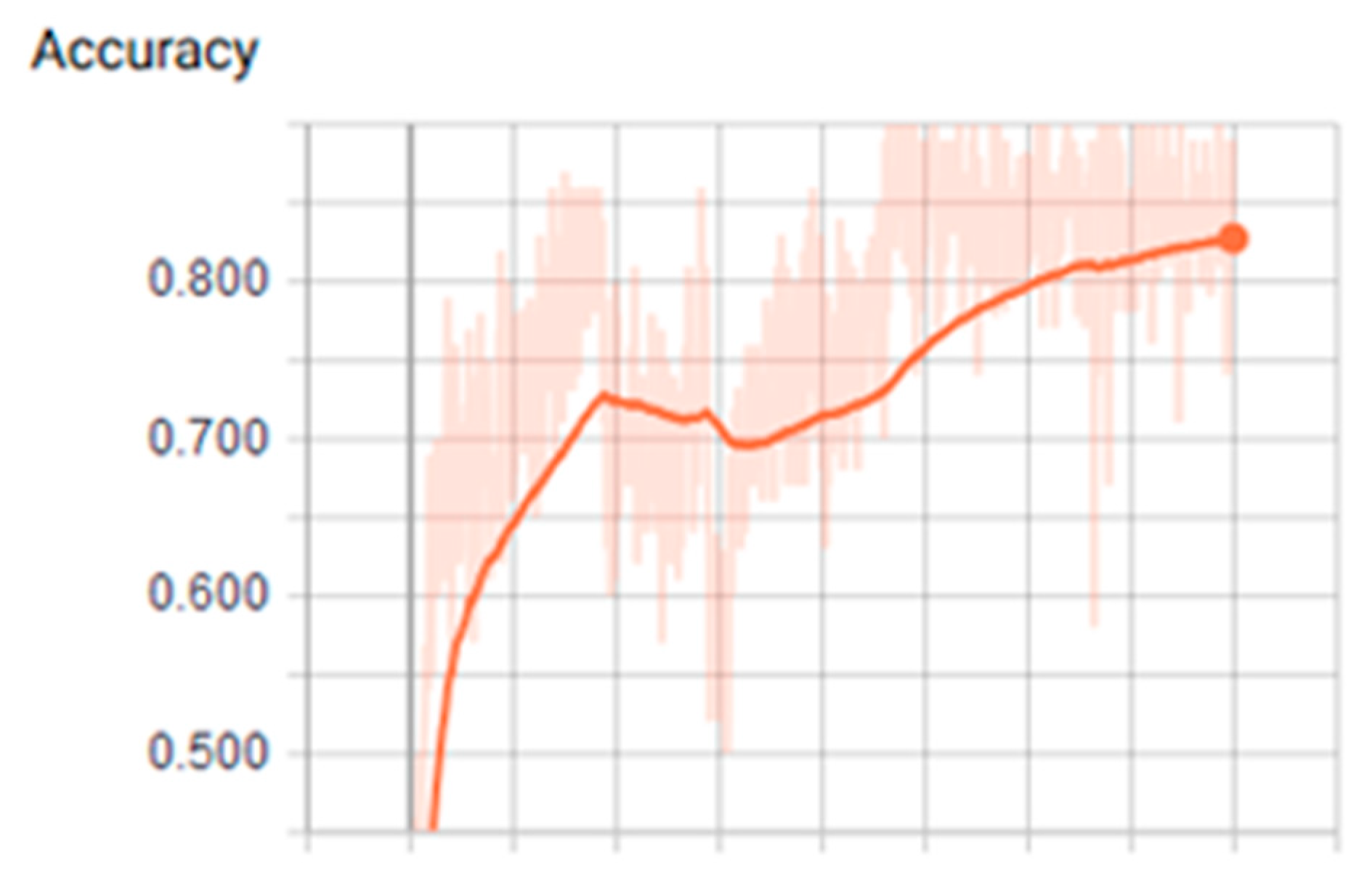
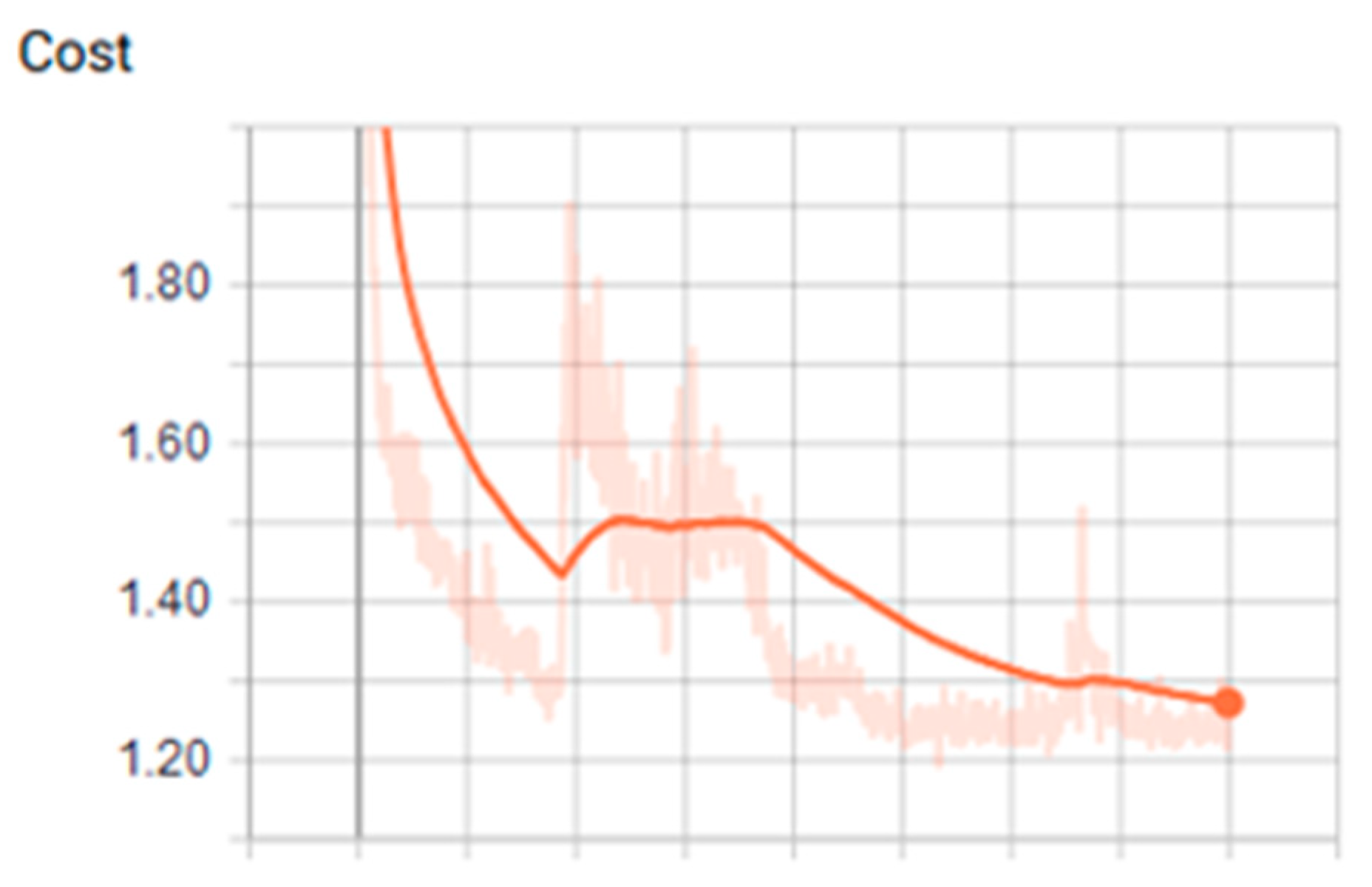
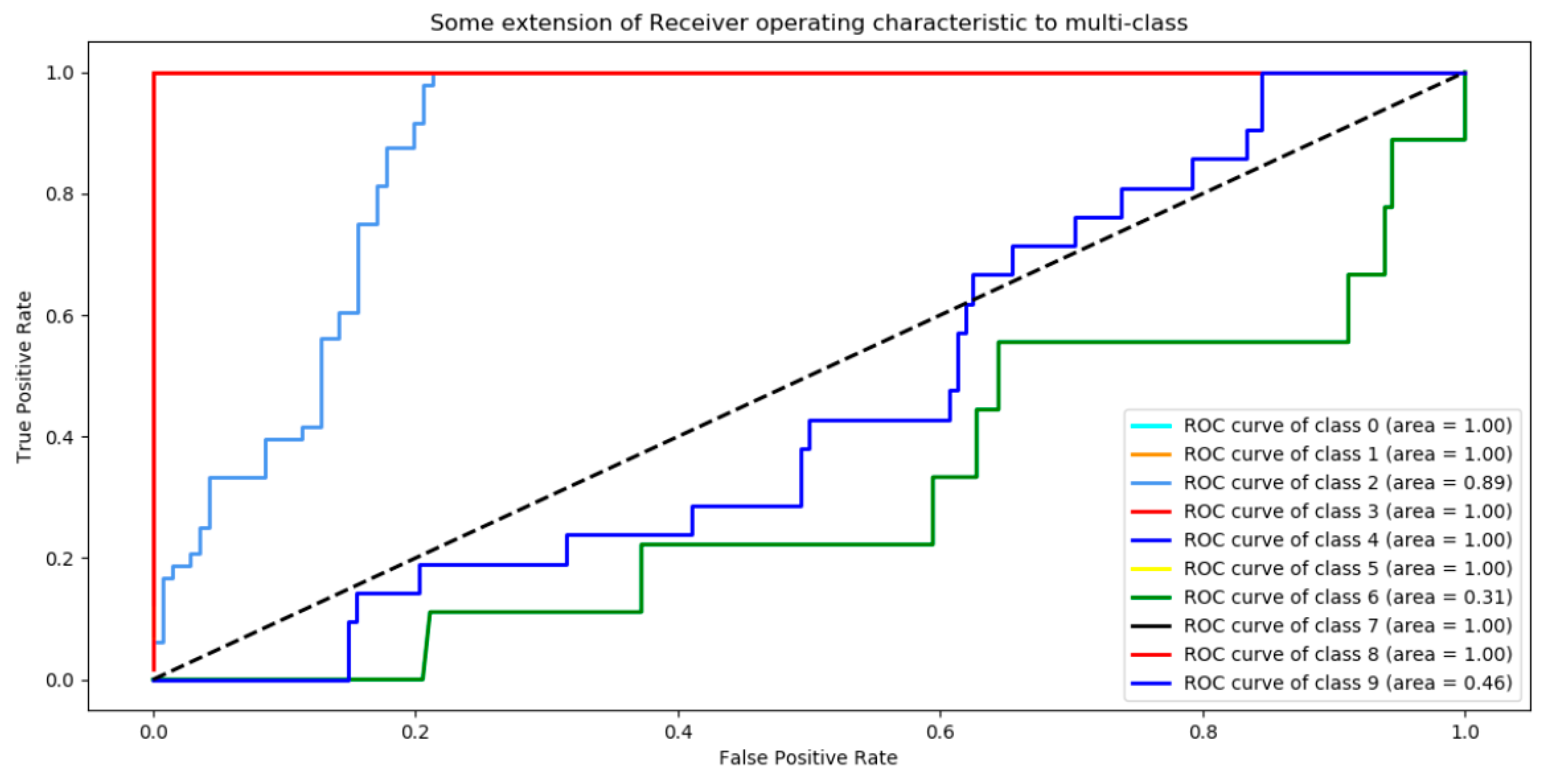
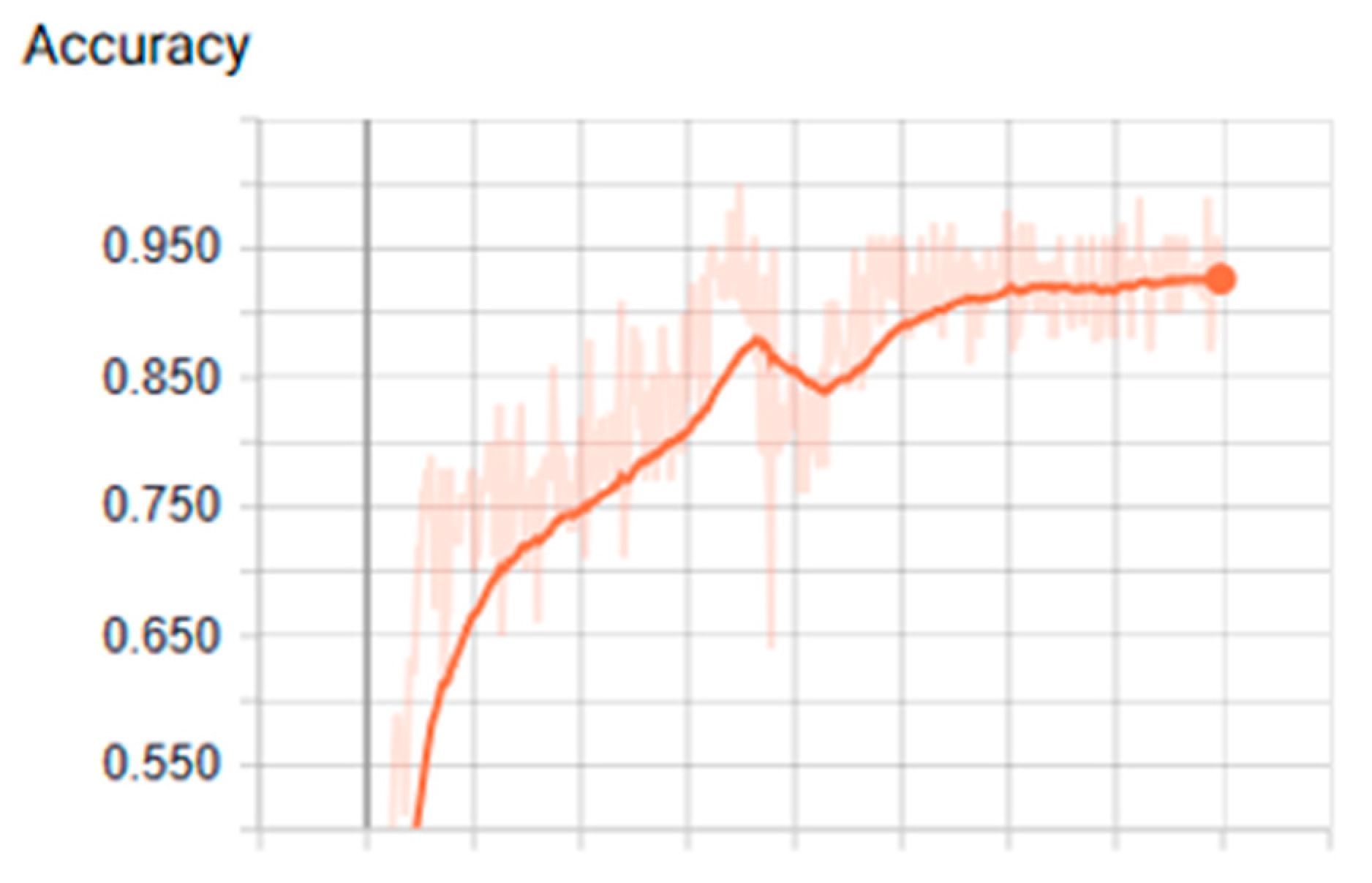
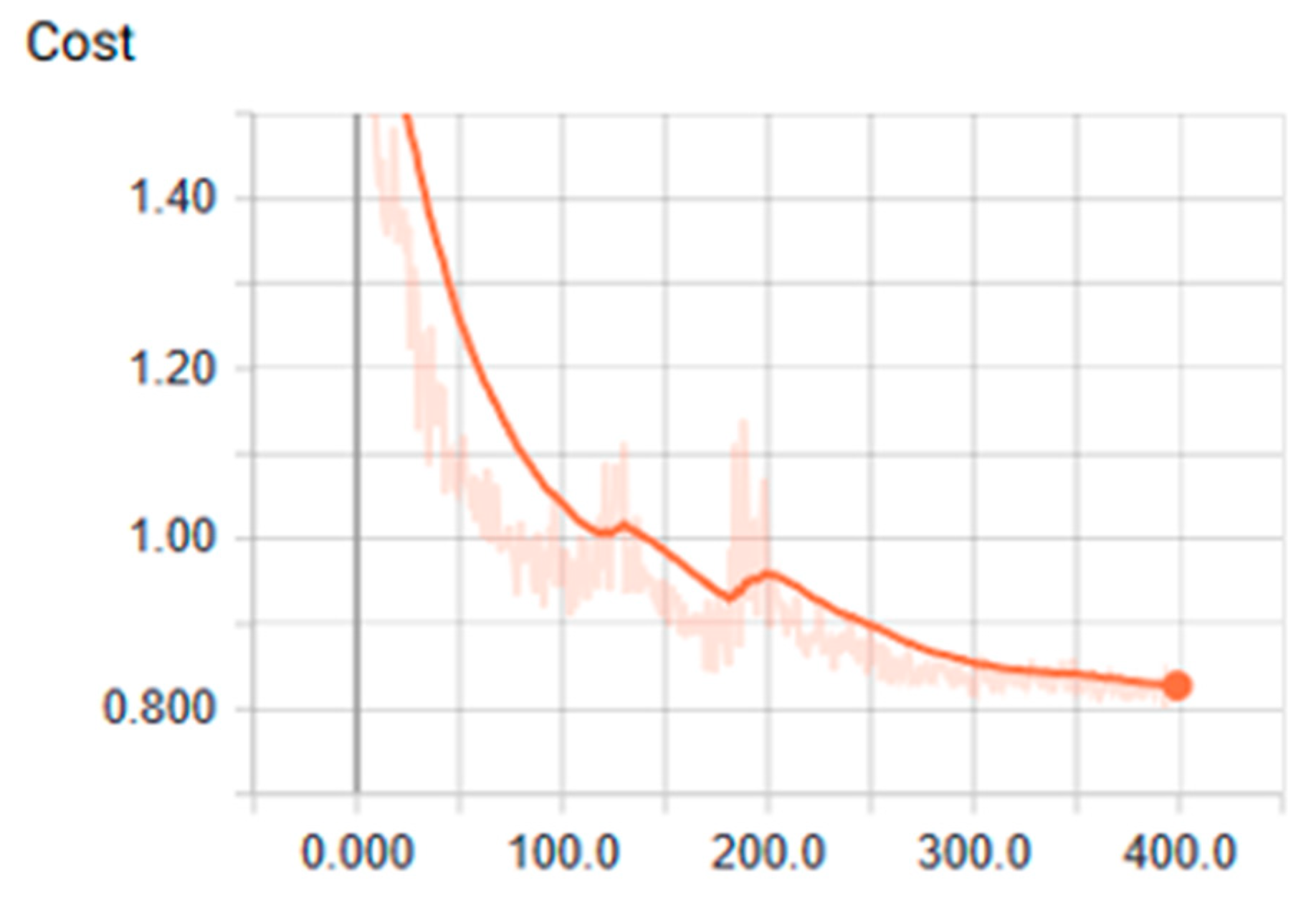
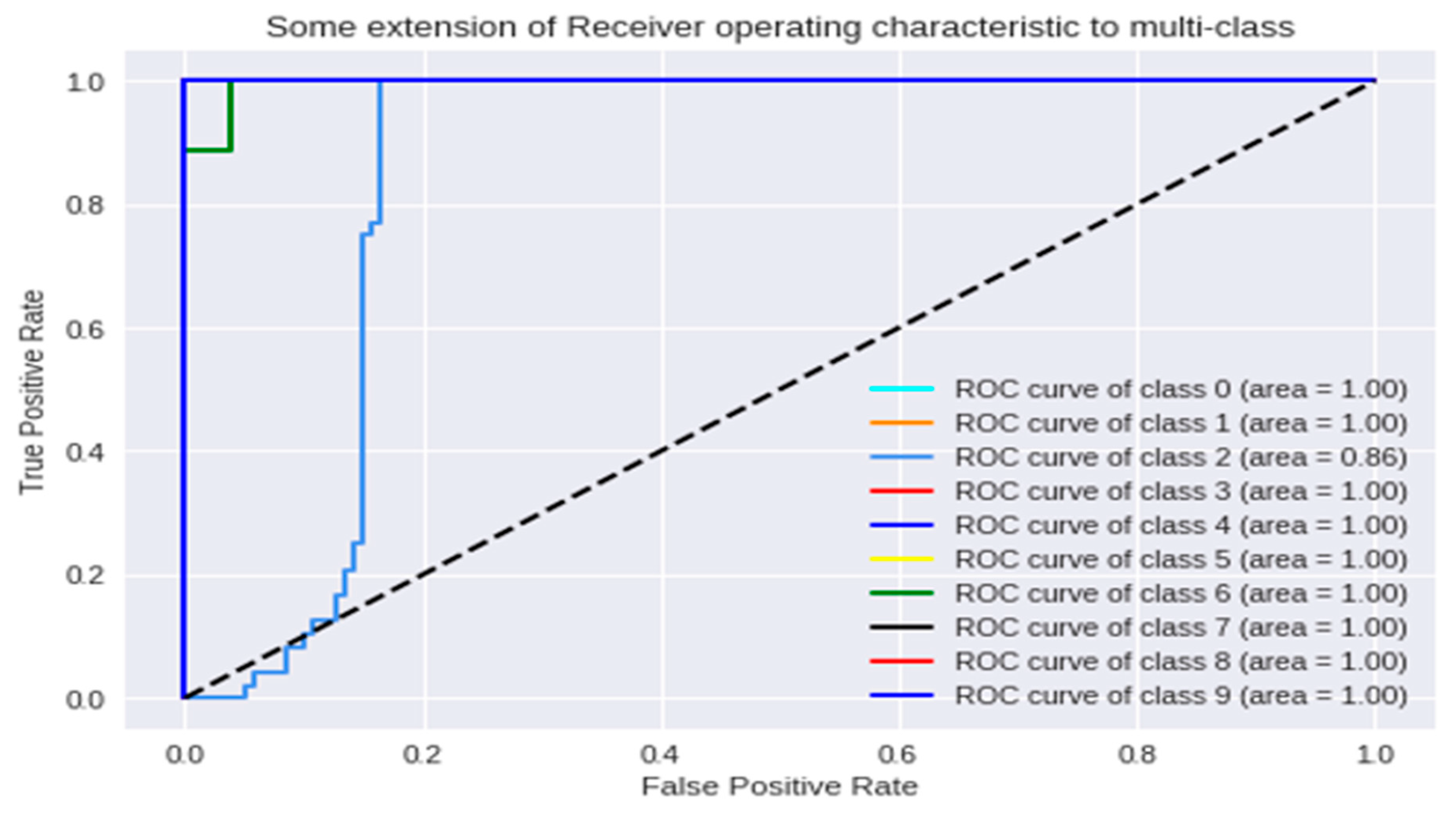
3.1. Analysis of the Graph Resulting from the ANN
3.2. Training Stage Over CPU an GPGPU
4. Discussion
4.1. Conclusions for the Tanh Activation Function
4.2. Conclusions About the Dataset
4.3. Perspective
Author Contributions
Funding
Conflicts of Interest
References
- Hoff, K.J.; Tech, M.; Lingner, T.; Daniel, R.; Morgenstern, B.; Meinicke, P. Gene prediction in metagenomic fragments: A large scale machine learning approach. BMC Bioinform. 2008, 9, 217. [Google Scholar] [CrossRef] [PubMed]
- Rasheed, Z.; Rangwala, H. Metagenomic Taxonomic Classification Using Extreme Learning Machines. J. Bioinform. Comput. Biol. 2012, 10, 1250015. [Google Scholar] [CrossRef] [PubMed]
- Soueidan, H.; Nikolski, M. Machine learning for metagenomics: Methods and tools. arXiv 2015, arXiv:1510.06621. [Google Scholar] [CrossRef]
- Cantón, R.; González-Alba, J.M.; Galán, J.C. CTX-M enzymes: origin and diffusion. Front. Microbiol. 2012, 3, 110. [Google Scholar] [CrossRef] [PubMed]
- Salazar, J.D.; Loaiza, S.; Ibáñez, J.P.; Hernandez, J.S. Primera mirada a la resistencia antibiótica de la ciudad de Manizales. Segundo Simposio Regional de Resistencia Antibiótica–Eje Cafetero, 2018. Universidad de Manizales, noviembre 3 de 2018.
- Thomas, T.; Gilbert, J.; Meyer, F. Metagenomics—A guide from sampling to data analysis. Microb. Inform. Exp. 2012, 2, 3. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.; Jain, K.; Madamwar, D. 2—Functional Metagenomics: Exploring Nature’s Gold Mine. In Current Developments in Biotechnology and Bioengineering; Elsevier: Amsterdam, The Netherlands, 2017; pp. 27–43. ISBN 9780444636676. Available online: http://www.sciencedirect.com/science/article/pii/B978044463667600002X (accessed on 11 October 2018).
- Ma, C.; Zhang, H.H.; Wang, X. Machine learning for Big Data analytics in plants. Trends Plant Sci. 2014, 19, 798–808. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, T.M. The Discipline of Machine Learning. CMU-ML-06-108; School of Computer Science, Carnegie Mellon University: Pittsburgh, PA, USA, 2006. [Google Scholar]
- Vervier, K.; Mahé, P.; Tournoud, M.; Veyrieras, J.B.; Vert, J.P. Large-scale Machine Learning for Metagenomics Sequence Classication. Bioinformatics 2015, 32, 1023–1032. [Google Scholar] [CrossRef] [PubMed]
- Lu, P.; Abedi, V.; Mei, Y.; Hontecillas, R.; Philipson, C.; Hoops, S.; Carbo, A.; Bassaganya-Riera, J. Emerging Trends in Computational Biology, Bioinformatics, and Systems Biology; Elsevier: Amsterdam, The Netherlands, 2015; ISBN 9780128025086. [Google Scholar]
- Nuñez, A. Anábioimutendifetide blaCTX-M. 2016. [Google Scholar]
- Krachunov, M.; Sokolova, M.; Simeonova, V.; Nisheva, M.; Avdjieva, I.; Vassilev, D. Quality of Different Machine Learning Models In Error Discovery For Parallel Genome Sequencing. Comptes Rendus De L Academie Bulgare Des Sciences 2017, 71, 922–929. [Google Scholar]
- Zeng, X.; Yeung, D.S. Sensitivity analysis of multilayer perceptron to input and weight perturbations. IEEE Trans. Neural Netw. 2001, 12, 1358–1366. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group CTX-M | Class |
|---|---|
| 1.0 | 0 |
| 9.0 | 1 |
| 14.0 | 2 |
| 15.0 | 3 |
| 22.0 | 4 |
| 24.0 | 5 |
| 27.0 | 6 |
| 55.0 | 7 |
| 59.0 | 8 |
| 65.0 | 9 |
| Activation Function | LEARNING_RATE | TRAINING_EPOCH | HIDDEN_SIZE | Initial Cost Value | Final Cost Value | Accuracy of Initial Training | Accuracy of Final Training | Precision Test |
|---|---|---|---|---|---|---|---|---|
| Tanh | 0.001 | 400 | 200 | 2.17 | 0.80 | 0.260 | 0.960 | 0.879 |
| Sigmoid | 0.001 | 400 | 200 | 2.19 | 1.61 | 0.030 | 0.680 | 0.698 |
| RELU | 0.001 | 300 | 200 | 2.19 | 0.00 | 0.110 | 1 | 1 |
| Activation Function | LEARNING_RATE | TRAINING_EPOCH | HIDDEN_SIZE | Initial Cost Value | Final Cost Value | Accuracy of Initial Training | Accuracy of Final Training | Precision Test |
|---|---|---|---|---|---|---|---|---|
| Tanh | 0.001 | 400 | 200 | 2.16 | 0.84 | 0.380 | 0.920 | 0.909 |
| Sigmoid | 0.001 | 400 | 200 | 2.20 | 1.67 | 0.440 | 0.560 | 0.628 |
| RELU | 0.001 | 300 | 200 | 1.90 | 1.00 | 0.590 | 1 | 1 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ceballos, D.; López-Álvarez, D.; Isaza, G.; Tabares-Soto, R.; Orozco-Arias, S.; Ferrin, C.D. A Machine Learning-based Pipeline for the Classification of CTX-M in Metagenomics Samples. Processes 2019, 7, 235. https://doi.org/10.3390/pr7040235
Ceballos D, López-Álvarez D, Isaza G, Tabares-Soto R, Orozco-Arias S, Ferrin CD. A Machine Learning-based Pipeline for the Classification of CTX-M in Metagenomics Samples. Processes. 2019; 7(4):235. https://doi.org/10.3390/pr7040235
Chicago/Turabian StyleCeballos, Diego, Diana López-Álvarez, Gustavo Isaza, Reinel Tabares-Soto, Simón Orozco-Arias, and Carlos D. Ferrin. 2019. "A Machine Learning-based Pipeline for the Classification of CTX-M in Metagenomics Samples" Processes 7, no. 4: 235. https://doi.org/10.3390/pr7040235
APA StyleCeballos, D., López-Álvarez, D., Isaza, G., Tabares-Soto, R., Orozco-Arias, S., & Ferrin, C. D. (2019). A Machine Learning-based Pipeline for the Classification of CTX-M in Metagenomics Samples. Processes, 7(4), 235. https://doi.org/10.3390/pr7040235