Degradation of Aqueous Polycyclic Musk Tonalide by Ultraviolet-Activated Free Chlorine



Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Experimental Procedures
2.3. Determination of UV Fluence Rate and UV Dosage
2.4. Analysis
2.5. Contributions of Different Radicals
2.6. Toxicity Evaluation of Samples
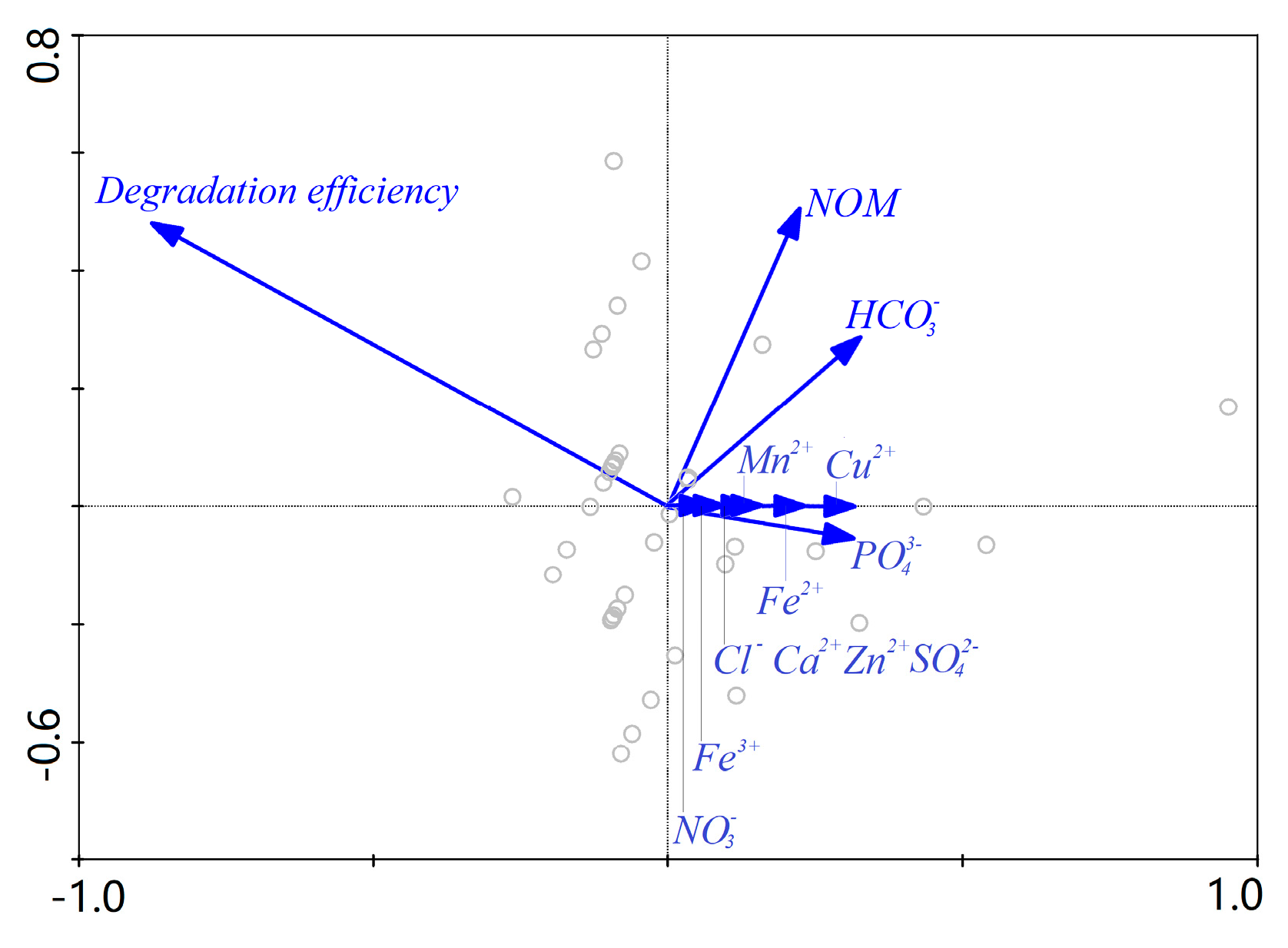
2.7. Principal Factor Analysis
3. Results and Discussion
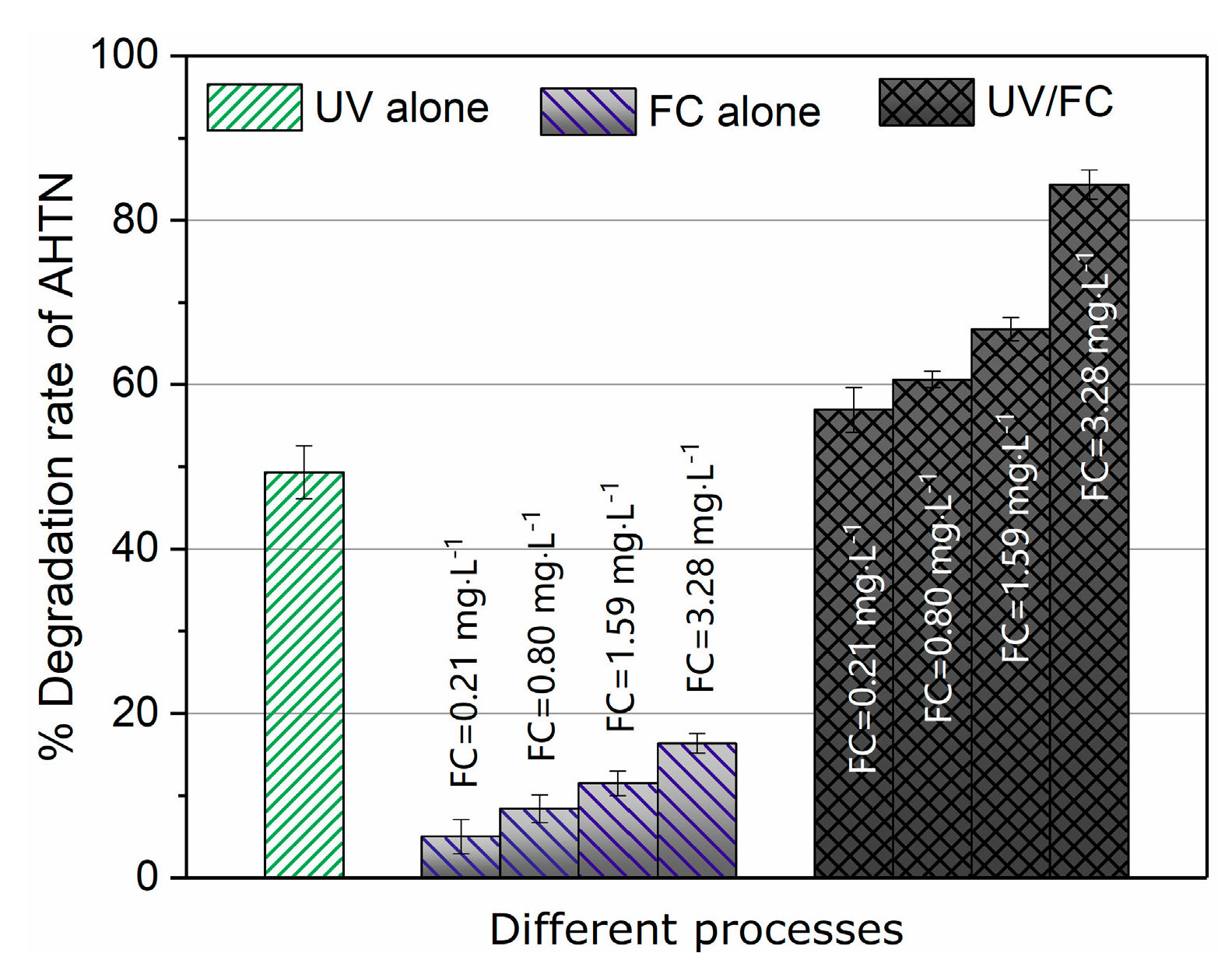
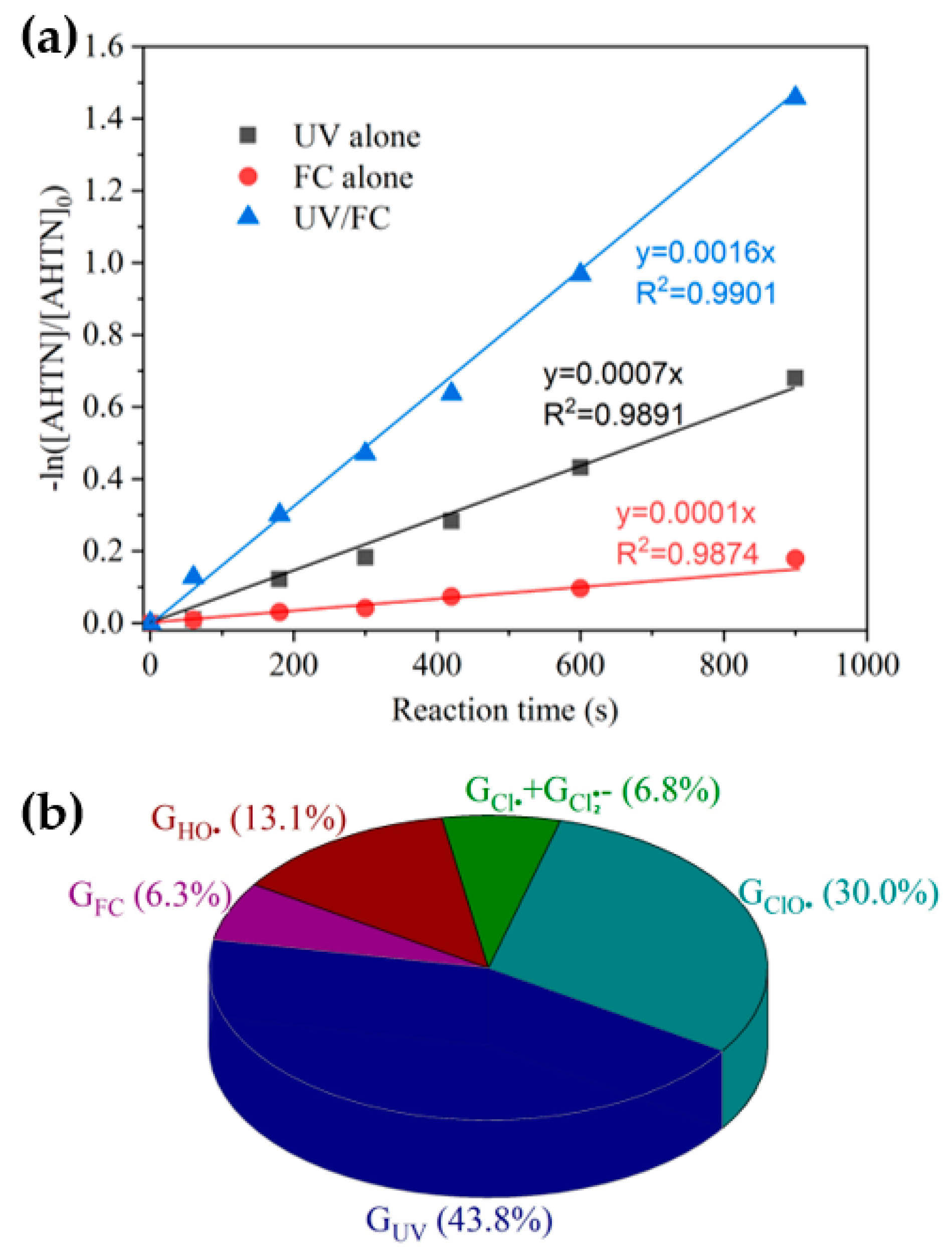
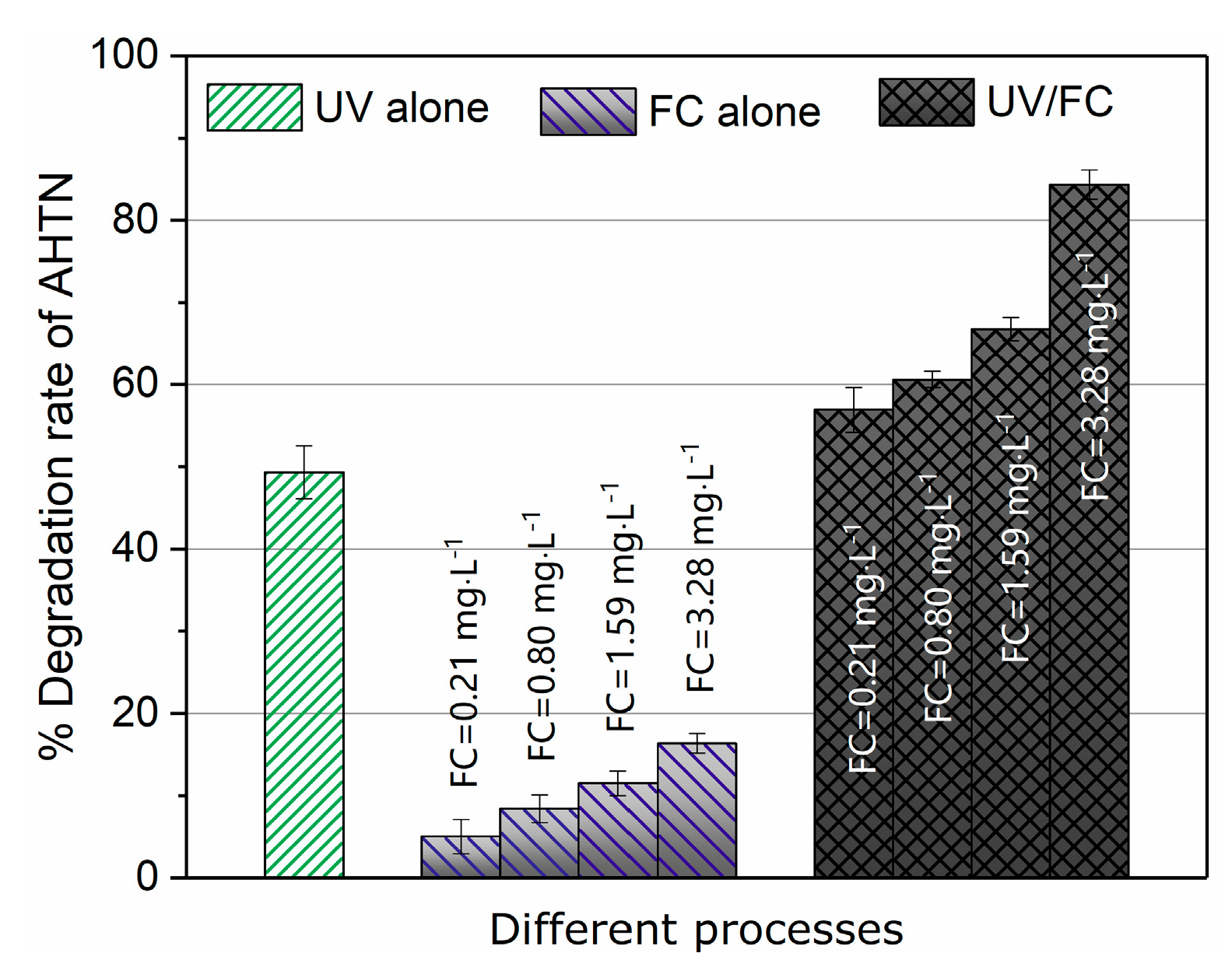
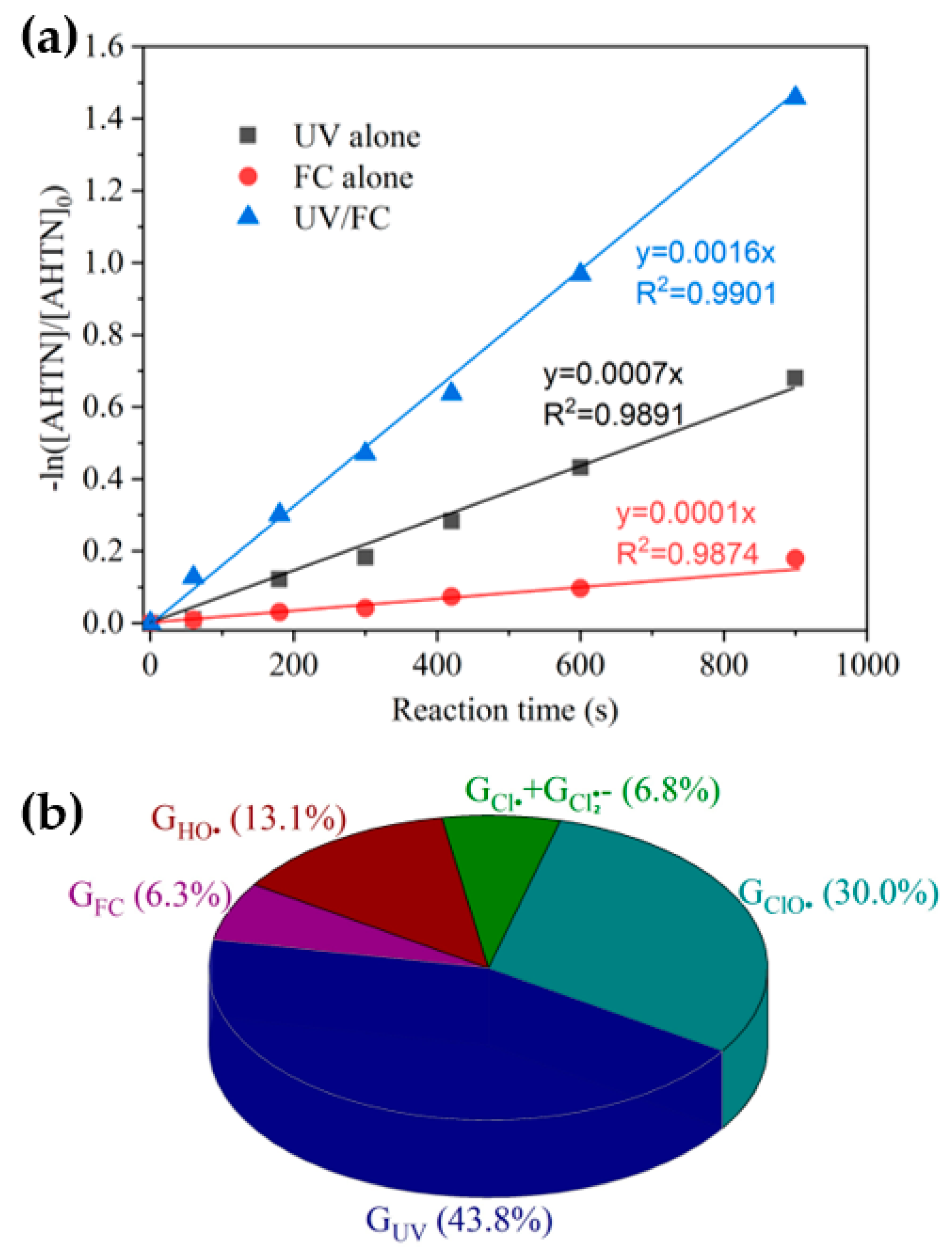
3.1. AHTN Degradation under UV/FC Conditions
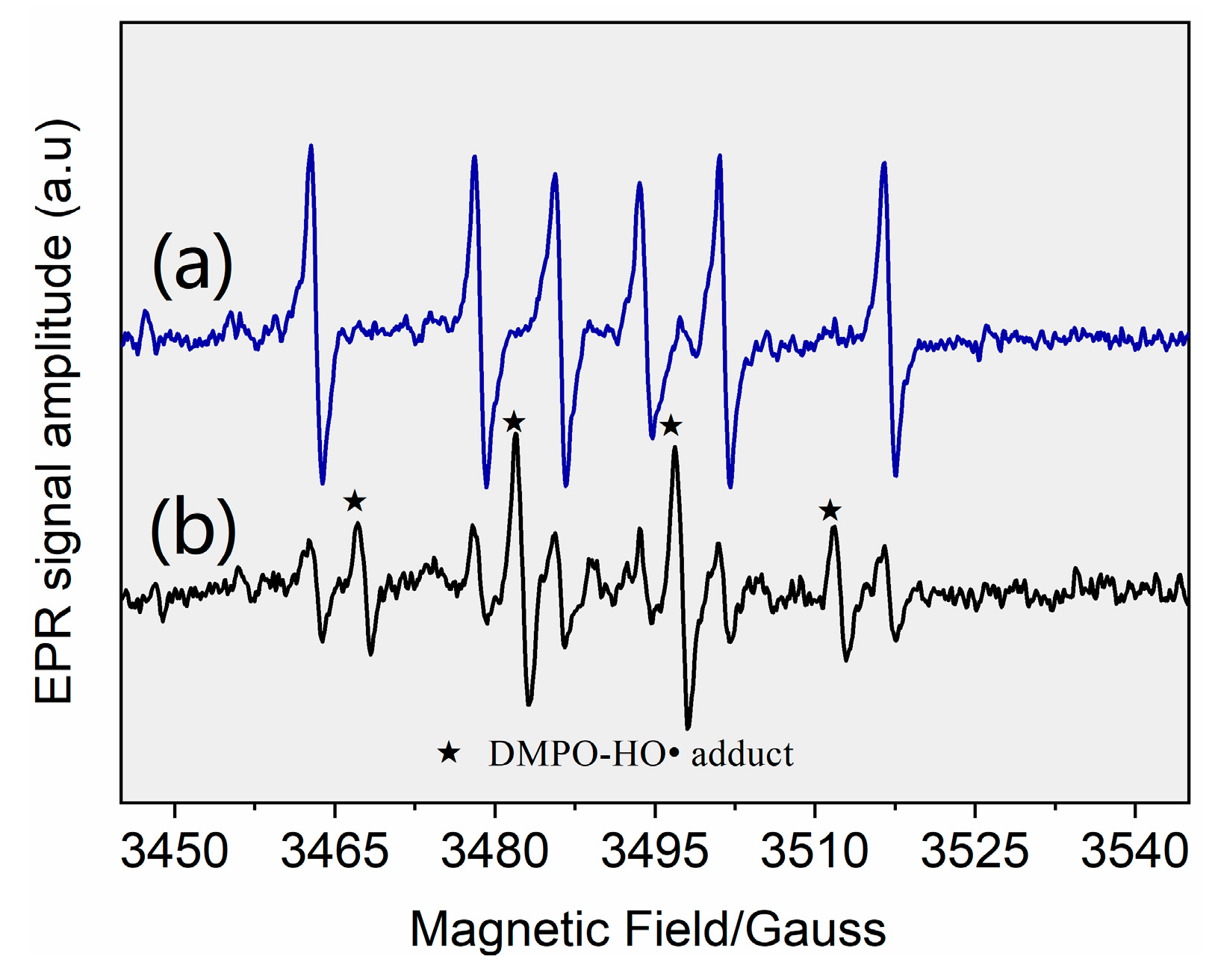
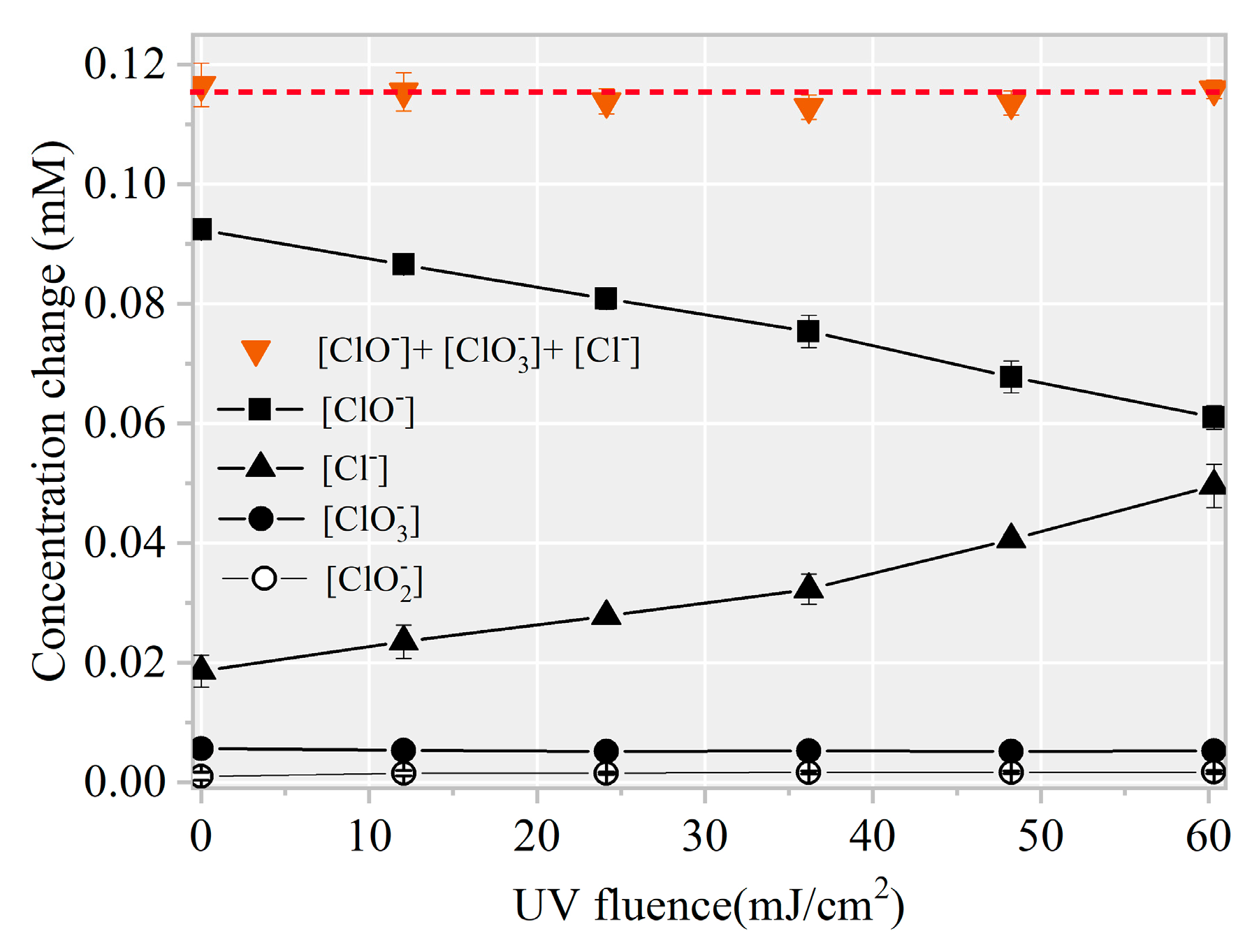
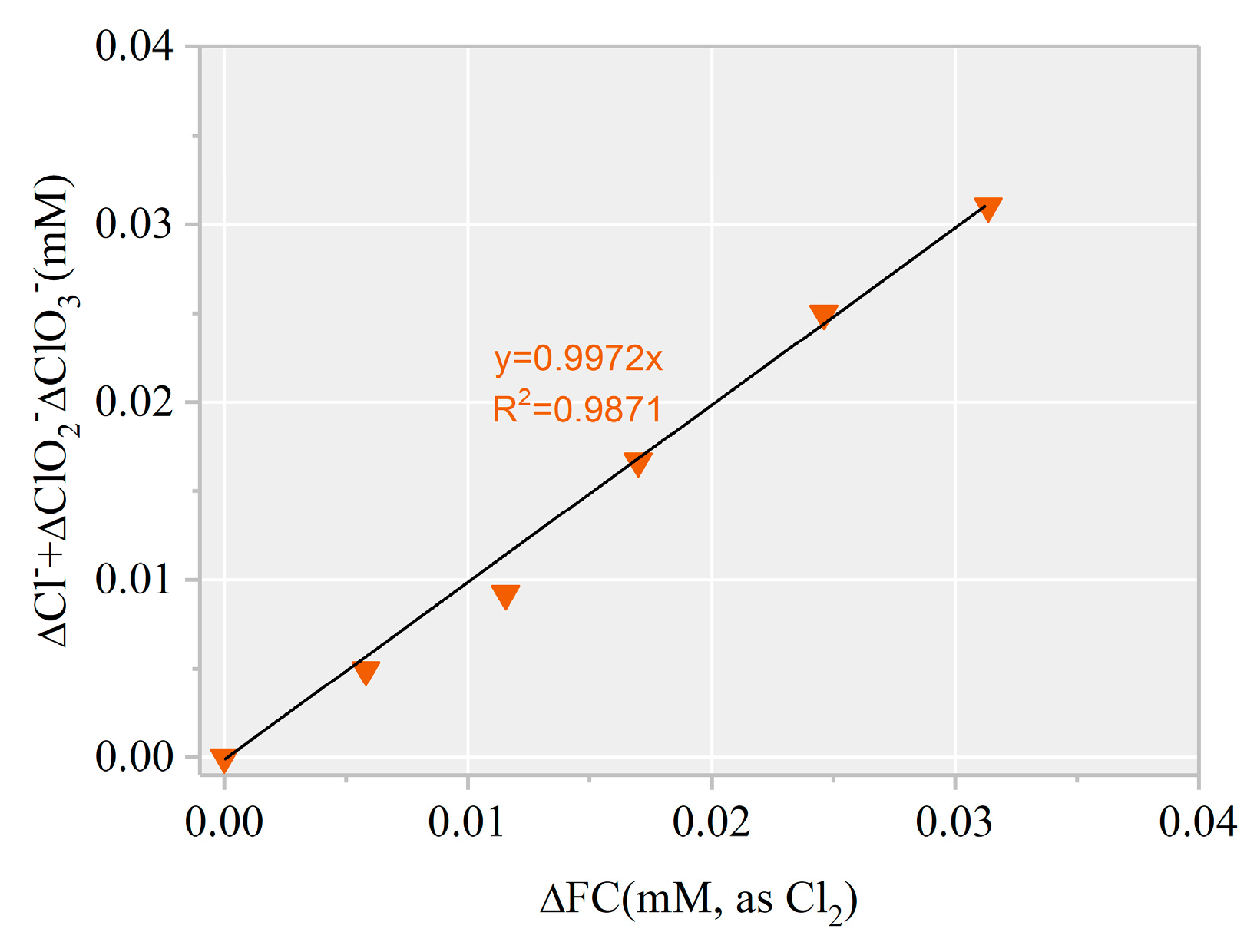
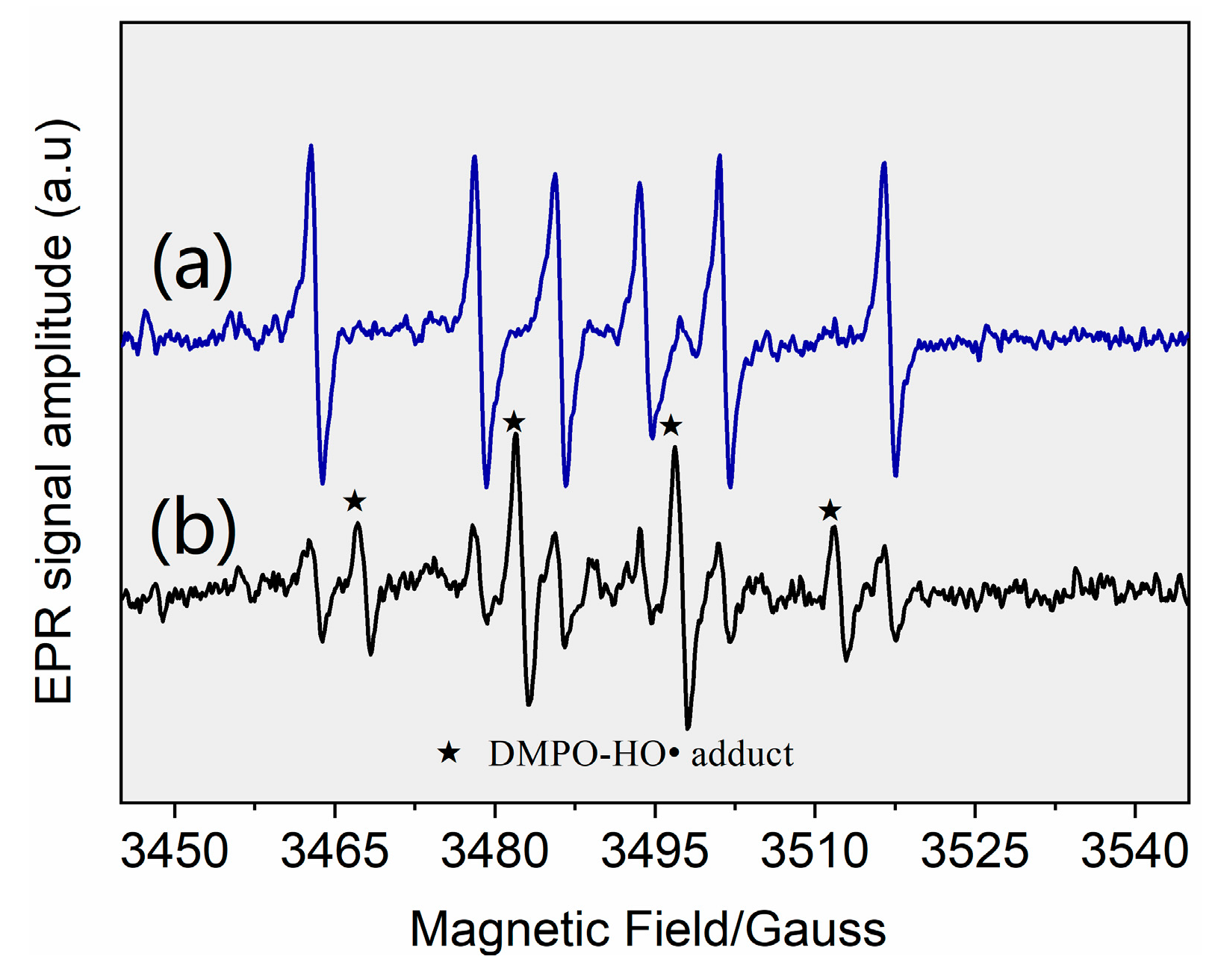
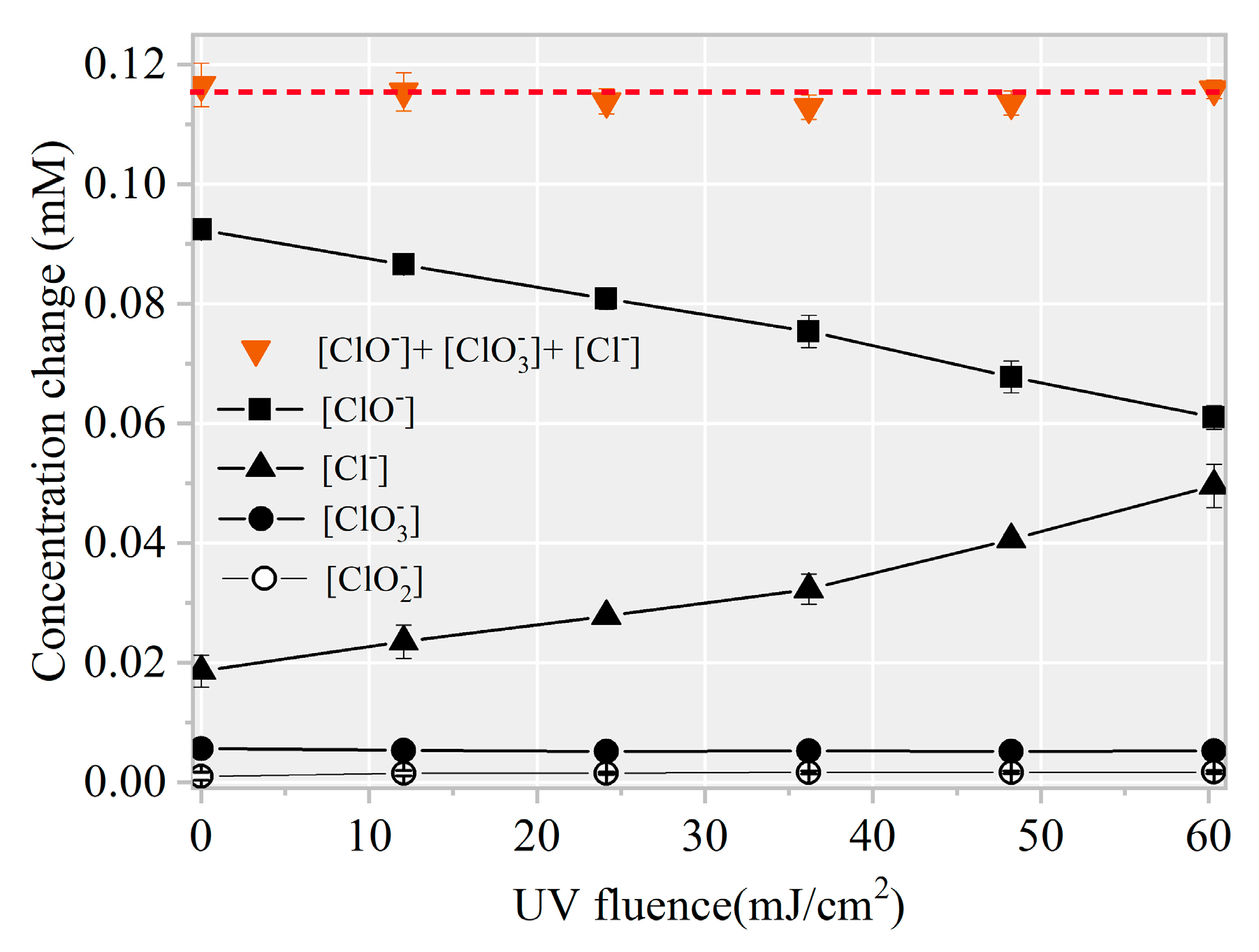
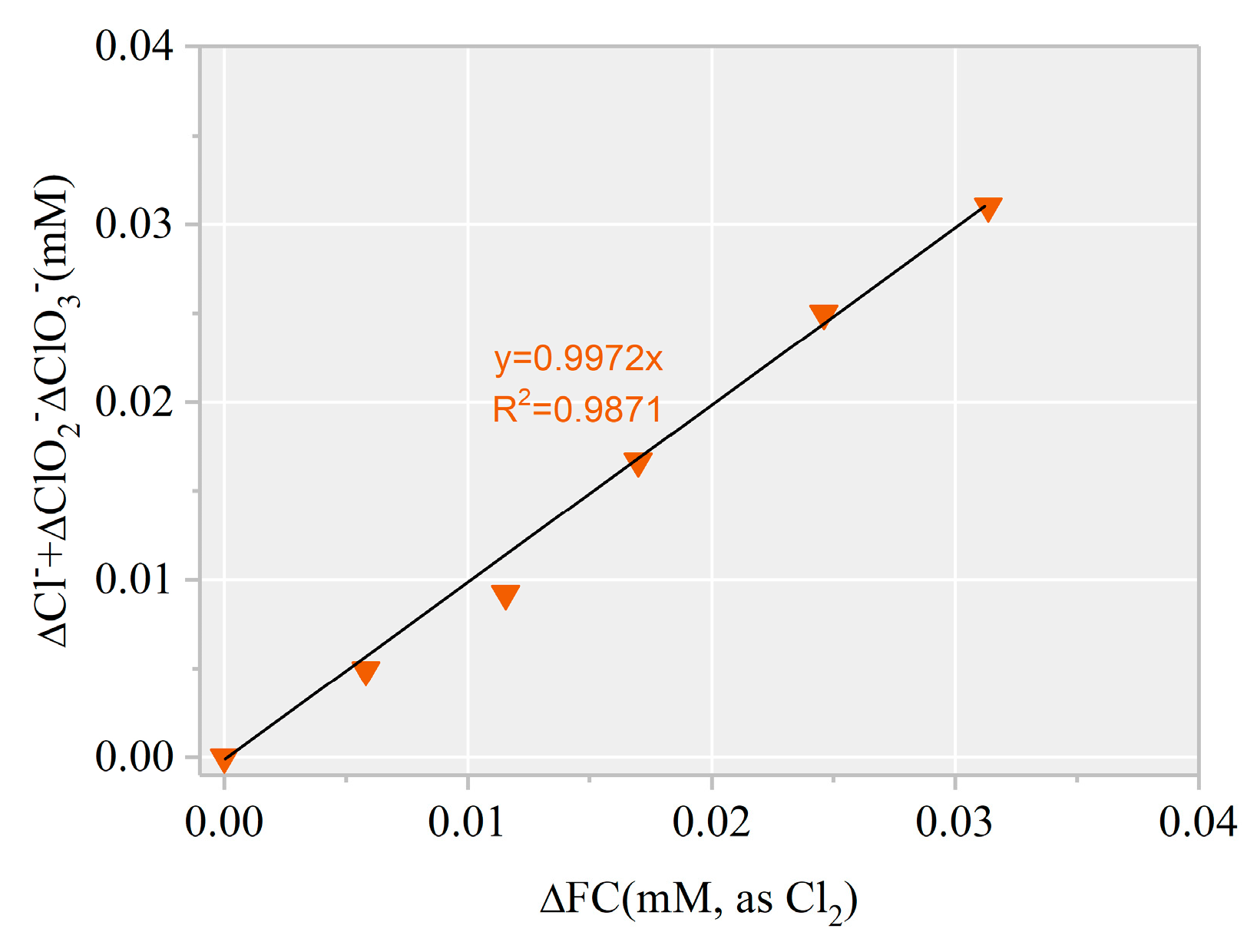
3.2. Identification of Reactive Species for AHTN Degradation by UV/FC
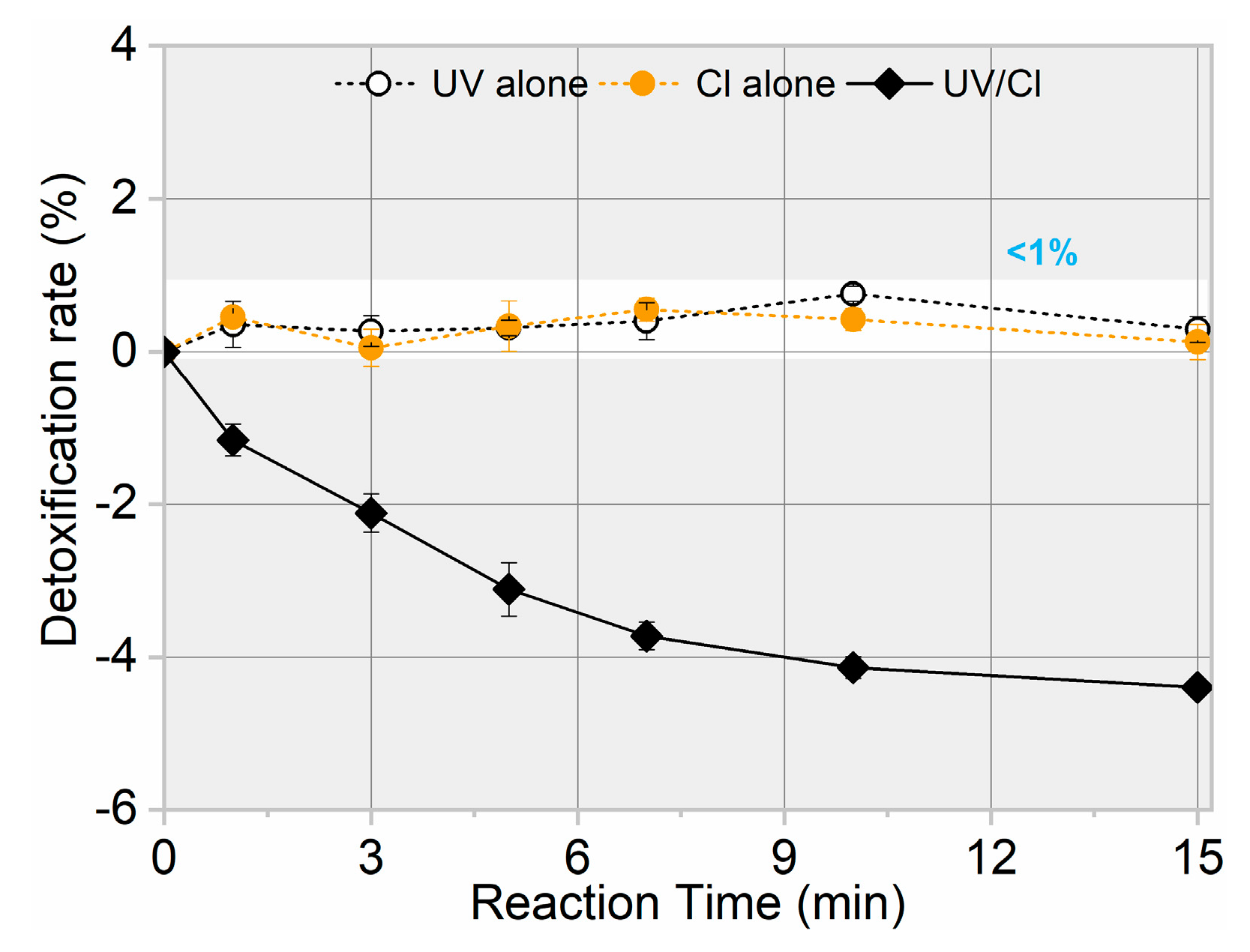
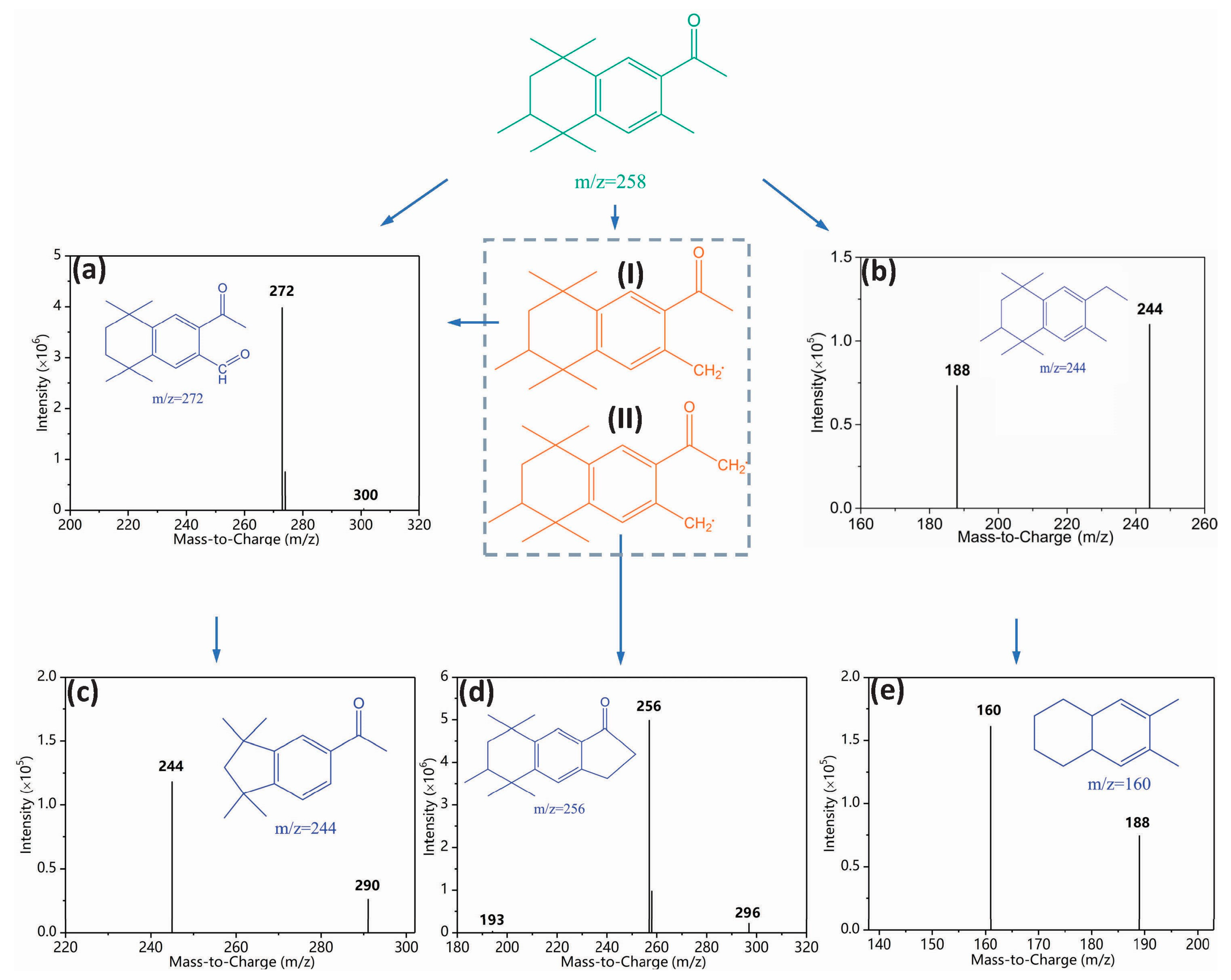
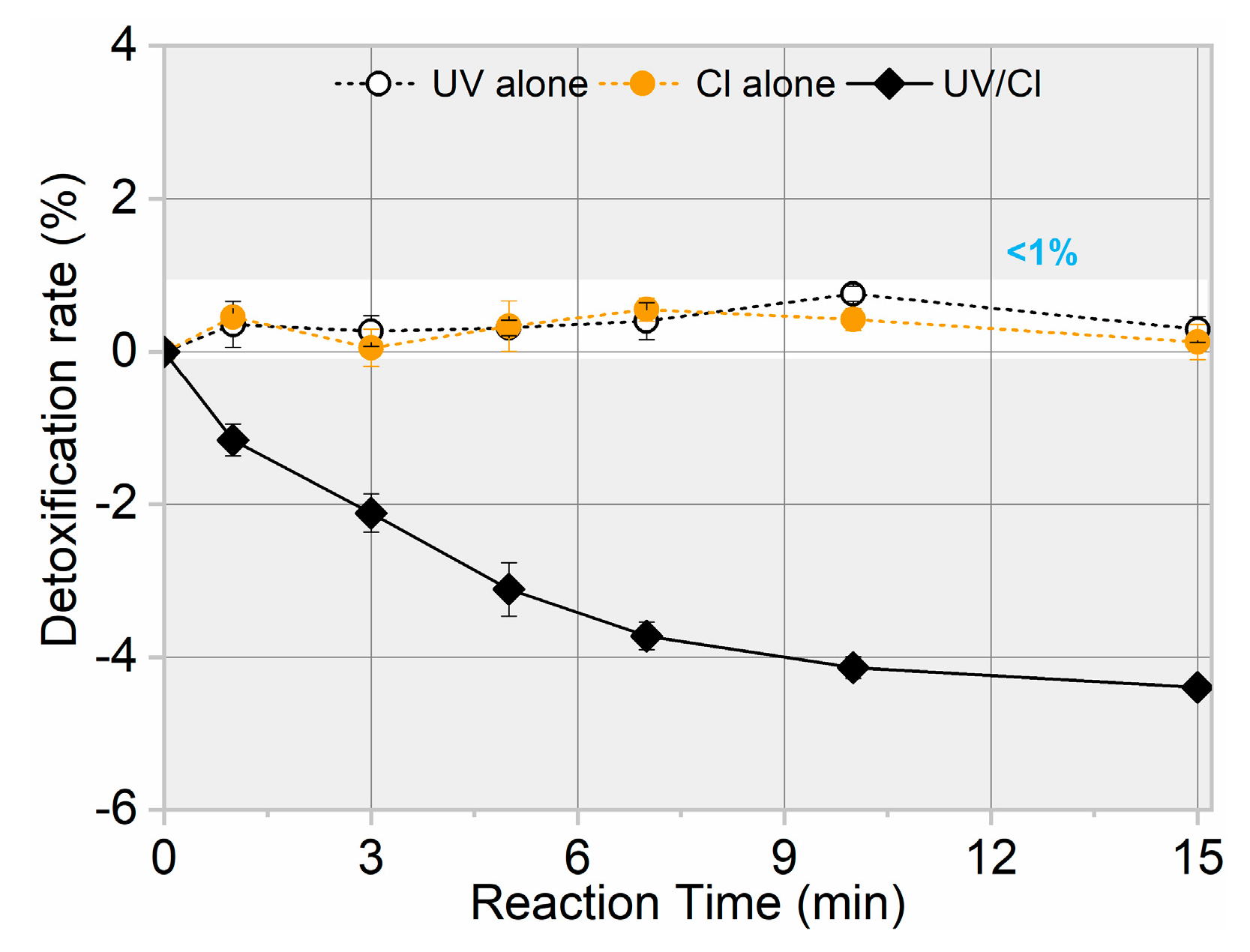
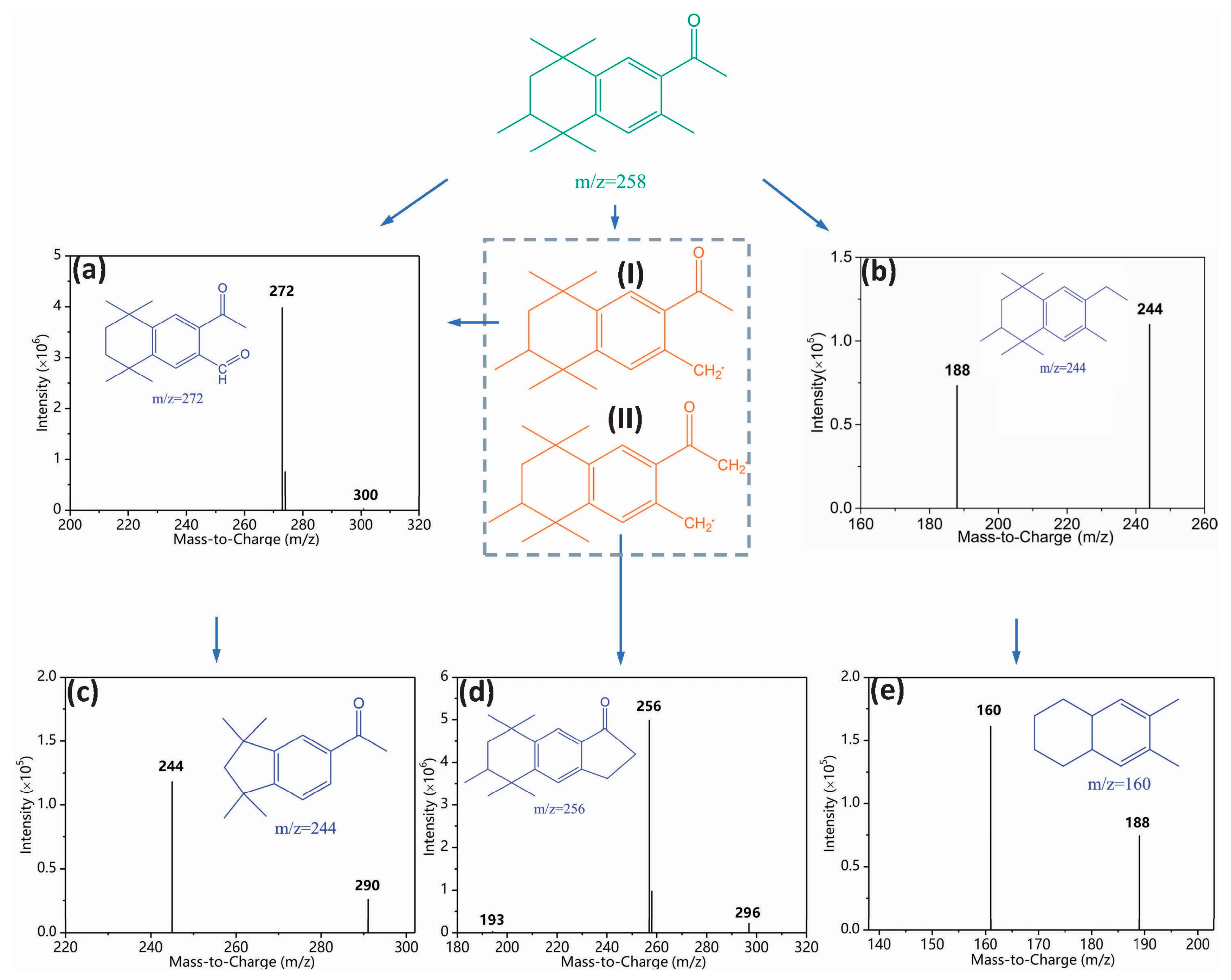
3.3. Toxicity Change, Intermediate Formation, and Degradation Pathway
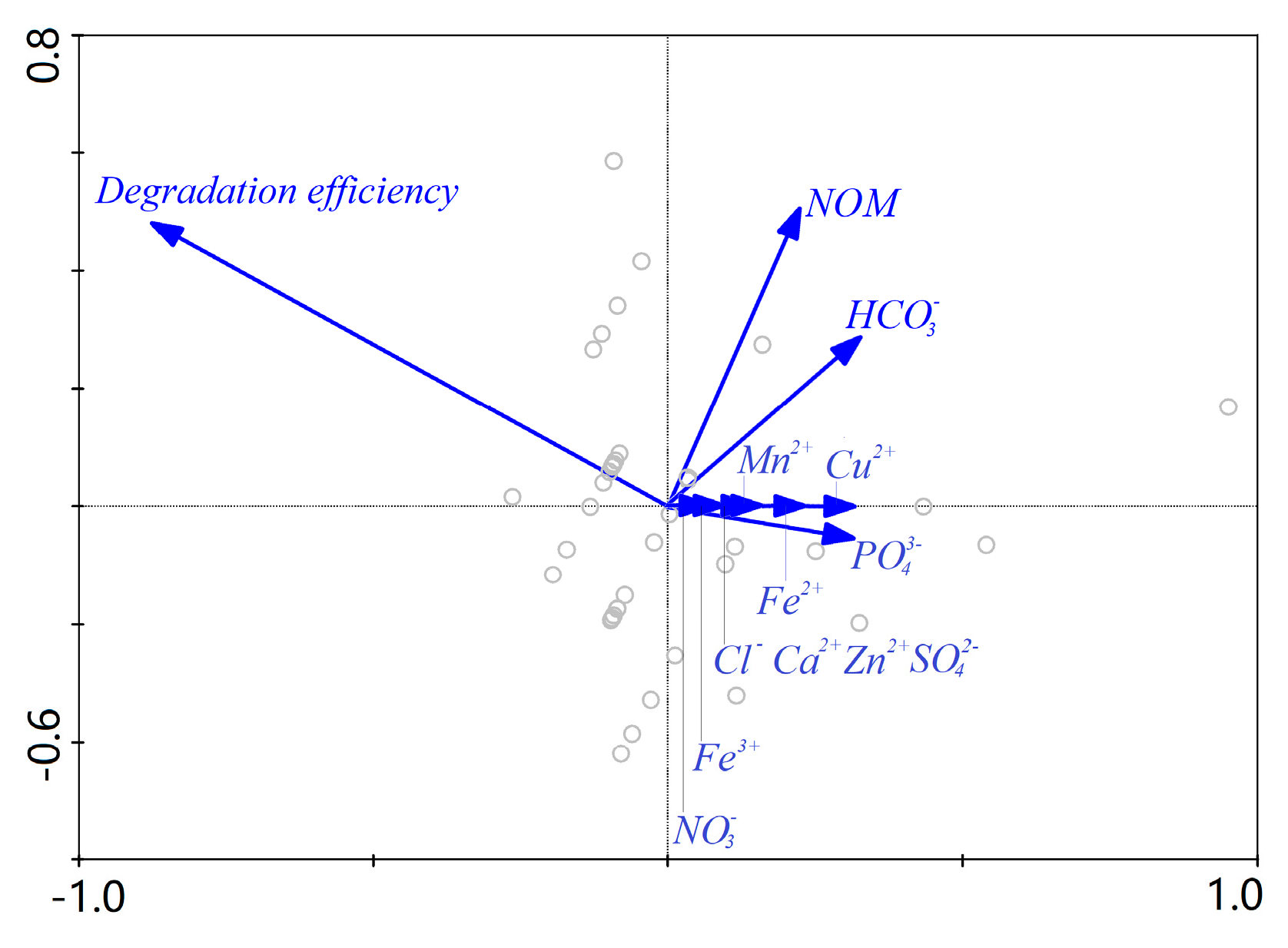
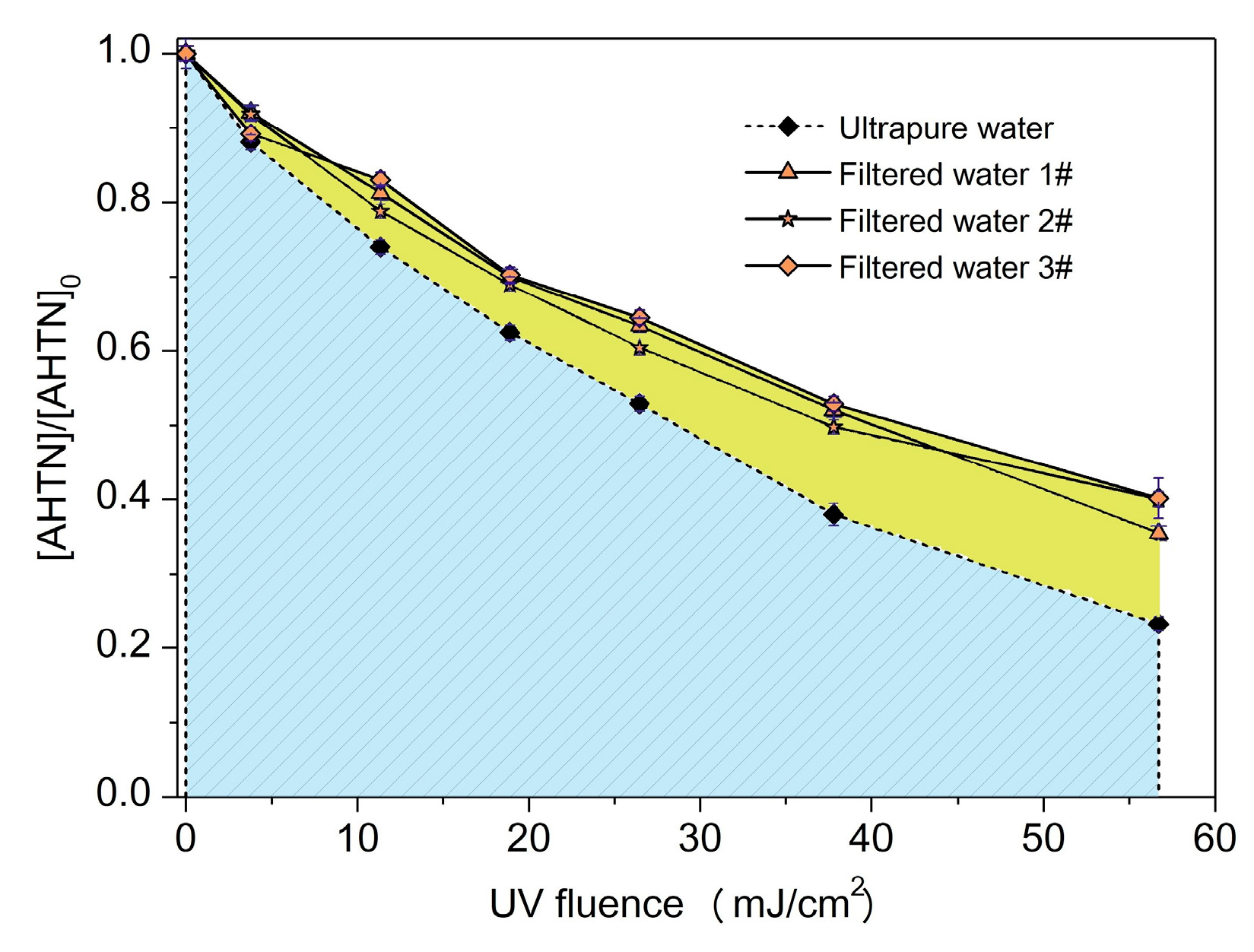
3.4. Effect of Water Background on AHTN Degradation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Parolini, M.; Magni, S.; Traversi, I.; Villa, S.; Finizio, A.; Binelli, A. Environmentally relevant concentrations of galaxolide (HHCB) and tonalide (AHTN) induced oxidative and genetic damage in Dreissena polymorpha. J. Hazard. Mater. 2015, 285, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Bitsch, N.; Dudas, C.; Korner, W.; Failing, K.; Biselli, S.; Rimkus, G.; Brunn, H. Estrogenic activity of musk fragrances detected by the E-screen assay using human MCF-7 cells. Arch. Environ. Contam. Toxicol. 2002, 43, 257–264. [Google Scholar] [CrossRef] [PubMed]
- Yamauchi, R.; Ishibashi, H.; Hirano, M.; Mori, T.; Kim, J.W.; Arizono, K. Effects of synthetic polycyclic musks on estrogen receptor, vitellogenin, pregnane X receptor, and cytochrome P450 3A gene expression in the livers of male medaka (Oryzias latipes). Aquat. Toxicol. 2008, 90, 261–268. [Google Scholar] [CrossRef] [PubMed]
- Stackelberg, P.E.; Gibs, J.; Furlong, E.T.; Meyer, M.T.; Zaugg, S.D.; Lippincott, R.L. Efficiency of conventional drinking-water-treatment processes in removal of pharmaceuticals and other organic compounds. Sci. Total. Environ. 2007, 377, 255–272. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Nanaboina, V.; Chen, F.; Korshin, G.V. Removal of polycyclic synthetic musks and antineoplastic drugs in ozonated wastewater: Quantitation based on the data of differential spectroscopy. J. Hazard. Mater. 2016, 304, 242–250. [Google Scholar] [CrossRef] [PubMed]
- Barışçı, S.; Dimoglo, A. Review on the stability of ferrate (vi) species in aqueous medium and oxidation of pharmaceuticals and personal care products (PPCPs) by ferrate (VI): Identification of transformation by-products. In Ferrites and Ferrates: Chemistry and Applications in Sustainable Energy and Environmental Remediation, 1st ed.; Sharma, V.K., Doong, R.A., Kim, H., Varma, R.S., Dionysiou, D.D., Eds.; American Chemical Society: Washington, DC, USA, 2016; Volume 1238, pp. 287–335. ISBN 9780841231870. [Google Scholar]
- Snyder, S.A.; Adham, S.; Redding, A.M.; Cannon, F.S.; DeCarolis, J.; Oppenheimer, J.; Wert, E.C.; Yoon, Y. Role of membranes and activated carbon in the removal of endocrine disruptors and pharmaceuticals. Desalination 2007, 202, 156–181. [Google Scholar] [CrossRef]
- Godayol, A.; Gonzalez-Olmos, R.; Sanchez, J.M.; Anticó, E. Assessment of the effect of UV and chlorination in the transformation of fragrances in aqueous samples. Chemosphere 2015, 125, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Kong, X.; Jiang, J.; Ma, J.; Yang, Y.; Liu, W.; Liu, Y. Degradation of atrazine by UV/chlorine: Efficiency, influencing factors, and products. Water Res. 2016, 90, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Remucal, C.K.; Manley, D. Emerging investigators series: The efficacy of chlorine photolysis as an advanced oxidation process for drinking water treatment. Environ. Sci. Water Res. Technol. 2016, 2, 565–579. [Google Scholar] [CrossRef]
- Felis, E.; Alder, A.C.; Surmacz-Gorska, J.; Miksch, K. Advanced oxidation of the polycyclic musk fragrances with using UV and UV/H2O2 processes. Arch. Environ. Prot. 2008, 34, 13–23. [Google Scholar] [CrossRef]
- Wang, D.; Bolton, J.R.; Andrews, S.A.; Hofmann, R. Formation of disinfection by-products in the ultraviolet/chlorine advanced oxidation process. Sci. Total. Environ. 2015, 518–519, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Lin, Y.; Xu, B.; Xia, S.; Tian, F.; Gao, N. Effect of UV irradiation on the proportion of organic chloramines in total chlorine in subsequent chlorination. Chemosphere 2016, 144, 940–947. [Google Scholar] [CrossRef] [PubMed]
- Deng, L.; Huang, C.; Wang, Y. Effects of combined UV and chlorine treatment on the formation of trichloronitromethane from amine precursors. Environ. Sci. Technol. 2014, 48, 2697–2705. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Qiang, Z.; Tian, F.; Zhang, T. Photodegradation of etridiazole by UV radiation during drinking water treatment. Chemosphere 2009, 76, 609–615. [Google Scholar] [CrossRef] [PubMed]
- Leenheer, J.A. Comprehensive approach to preparative isolation and fractionation of dissolved organic carbon from natural waters and wastewaters. Environ. Sci. Technol. 1981, 15, 578–587. [Google Scholar] [CrossRef] [PubMed]
- Swietlik, J.; Dabrowska, A.; Raczyk-Stanislawiak, U.; Nawrocki, J. Reactivity of natural organic matter fractions with chlorine dioxide and ozone. Water Res. 2004, 38, 547–558. [Google Scholar] [CrossRef] [PubMed]
- Rosenfeldt, E.; Linden, K.; Canonica, S.; von Gunten, U. Comparison of the efficiency of OH radical formation during ozonation and the advanced oxidation processes O3/H2O2 and UV/H2O2. Water Res. 2006, 40, 3695–3704. [Google Scholar] [CrossRef] [PubMed]
- Guan, Y.; Ma, J.; Li, X.; Fang, J.; Chen, L. Influence of pH on the formation of sulfate and hydroxyl radicals in the UV/peroxymonosulfate system. Environ. Sci. Technol. 2011, 45, 9308–9314. [Google Scholar] [CrossRef] [PubMed]
- Fang, J.; Fu, Y.; Shang, C. The roles of reactive species in micropollutant degradation in the UV/free chlorine system. Environ. Sci. Technol. 2014, 48, 1859–1868. [Google Scholar] [CrossRef] [PubMed]
- Watts, M.J.; Linden, K.G. Chlorine photolysis and subsequent OH radical production during UV treatment of chlorinated water. Water Res. 2007, 41, 2871–2878. [Google Scholar] [CrossRef] [PubMed]
- Zehavi, D.; Rabani, J. Oxidation of aqueous bromide ions by hydroxyl radicals. Pulse radiolytic investigation. J. Phys. Chem. 1972, 76, 312–319. [Google Scholar] [CrossRef]
- Kläning, U.K.; Wolff, T. Laser flash photolysis of HCIO, CIO−, HBrO, and BrO− in aqueous solution. Reactions of Cl− and Br− Atoms. Berichte der Bunsengesellschaft für Physikalische Chemie 1985, 89, 243–245. [Google Scholar] [CrossRef]
- Sun, P.; Lee, W.; Zhang, R.; Huang, C. Degradation of DEET and caffeine under UV/Chlorine and simulated sunlight/chlorine conditions. Environ. Sci. Technol. 2016, 50, 13265–13273. [Google Scholar] [CrossRef] [PubMed]
- Buxton, G.V.; Greenstock, C.L.; Helman, W.P.; Ross, A.B. Critical review of rate constants for reactions of hydrated electrons, hydrogen atoms and hydroxyl radicals (•OH/•O−) in aqueous solution. J. Phys. Chem. Ref. Data 1988, 17, 513–886. [Google Scholar] [CrossRef]
- Molina, M.J.; Ishiwata, T.; Molina, L.T. Production of hydroxyl from photolysis of hypochlorous acid at 307–309 nm. J. Phys. Chem. 1980, 84, 821–826. [Google Scholar] [CrossRef]
- Kläning, U.K.; Sehested, K.; Wolff, T. Ozone formation in laser flash photolysis of oxoacids and oxoanions of chlorine and bromine. J. Chem. Soc. Faraday Trans. 1984, 80, 2969–2979. [Google Scholar] [CrossRef]
- Jayson, G.; Parsons, B.; Swallow, A.J. Some simple, highly reactive, inorganic chlorine derivatives in aqueous solution. Their formation using pulses of radiation and their role in the mechanism of the Fricke dosimeter. J. Chem. Soc. Faraday Trans. 1973, 69, 1597–1607. [Google Scholar] [CrossRef]
- Grigor’ev, A.; Makarov, I.; Pikaev, A. Formation of Cl2− in the bulk of solution during radiolysis of concentrated aqueous solutions of chlorides. Khimiya Vysokikh Ehnergij 1987, 21, 99–102. [Google Scholar]
- Grebel, J.E.; Pignatello, J.J.; Mitch, W.A. Effect of halide ions and carbonates on organic contaminant degradation by hydroxyl radical-based advanced oxidation processes in saline Waters. Environ. Sci. Technol. 2010, 44, 6822–6828. [Google Scholar] [CrossRef] [PubMed]
- Zuo, Z.; Katsumura, Y.; Ueda, K.; Ishigure, K. Reactions between some inorganic radicals and oxychlorides studied by pulse radiolysis and laser photolysis. J. Chem. Soc. Faraday Trans. 1997, 93, 1885–1891. [Google Scholar] [CrossRef]
- Kuhlich, P.; Göstl, R.; Teichert, P.; Piechotta, C.; Nehls, I. Transformations of polycyclic musks AHTN and HHCB upon disinfection with hypochlorite: Two new chlorinated disinfection by-products (CDBP) of AHTN and a possible source for HHCB-lactone. Anal. Bioanal. Chem. 2011, 399, 3579–3588. [Google Scholar] [CrossRef] [PubMed]
- Radjenovic, J.; Godehardt, M.; Hein, A.; Farre, M.; Jekel, M.; Barcelo, D. Evidencing generation of persistent ozonation products of antibiotics roxithromycin and trimethoprim. Environ. Sci. Technol. 2009, 43, 6808–6815. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Linden, K.G.; Hinton, D.E.; Kashiwada, S.; Rosenfeldt, E.J.; Kullman, S.W. Biological assessment of bisphenol A degradation in water following direct photolysis and UV advanced oxidation. Chemosphere 2006, 65, 1094–1102. [Google Scholar] [CrossRef] [PubMed]
- Alfassi, Z.B.; Huie, R.E.; Mosseri, S.; Neta, P. Kinetics of one-electron oxidation by the ClO radical. Int. J. Radiat. Appl. Instrum. C 1988, 32, 85–88. [Google Scholar] [CrossRef]
- von Sonntag, C. Advanced oxidation processes: Mechanistic aspects. Water Sci. Technol. 2008, 58, 1015–1021. [Google Scholar] [CrossRef] [PubMed]
- Cooper, W.J.; Cramer, C.J.; Martin, N.H.; Mezyk, S.P.; O’Shea, K.E.; Sonntag, C.V. Free radical mechanisms for the treatment of methyl tert-butyl ether (MTBE) via advanced oxidation/reductive processes in aqueous solutions. Chem. Rev. 2009, 109, 1302–1345. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Pignatello, J.J.; Ma, J.; Mitch, W.A. Comparison of halide impacts on the efficiency of contaminant degradation by sulfate and hydroxyl radical-based advanced oxidation processes (AOPs). Environ. Sci. Technol. 2014, 48, 2344–2351. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Fang, J.; Xiang, Y.; Shang, C.; Li, X.; Meng, F.; Yang, X. Roles of reactive chlorine species in trimethoprim degradation in the UV/chlorine process: Kinetics and transformation pathways. Water Res. 2016, 104, 272–282. [Google Scholar] [CrossRef] [PubMed]
- Bard, A.J.; Parsons, R.; Jordan, J. (Eds.) Standard Potentials in Aqueous Solution, 1st ed.; Marcel Dekker: New York, NY, USA, 1985; pp. 127–413. ISBN 0-8247-7291-1. [Google Scholar]
- Neta, P.; Huie, R.E.; Ross, A.B. Rate constants for reactions of inorganic radicals in aqueous solution. J. Phys. Chem. Ref. Data 1988, 17, 1027–1284. [Google Scholar] [CrossRef]
- Guo, K.; Wu, Z.; Shang, C.; Yao, B.; Hou, S.; Yang, X.; Song, W.; Fang, J. Radical chemistry and structural relationships of PPCP degradation by UV/Chlorine treatment in simulated drinking water. Environ. Sci. Technol. 2017, 51, 10431–10439. [Google Scholar] [CrossRef] [PubMed]
- Piscopo, A.; Robert, D.; Weber, J.V. Influence of pH and chloride anion on the photocatalytic degradation of organic compounds. Appl. Catal. B Environ. 2001, 35, 117–124. [Google Scholar] [CrossRef]
- Abe, K.; Tanaka, K. Effect of Fe3+ on UV-illuminated ozonation of nitrophenolic compounds. Chemosphere 1999, 38, 2747–2752. [Google Scholar] [CrossRef]
- Wang, H.; Yao, H.; Sun, P.; Li, D.; Huang, C. Transformation of tetracycline antibiotics and Fe (II) and Fe (III) species induced by their complexation. Environ. Sci. Technol. 2015, 50, 145–153. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Equations | Reaction | Rate Constant | Reference |
|---|---|---|---|
| (I) | HOCl ⇌ + | pKa1 = 7.5 | [20] |
| (II) | HOCl + hv → HO• + Cl• | ΦHO• = 1.45 | [21] |
| (III) | + hv → + Cl• | ΦOCl− = 0.97 | [20] |
| (IV) | + hv → O(1D) + Cl− | ΦO(1D) = 0.133 | [10] |
| (V) | + hv → O(3P) + Cl− | ΦO(3P)• = 0.074 | [10] |
| (VI) | HO• → + | pKa1 = 11.9 | [20] |
| (VII) | HO• + HOCl → ClO• + H2O | k7 = 2.0 × 109 M−1·s−1 | [20] |
| (VIII) | HO• + → ClO• + | k8 = 8.8 × 109 M−1·s−1 | [20] |
| (IX) | HO• + → | k9 = 4.3 × 109 M−1·s−1 | [10] |
| (X) | HO• + → + H2O | k10 = 1.3 × 1010 M−1·s−1 | [20] |
| (XI) | Cl• + HOCl → ClO• + + | k11 = 3.0 × 109 M−1·s−1 | [20] |
| (XII) | Cl• + → ClO• + | k12 = 8.2 × 109 M−1·s−1 | [22] |
| (XIII) | Cl• + → | k13 = 6.5 × 109 M−1·s−1 | [23] |
| (XIV) | Cl• + H2O → + | k14 = 4.5 × 103 M−1·s−1 | [24] |
| (XV) | + H2O → HO• + | k15 = 1.8 × 106 M−1·s−1 | [25] |
| (XVI) | O(1D) + H2O → 2 HO• | k16 = 1.2 × 1011 M−1·s−1 | [26] |
| (XVII) | O(3P) + O2 →O3 | k17 = 4.0 × 109 M−1·s−1 | [27] |
| (XVIII) | O(3P) + → | k18 = 9.4 × 109 M−1·s−1 | [27] |
| (XIX) | → Cl• + | K19 = 1.1 × 105 M−1·s−1 | [28] |
| (XX) | → HO• + | k20 = 6.1 × 109 M−1·s−1 | [23] |
| (XXI) | + → + | k21 = 1.0 × 105 M−1·s−1 | [29] |
| (XXII) | + → Cl• + H2O | K22 = 2.1 × 1010 M−1·s−1 | [23] |
| (XXIII) | 2ClO• + H2O →HOCl + + | k23 = 2.5 × 109· s−1 | [23] |
| (XXIV) | HO• + → + | k24 = 3.9 × 108 M−1·s−1 | [30] |
| (XXV) | HO• + → + H2O | k25 = 8.6 × 106 M−1·s−1 | [25] |
| (XXVI) | HO• + → + | k26 = 6.3 × 109 M−1·s−1 | [31] |
| (XXVII) | HO• + → + | k27 = 4.0 × 109 M−1·s−1 | [31] |
| Water Matrices | Units | FW1# | FW2# | FW3# |
|---|---|---|---|---|
| pH | -- | 7.15 | 6.68 | 7.47 |
| TOC | mg·L−1 | 1.93 | 4.00 | 8.77 |
| mg·L−1 | 7.89 | 5.89 | 12.83 | |
| mg·L−1 | ND | ND | ND | |
| mg·L−1 | 15.71 | 8.91 | 34.69 | |
| mg·L−1 | 9.39 | 8.49 | 3.90 | |
| UV254 | cm−1·(mg·L−1)−1 | 0.015 | 0.003 | 0.029 |
| mg·L−1 | 18.33 | 11.35 | 22.41 | |
| mg·L−1 | 7.22 | 9.52 | 10.36 | |
| mg·L−1 | 0.012 | 0.007 | 0.006 | |
| mg·L−1 | 0.076 | 0.001 | ND | |
| mg·L−1 | 1.23 | 0.034 | 0.022 | |
| Total Fe | mg·L−1 | 0.16 | 0.14 | 0.061 |
| Varying Parameters | Concentration | * Ionic Strength (×10−5 M) | ∆pH | k (s−1) | R2 |
|---|---|---|---|---|---|
| (mM, as CaCl) | 0.2 | 60 | 0.01 | 0.0014 | 0.997 |
| 1.0 | 300 | 0.01 | 0.0013 | 0.997 | |
| 2.0 | 900 | 0.01 | 0.0014 | 0.999 | |
| (mM, as NaCl) | 0.5 | 50 | 0.01 | 0.0014 | 0.999 |
| 1.0 | 100 | 0.01 | 0.0013 | 0.998 | |
| 4.0 | 400 | 0.01 | 0.0014 | 0.999 | |
| (μM, as CuCl2) | 0.6 | 0.18 | 0.01 | 0.0015 | 0.994 |
| 1.8 | 0.54 | 0.01 | 0.0014 | 0.997 | |
| 3.0 | 0.9 | 0.01 | 0.0015 | 0.998 | |
| NOM (mg·L−1) | 0.5 | – | 0 | 0.0012 | 0.999 |
| 1.0 | – | 0 | 0.0011 | 0.998 | |
| 2.0 | – | 0 | 0.0010 | 0.999 | |
| 5.0 | – | 0 | 0.0007 | 0.997 | |
| (mM, as FeCl2) | 0.001 | 0.3 | 0.01 | 0.0015 | 0.998 |
| 0.002 | 0.6 | 0.01 | 0.0014 | 0.998 | |
| 0.004 | 1.2 | 0.01 | 0.0014 | 0.999 | |
| (mM, as FeCl3) | 0.001 | 0.6 | 0.01 | 0.0015 | 0.999 |
| 0.002 | 1.2 | −0.04 | 0.0013 | 0.996 | |
| 0.004 | 2.4 | 0.01 | 0.0012 | 0.995 | |
| (mM, as NaHCO3) | 0.1 | 10 | 0.01 | 0.0013 | 0.998 |
| 0.5 | 50 | 0.01 | 0.0012 | 0.994 | |
| 1.0 | 100 | 0.01 | 0.0012 | 0.993 | |
| 2.0 | 200 | 0.01 | 0.0013 | 0.998 | |
| 4.0 | 400 | 0.01 | 0.0013 | 0.990 | |
| 10.0 | 1000 | 0.01 | 0.0009 | 0.996 | |
| (μM, as MnCl2) | 0.5 | 0.15 | 0.01 | 0.0015 | 0.999 |
| 1.0 | 0.3 | 0.01 | 0.0014 | 0.998 | |
| 2.0 | 0.6 | 0.01 | 0.0014 | 0.999 | |
| (mM, as NaNO3) | 0.05 | 5 | 0.01 | 0.0015 | 0.996 |
| 0.1 | 10 | 0.01 | 0.0012 | 0.998 | |
| 0.15 | 15 | 0.01 | 0.0013 | 0.999 | |
| 1.0 | 100 | 0.01 | 0.0013 | 0.999 | |
| (μM, as Na3PO4) | 0.1 | 0.06 | 0.01 | 0.0013 | 0.999 |
| 0.2 | 0.12 | 0.01 | 0.0014 | 0.998 | |
| 0.4 | 0.24 | 0.01 | 0.0014 | 0.997 | |
| 1.0 | 0.6 | 0.01 | 0.0010 | 0.996 | |
| (mM, as Na2SO4) | 0.2 | 60 | 0.01 | 0.0013 | 0.996 |
| 0.4 | 120 | 0.01 | 0.0014 | 0.998 | |
| 0.6 | 180 | 0.01 | 0.0012 | 0.997 | |
| 1.0 | 300 | 0.01 | 0.0010 | 0.994 | |
| (μM as ZnCl2) | 4.0 | 1.2 | 0.01 | 0.0013 | 0.998 |
| 8.0 | 2.4 | 0.01 | 0.0014 | 0.998 | |
| 12.0 | 3.6 | 0.01 | 0.0014 | 0.999 |
| Water Matrices | Concentration (C) | Reaction Rate Constant with HO• (k) | Scavenging Rate (C × k) |
|---|---|---|---|
| AHTN | 1.0 mg·L−1 | 8.3 × 109 M−1·s−1 (this work) | 3.2 × 104 s−1 |
| 0.4–4 mM | 1.4 × 109 M−1·s−1 ([10]) | (0.6–5.6) × 106 s−1 | |
| * | 0.1–1000 μM | 1.5 × 105 M−1·s−1 (HPO42−, [25]) | (0.007–1.0) s−1 |
| 2.0 × 104 M−1·s−1 (H2PO4−, [25]) | |||
| 0.05–1 mM | < 1.0 × 105 M−1·s−1 | <(0.05–1.0) × 102 s−1 | |
| ** | 0.1–4 mM | 8.6 × 106 M−1·s−1 (, [41]) | (0.07–2.9) × 104 s−1 |
| <1.0 × 106 M−1·s−1 (, [25]) | |||
| 0.2–1 mM | 6.9 × 105 M−1·s−1 (, [25]) | ≤ ~102 s−1 | |
| NOM | 0.5–5 mg·L−1 | 2.5 × 104 (mg·L−1)−1·s−1 ([42]) | (1.3–13) × 104 s−1 |
| 0.6–3 μM | 3.5 × 108 M−1·s−1 ([25]) | (0.21–1.1) × 103 s−1 | |
| 1–3 μM | 3.2 × 108 M−1·s−1 ([25]) | (3.2–9.6) × 102 s−1 | |
| 1–4 μM | NA | ||
| 0.5–2 μM | 2.9 × 107 M−1·s−1 ([25]) | (1.5–5.8) × 101 s−1 | |
| 4–12 μM | <5.0 × 105 M−1·s−1 ([25]) | <(2–6) s−1 | |
| 0.2–3 mM | NA | ||
| HOCl *** | 46.2 μM | 2.0 × 109 M−1·s−1 (HOCl, [20]) | 1.7 × 105 M−1·s−1 |
| 8.8 × 109 M−1·s−1 (OCl−, [20]) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, L.; Liu, X. Degradation of Aqueous Polycyclic Musk Tonalide by Ultraviolet-Activated Free Chlorine. Processes 2019, 7, 95. https://doi.org/10.3390/pr7020095
Wang L, Liu X. Degradation of Aqueous Polycyclic Musk Tonalide by Ultraviolet-Activated Free Chlorine. Processes. 2019; 7(2):95. https://doi.org/10.3390/pr7020095
Chicago/Turabian StyleWang, Lili, and Xiaowei Liu. 2019. "Degradation of Aqueous Polycyclic Musk Tonalide by Ultraviolet-Activated Free Chlorine" Processes 7, no. 2: 95. https://doi.org/10.3390/pr7020095
APA StyleWang, L., & Liu, X. (2019). Degradation of Aqueous Polycyclic Musk Tonalide by Ultraviolet-Activated Free Chlorine. Processes, 7(2), 95. https://doi.org/10.3390/pr7020095