Effect of Ammonia Activation and Chemical Vapor Deposition on the Physicochemical Structure of Activated Carbons for CO2 Adsorption


Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Pretreatment
2.2. Samples Preparation
2.3. Measurement of Physicochemical Structure of Samples
2.4. CO2 Adsorption Test of Typical Samples
3. Results and Discussion
3.1. Surface Morphology and Microstructure Analysis of Samples at Different Ammonia Activation Conditions
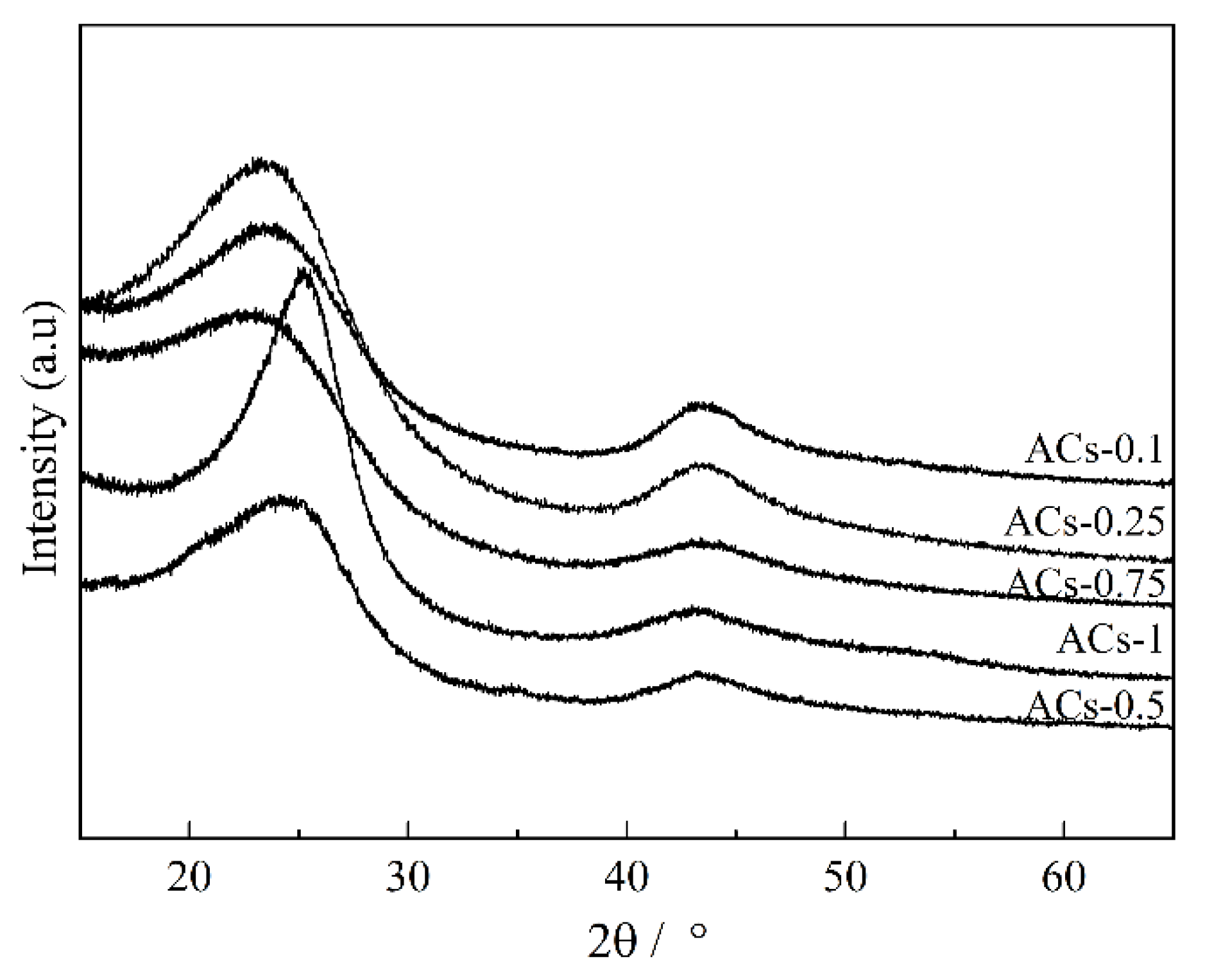
3.2. Crystal Structure Analysis of Samples at Different Ammonia Activation Conditions
3.3. Carbon Structure Analysis of Samples at Different Ammonia Activation Conditions
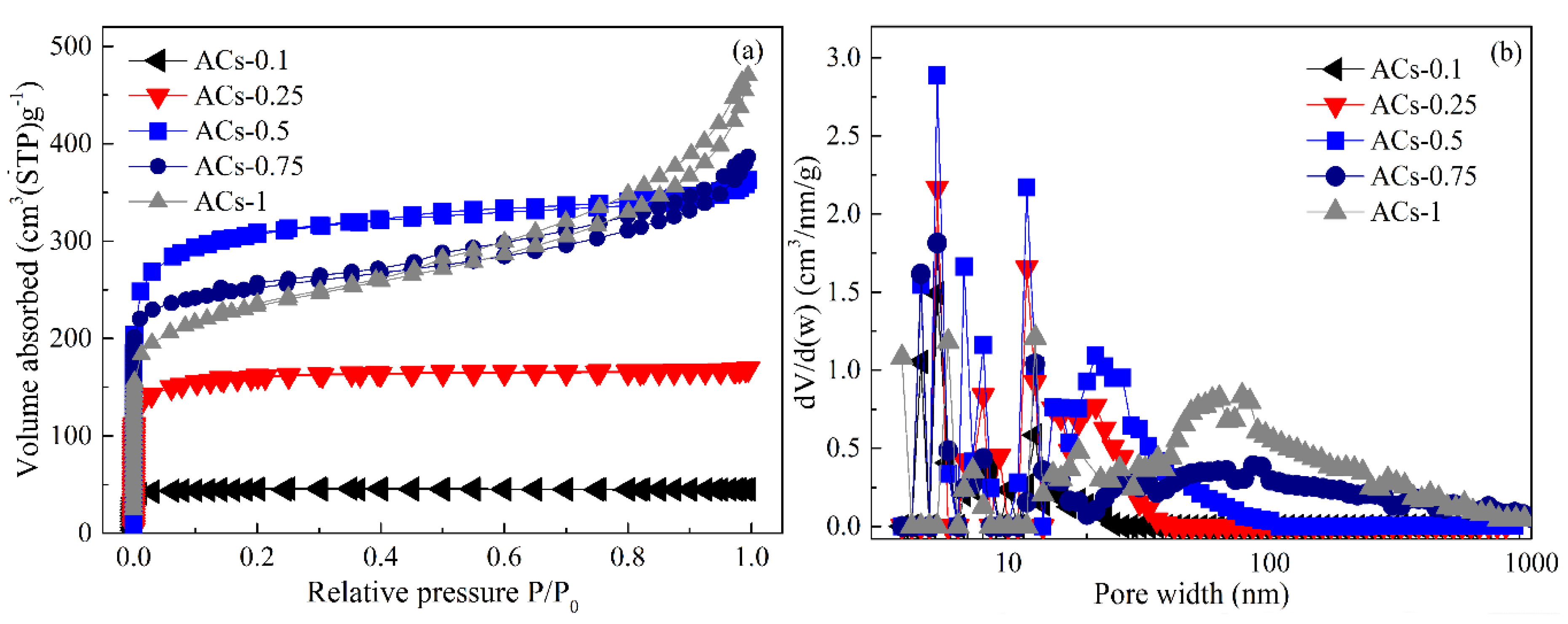
3.4. Pore Structure Analysis of Samples at Different Ammonia Activation Conditions
3.5. Surface Chemical Structure Analysis of Samples at Different Ammonia-Activation Conditions
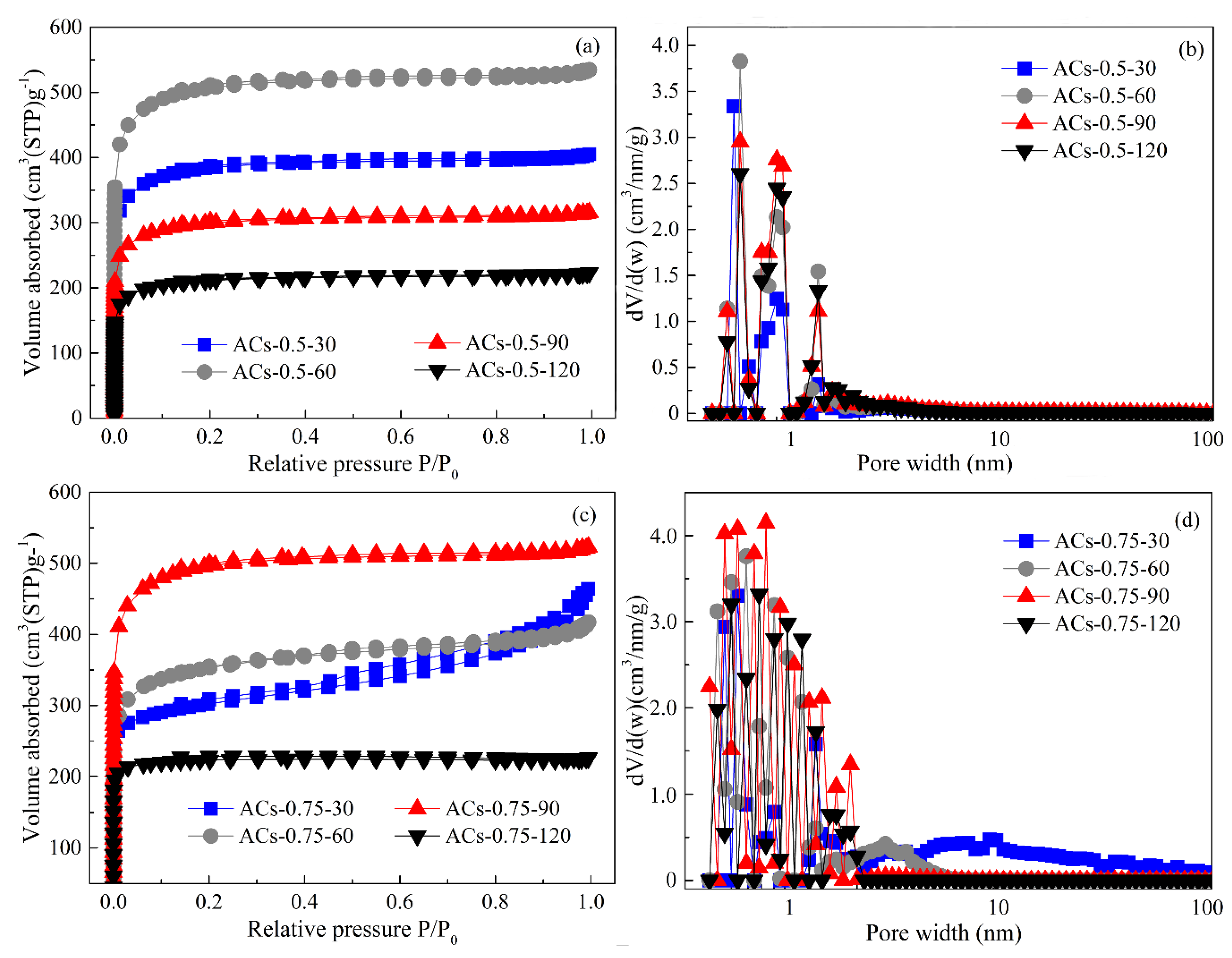
3.6. Pore Structure Analysis of Typical Samples at Different Deposition Times
3.7. Study of CO2 Adsorption
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- D’Alessandro, D.M.; Smit, B.; Long, J.R. Carbon dioxide capture: Prospects for new materials. Angew. Chem. 2010, 49, 6058–6082. [Google Scholar] [CrossRef]
- Chen, B.L.; Wen, H.M.; Liao, C.J.; Li, L.B.; Alothman, Z.A.; Krishna, R.; Wu, H.; Zhou, W.; Hu, J. A metal−organic framework with suitable pore size and dual functionalities for highly efficient post-combustion CO2 capture. J. Mater. Chem. A 2019, 7, 3128–3134. [Google Scholar] [CrossRef]
- Li, H.; Wang, K.C.; Hu, Z.G.; Chen, Y.P.; Verdegaal, W.M.; Zhao, D.; Zhou, H.C. Harnessing solvent effects to integrate alkylamine into metal–organic frameworks for exceptionally high CO2 uptake. J. Mater. Chem. A 2019, 7, 7867–7874. [Google Scholar] [CrossRef]
- Drage, T.C.; Blackman, J.M.; Pevida, C.; Snape, C.E. Evaluation of activated carbon adsorbents for CO2 capture in gasification. Energy Fuels 2009, 23, 2790–2796. [Google Scholar] [CrossRef]
- Liu, Z.; Yang, Y.; Du, Z.Y.; Xing, W.; Komarneni, S.; Zhang, Z.D.; Gao, X.H.; Yan, Z.F. Furfuralcohol co-polymerized urea formaldehyde resin-derived N-doped microporous carbon for CO2 capture. Nanoscale Res. Lett. 2015, 10, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Yi, H.H.; Li, Y.D.; Tang, X.L.; Li, F.R.; Li, K.; Yuan, Q.; Sun, X. Effect of the adsorbent pore structure on the separation of carbon dioxide and methane gas mixtures. J. Chem. Eng. Data 2015, 60, 1388–1395. [Google Scholar] [CrossRef]
- Zhao, Y.L.; Feng, Y.H.; Zhang, X.X. Molecular simulation of CO2/CH4 self-and transport diffusion coefficients in coal. Fuel 2016, 165, 19–27. [Google Scholar] [CrossRef]
- Wickramaratne, N.P. Importance of small micropores in CO2 capture by phenolic resin-based activated carbon spheres. J. Mater. Chem. A 2012, 1, 112–116. [Google Scholar] [CrossRef]
- Presser, V.; Mcdonough, J.; Yeon, S.H.; Gogotsi, Y. Effect of pore size on carbon dioxide sorption by carbide derived carbon. Energy Environ. Sci. 2011, 4, 3059–3066. [Google Scholar] [CrossRef]
- Hohm, U. Polarizabilities of atoms and molecules, new insights into an old subject. Vacuum 2000, 58, 117–134. [Google Scholar] [CrossRef]
- Khoerunnisa, F.; Minami, D.; Fujimori, T.; Hong, S.Y.; Choi, Y.C.; Sakamoto, H.; Endo, M.; Kaneko, K. Enhanced CO2 adsorptivity of SWCNT by polycyclic aromatic hydrocarbon intercalation. Adsorption 2014, 20, 301–309. [Google Scholar] [CrossRef]
- Gong, K.P.; Du, F.; Xia, Z.H.; Durstock, M.; Dai, L.M. Nitrogen-doped carbon nanotube arrays with high electrocatalytic activity for oxygen reduction. Science 2009, 323, 760–764. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.X.; Webley, P.A.; Zhao, D.Y. Post-enrichment of nitrogen in soft-templated ordered mesoporous carbon materials for highly efficient phenol removal and CO2 capture. J. Mater. Chem. 2012, 22, 11379–11389. [Google Scholar] [CrossRef]
- Wilke, A.; Weber, J. Hierarchical nanoporous melamine resin sponges with tunable porosity-porosity analysis and CO2 sorption properties. J. Mater. Chem. 2011, 21, 5226–5229. [Google Scholar] [CrossRef]
- Zhai, Y.B.; Xu, B.B.; Zhu, Y.; Qing, R.P.; Peng, C.; Wang, T.F.; Li, C.T.; Zeng, G.M. Nitrogen-doped porous carbon from Camellia oleifera, shells with enhanced electrochemical performance. Mater. Sci. Eng. 2016, 61, 449–456. [Google Scholar] [CrossRef]
- Jin, H.; Zhang, H.M.; Zhong, H.X.; Zhang, J.L. Nitrogen-doped carbon xerogel: A novelcarbon-based electrocatalyst for oxygen reduction reaction in proton exchange membrane (PEM) fuel cells. Energy Environ. Sci. 2011, 4, 3389–3394. [Google Scholar] [CrossRef]
- Kim, N.D.; Kim, W.; Joo, J.B.; Oh, S.; Kim, P.; Kim, Y.; Yi, J. Electrochemical capacitor performance of N-doped mesoporous carbons prepared by ammoxidation. J. Power Sources 2008, 180, 671–675. [Google Scholar] [CrossRef]
- Shiratori, N.; Lee, K.J.; Miyawaki, J.; Hong, S.H.; Mochida, I.; An, B.; Yokogawa, K.; Jang, J.; Yoon, S.H. Pore structure analysis of activated carbon fiber by microdomain-based model. Langmuir 2009, 25, 7631–7637. [Google Scholar] [CrossRef]
- Liu, D.D.; Gao, J.H.; Wu, S.H.; Qin, Y.K. Effect of char structures caused by varying the amount of FeCl3 the pore development during activation. RSC Adv. 2016, 6, 87478–87485. [Google Scholar] [CrossRef]
- Srinivas, G.; Yue, L.; Neal, S.; Taner, Y.; Zheng, X.G. Design of hyperporous graphene networks and their application in solid-amine based carbon capture systems. J. Mater. Chem. A 2017, 5, 17833–17840. [Google Scholar] [CrossRef]
- Hu, B.; Wang, K.; Wu, L.H.; Yu, S.H.; Antonietti, M.; Titirici, M.M. Engineering carbon materials from the hydrothermal carbonization process of biomass. Adv. Mater. 2010, 22, 813–828. [Google Scholar] [CrossRef] [PubMed]
- Lee, F.Y.M.; Tran, N.H.; Russell, J.J.; Lamb, R.N. Cathodoluminescence of Zinc Sulfide Films Grown by Single Source Chemical Vapor Deposition. J. Phys. Chem. B 2019, 108, 8355–8358. [Google Scholar] [CrossRef]
- Mohammadi, M.; Najafpour, G.D.; Mohamed, A.R. Production of carbon molecular sieves from palm shell through carbon deposition from methane. Chem. Ind. Chem. Eng. Q. 2011, 17, 525–533. [Google Scholar] [CrossRef]
- Zhang, T.; Walawender, W.P.; Fan, L. Preparation of carbon molecular sieves by carbon deposition from methane. Bioresour. Technol. 2005, 96, 1929–1935. [Google Scholar] [CrossRef]
- Cansado, I.P.P.; Mouräo, P.A.M.; Ribeiro Carrott, M.M.L.; Carrott, P. Activated carbons prepared from natural and synthetic raw materials with potential applications in gas separations. Adv. Mater. Res. 2010, 107, 1–7. [Google Scholar] [CrossRef]
- Kang, H.U.; Kim, W.; Kim, S.H. Pore size control through benzene vapor deposition on activated carbon. Chem. Eng. J. 2008, 144, 167–174. [Google Scholar] [CrossRef]
- Smith, M.T.; Yager, J.W.; Steinmetz, K.L.; Eastmond, D.A. Peroxidase-dependent metabolism of benzene’s phenolic metabolites and its potential role in benzene toxicity and carcinogenicity. Environ. Health Perspect. 1989, 82, 23–29. [Google Scholar] [CrossRef]
- Villar-Rodil, S.; Navarrete, R.; Denoyel, R.; Albiniak, A.; Paredes, J.I.; Martínez-Alonso, A.; Tascón, J.M.D. Carbon molecular sieve cloths prepared by chemical capour deposition of methane for separation of gas mixtures. Microporous Mesoporous Mater. 2005, 77, 109–118. [Google Scholar] [CrossRef]
- Sathe, C.; Pang, Y.; Li, C.Z. Effects of heating rate and ion-exchangeable cations on the pyrolysis yields from a Victorian brown coal. Energy Fuels 1999, 13, 748–755. [Google Scholar] [CrossRef]
- Pietrzak, R. XPS study and physico-chemical properties of nitrogen-enriched microporous activated carbon from high volatile bituminous coal. Fuel 2009, 88, 1871–1877. [Google Scholar] [CrossRef]
- Gong, X.Z.; Guo, Z.C.; Wang, Z. Variation of char structure during anthracite pyrolysis catalyzed by Fe2O3 and its influence on char combustion reactivity. Energy Fuels 2009, 23, 4547–4552. [Google Scholar] [CrossRef]
- Belhachemi, M.; Rios, R.V.R.A.; Addoun, F.; Silvestre-Albero, J.; Sepulveda-Escribano, A.; Rodrìguez-Reinoso, F. Preparation of activated carbon from date pits: Effect of the activation agent and liquid phase oxidation. J. Anal. Appl. Pyrolysis 2009, 86, 168–172. [Google Scholar] [CrossRef]
- Yang, K.B.; Peng, J.H.; Xia, H.Y.; Zhang, L.B.; Srinivasakannan, C.; Guo, S.H. Textural characteristics of activated carbon by single step CO2 activation from coconut shells. J. Taiwan Inst. Chem. Eng. 2010, 41, 367–372. [Google Scholar] [CrossRef]
- Heras, F.; Alonso-Morales, N.; Jimenez-Cordero, D.; Gilarranz, M.A.; Rodriguez, J.J. Granular mesoporous activated carbons from waste tires by cyclic oxygen chemisorption-desorption. Ind. Eng. Chem. Res. 2012, 51, 2609–2614. [Google Scholar] [CrossRef][Green Version]
- Zhu, Y.W.; Gao, J.H.; Li, Y.; Sun, F. Preparation of activated carbons for SO2 adsorption by CO2 and steam activation. J. Taiwan Inst. Chem. Eng. 2012, 43, 112–119. [Google Scholar] [CrossRef]
- He, X.F.; Jin, L.J.; Wang, D.; Zhao, Y.P.; Zhu, S.W.; Hu, H.Q. Integrated process of coal pyrolysis with CO2 reforming of methane by dielectric barrier discharge plasma. Energy Fuels 2011, 25, 4036–4042. [Google Scholar] [CrossRef]
- Li, W.; Zhu, Y.M. Structural characteristics of coal vitrinite during pyrolysis. Energy Fuels 2014, 28, 3645–3654. [Google Scholar] [CrossRef]
- Li, T.; Zhang, L.; Li, D. Effects of gasification atmosphere and temperature on char structural evolution during the gasification of collie sub-bituminous coal. Fuel 2014, 117, 1190–1195. [Google Scholar] [CrossRef]
- Sasezky, A.; Muckenhuber, H.; Grothe, H. Raman microspectroscopy of soot and related carbonaceous materials: Spectral analysis and structural information. Carbon 2005, 43, 1731–1742. [Google Scholar] [CrossRef]
- Li, Y.; Yang, H.P.; Hu, J.H.; Wang, X.H.; Chen, H.P. Effect of catalysts on the reactivity and structure evolution of char in petroleum coke steam gasification. Fuel 2014, 117, 1174–1180. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Vad | FCad | Aad | Mad | Cdaf | Hdaf | Odaf * | Ndaf | Sdaf |
|---|---|---|---|---|---|---|---|---|
| 39.66 | 56.60 | 0.12 | 3.62 | 74.81 | 19.49 | 4.01 | 1.31 | 0.38 |
| Samples | La (Å) | Lc (Å) | d002 (Å) | N= Lc/d002 |
|---|---|---|---|---|
| ACs-0.1 | 24.57 | 13.15 | 3.61 | 3.70 |
| ACs-0.25 | 24.24 | 13.02 | 3.74 | 3.48 |
| ACs-0.5 | 23.87 | 12.75 | 3.95 | 3.23 |
| ACs-0.75 | 23.01 | 12.04 | 4.28 | 2.81 |
| ACs-1 | 25.15 | 13.44 | 3.57 | 3.76 |
| Samples | AD1/AG | AD3/AG | AD4/AG |
|---|---|---|---|
| ACs-0.1 | 3.216 | 1.832 | 0.508 |
| ACs-0.25 | 3.271 | 1.762 | 0.488 |
| ACs-0.5 | 3.394 | 1.662 | 0.471 |
| ACs-0.75 | 3.609 | 1.534 | 0.459 |
| ACs-1 | 1.891 | 1.022 | 0.103 |
| Samples | SBET (m2/g) a | Vt (m3/g) b | Vmic (m3/g) c | Non-Vmic (%) d |
|---|---|---|---|---|
| ACs-0.1 | 60.11 | 0.051 | 0.045 | 11.76 |
| ACs-0.25 | 317.65 | 0.178 | 0.131 | 26.40 |
| ACs-0.5 | 1048.65 | 0.471 | 0.318 | 32.48 |
| ACs-0.75 | 814.36 | 0.516 | 0.271 | 47.48 |
| ACs-1 | 442.14 | 0.588 | 0.198 | 66.32 |
| Samples | N-6 (%) | N-5 (%) | N-Q (%) | N-O (%) |
|---|---|---|---|---|
| ACs-0.1 | 33.7 | 30.3 | 31.9 | 4.1 |
| ACs-0.25 | 45.9 | 24.1 | 23.8 | 6.2 |
| ACs-0.5 | 60.9 | 15.1 | 14.7 | 9.3 |
| ACs-0.75 | 15.4 | 35.9 | 37.6 | 11.1 |
| ACs-1 | 7.6 | 18.9 | 61.2 | 12.3 |
| Samples | SBET (m2/g) a | Vt (m3/g) b | Vmic (m3/g) c | Non-Vmic (%) d |
|---|---|---|---|---|
| ACs-0.5-30 | 1178.63 | 0.455 | 0.338 | 25.71 |
| ACs-0.5-60 | 1897.25 | 0.421 | 0.367 | 12.826 |
| ACs-0.5-90 | 787.98 | 0.304 | 0.268 | 11.84 |
| ACs-0.5-120 | 514.87 | 0.147 | 0.133 | 9.52 |
| ACs-0.75-30 | 907.14 | 0.509 | 0.298 | 41.45 |
| ACs-0.75-60 | 1244.78 | 0.479 | 0.326 | 31.94 |
| ACs-0.75-90 | 1971.57 | 0.441 | 0.380 | 13.83 |
| ACs-0.75-120 | 547.48 | 0.154 | 0.136 | 11.69 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, D.; Li, J.; Dong, J.; Li, S.; Feng, W.; Jia, B. Effect of Ammonia Activation and Chemical Vapor Deposition on the Physicochemical Structure of Activated Carbons for CO2 Adsorption. Processes 2019, 7, 801. https://doi.org/10.3390/pr7110801
Liu D, Li J, Dong J, Li S, Feng W, Jia B. Effect of Ammonia Activation and Chemical Vapor Deposition on the Physicochemical Structure of Activated Carbons for CO2 Adsorption. Processes. 2019; 7(11):801. https://doi.org/10.3390/pr7110801
Chicago/Turabian StyleLiu, Dongdong, Jinming Li, Jiaqi Dong, Song Li, Weizhi Feng, and Boyin Jia. 2019. "Effect of Ammonia Activation and Chemical Vapor Deposition on the Physicochemical Structure of Activated Carbons for CO2 Adsorption" Processes 7, no. 11: 801. https://doi.org/10.3390/pr7110801
APA StyleLiu, D., Li, J., Dong, J., Li, S., Feng, W., & Jia, B. (2019). Effect of Ammonia Activation and Chemical Vapor Deposition on the Physicochemical Structure of Activated Carbons for CO2 Adsorption. Processes, 7(11), 801. https://doi.org/10.3390/pr7110801