Diffusion in Nanoporous Materials: Novel Insights by Combining MAS and PFG NMR


Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Experimental Procedure
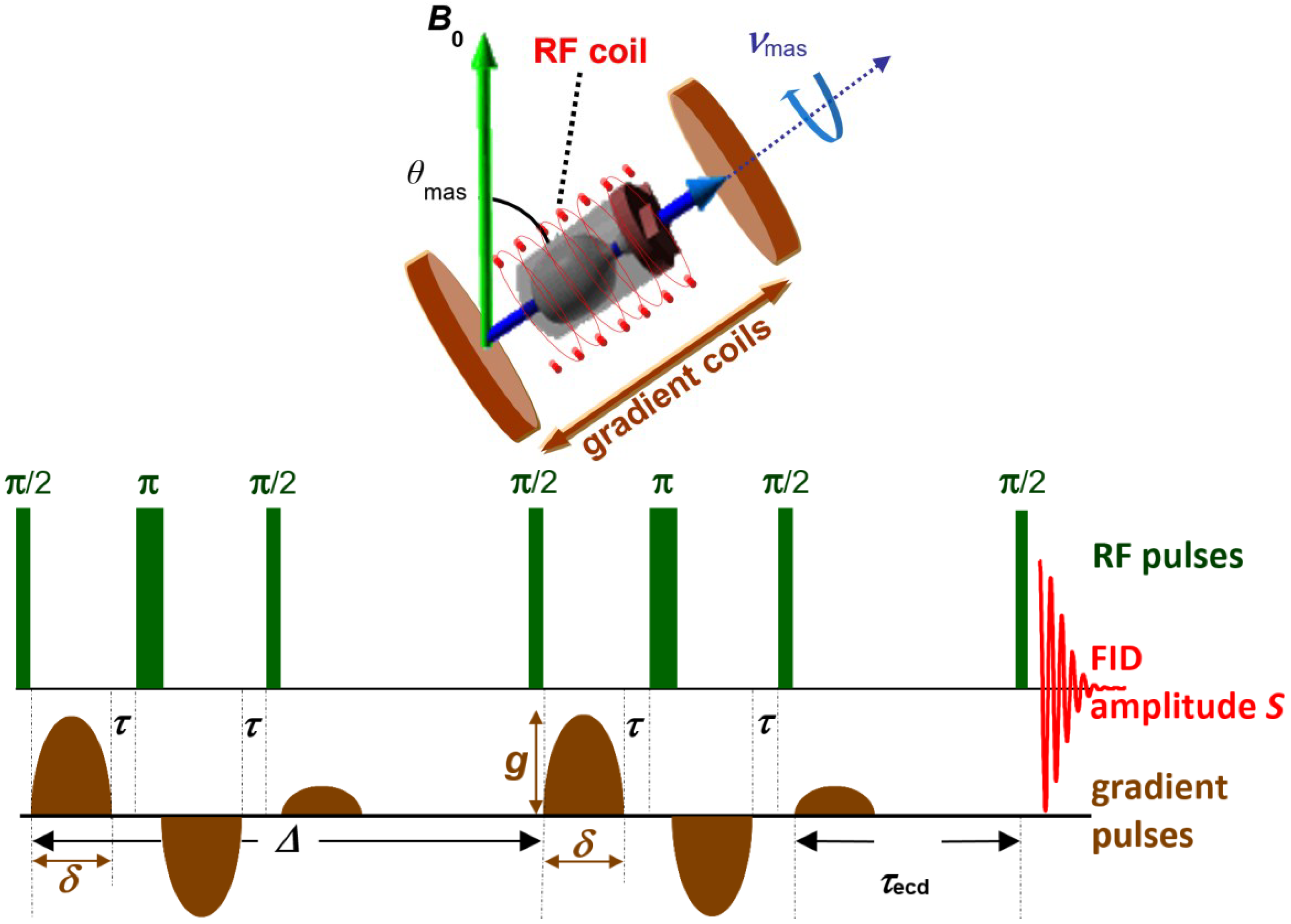
3. Diffusion Measurement by MAS PFG NMR
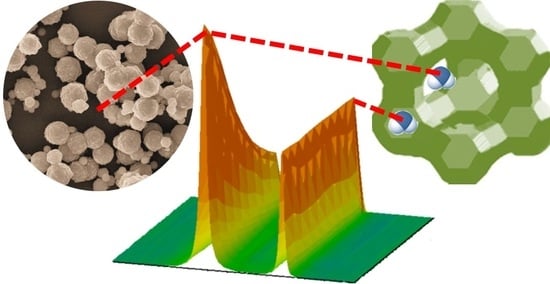
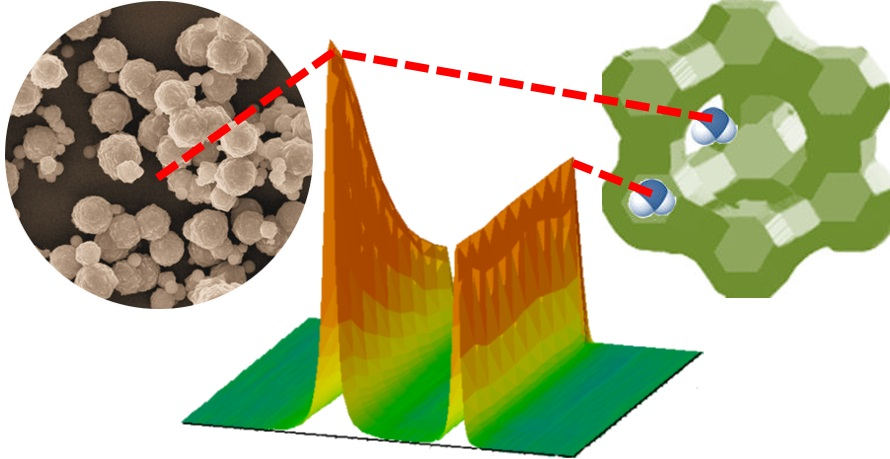
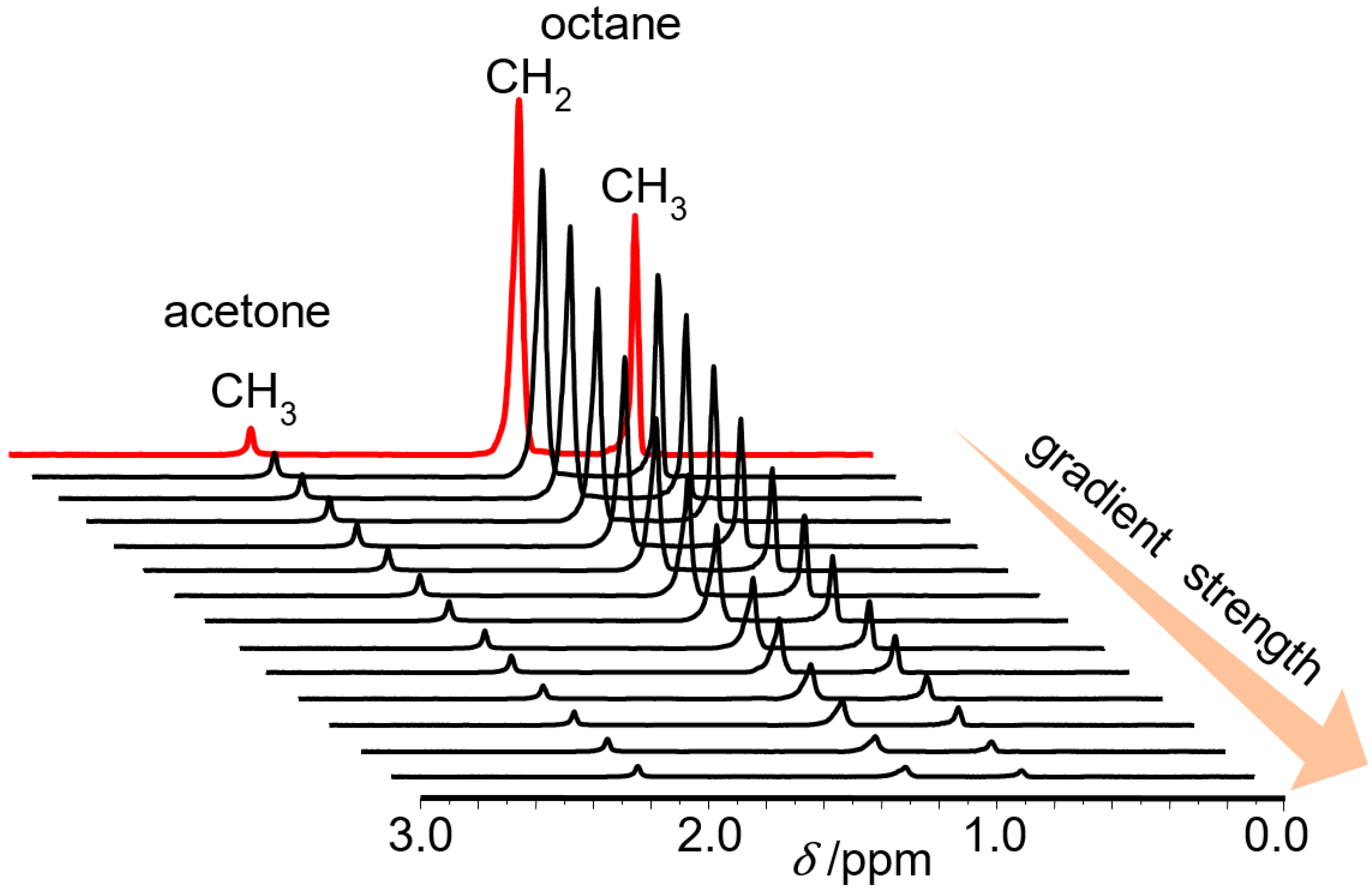
3.1. Mixture Diffusion in Microporous Materials
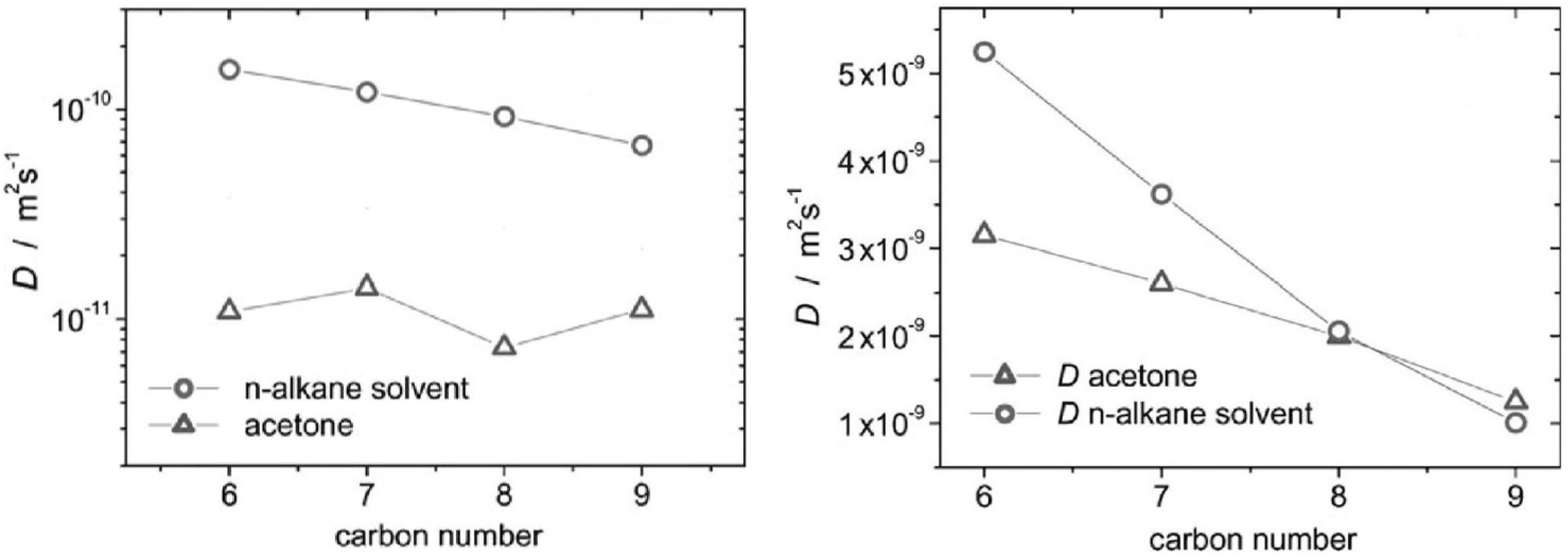
3.2. Complex Formation in Acetone–n-Alkane Mixtures Revealed via MAS PFG NMR
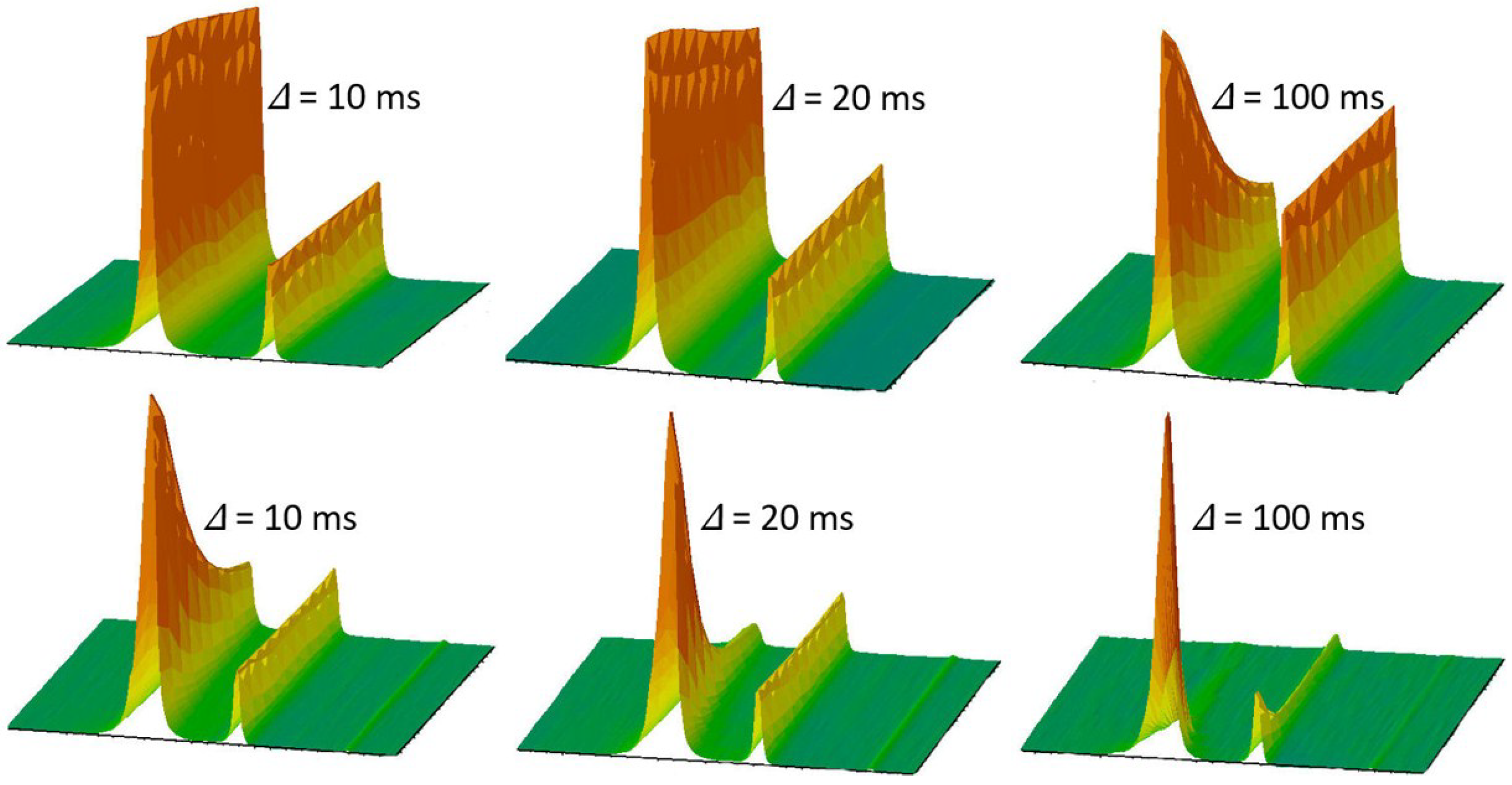
3.3. Diffusion Studies with Nematic Liquid Crystals in Confining Pore Spaces
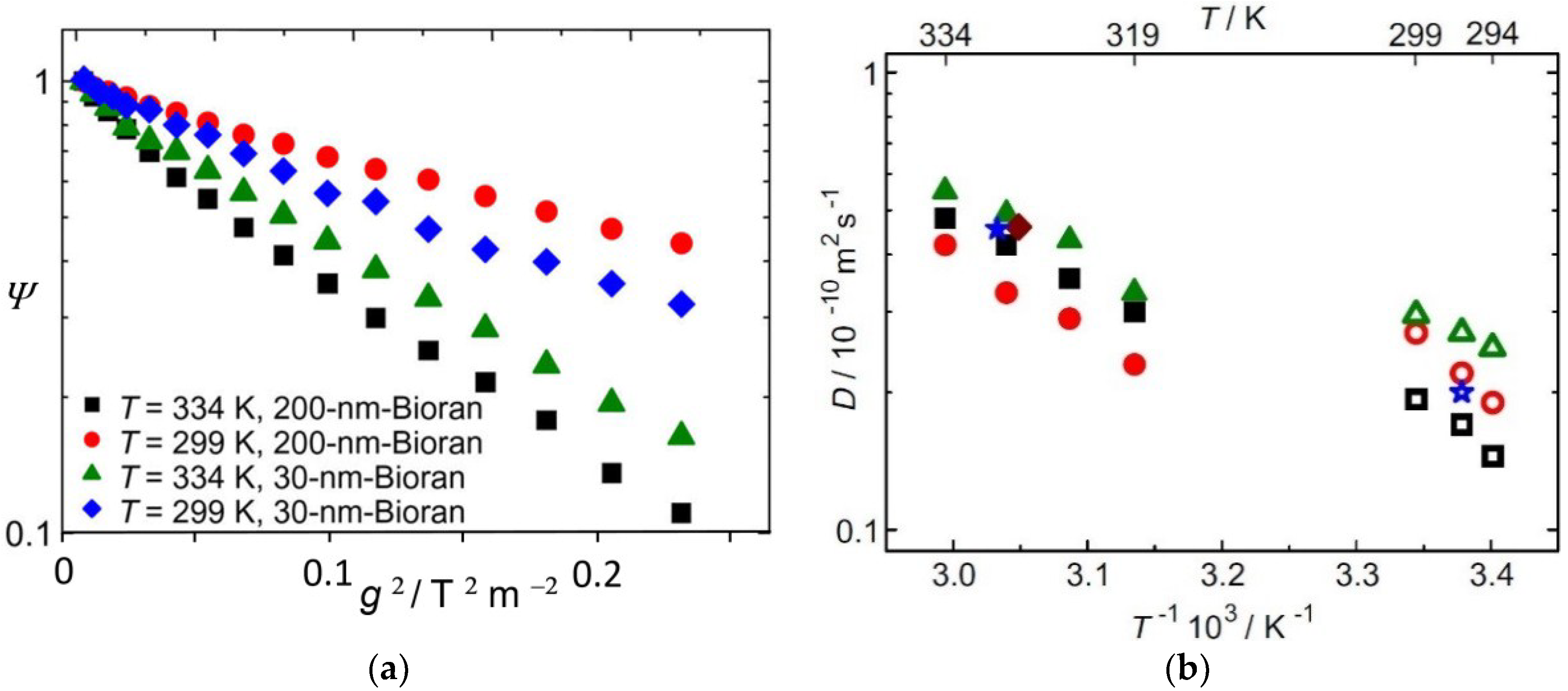
3.4. Water Diffusion in Lithium-Exchanged Low-Silica X-Type Zeolites
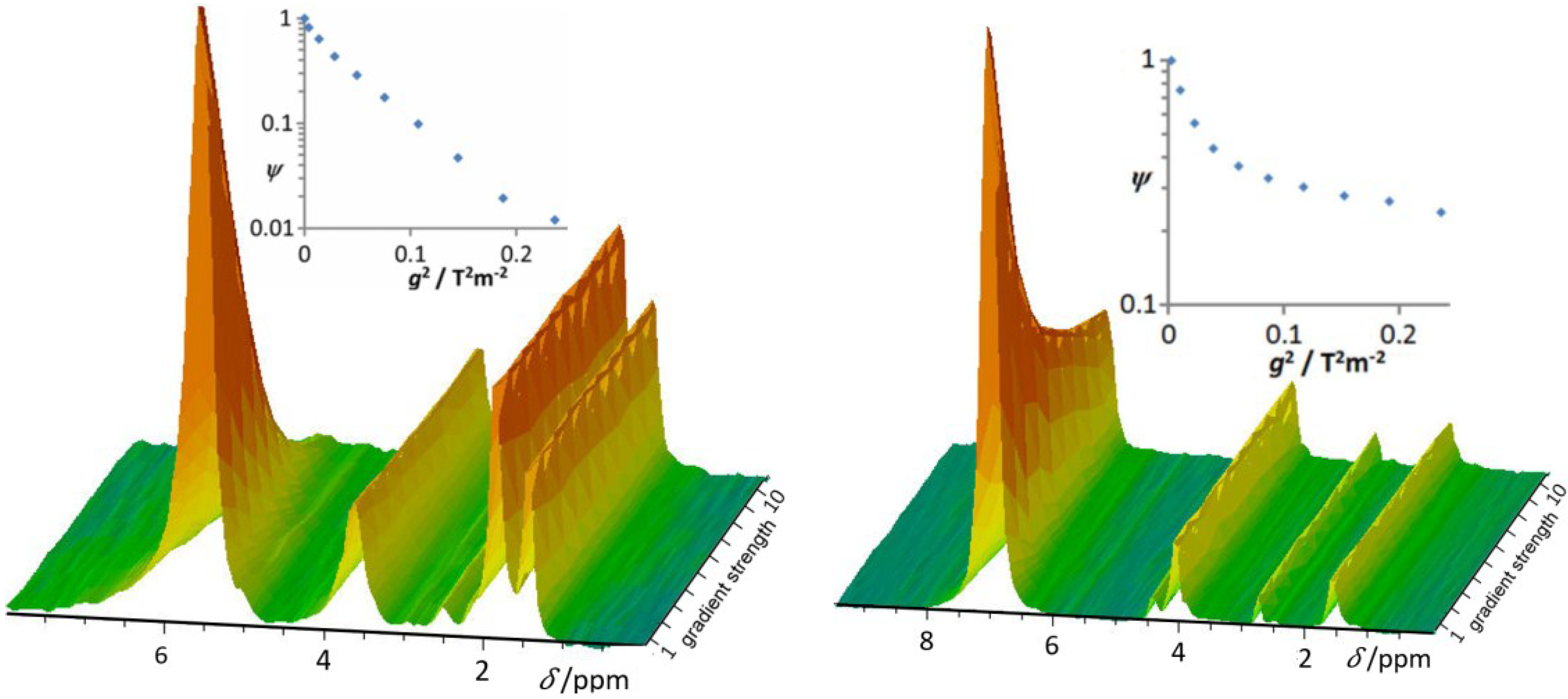
3.5. Proton Mobility in Functionalized Mesoporous Materials
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Bunde, A.; Caro, J.; Kärger, J.; Vogl, G. (Eds.) Diffusive Spreading in Nature, Technology and Society; Springer International Publishing: Cham, Switzerland, 2018. [Google Scholar]
- Ruthven, D.M.; Farooq, S.; Knaebel, K.S. Pressure Swing Adsorption; VCH: New York, NY, USA, 1994. [Google Scholar]
- Schüth, F.; Sing, K.S.W.; Weitkamp, J. (Eds.) Handbook of Porous Solids; Wiley-VCH: Weinheim, Germany, 2002. [Google Scholar]
- Ertl, G.; Knözinger, H.; Schüth, F.; Weitkamp, J. (Eds.) Handbook of Heterogeneous Catalysis, 2nd ed.; Wiley-VCH: Weinheim, Germany, 2008. [Google Scholar]
- Van de Voorde, M.H.; Sels, B. (Eds.) Nanotechnology in Catalysis: Applications in the Chemical Industry, Energy Development, and Environment Protection; Wiley-VCH: Weinheim, Germany, 2017. [Google Scholar]
- Chen, N.Y.; Degnan, T.F.; Smith, C.M. Molecular Transport and Reaction in Zeolites; VCH: New York, NY, USA, 1994. [Google Scholar]
- Kärger, J.; Freude, D. Mass transfer in micro- and mesoporous materials. Chem. Eng. Technol. 2002, 25, 769–778. [Google Scholar] [CrossRef]
- Kärger, J.; Ruthven, D.M.; Theodorou, D.N. Diffusion in Nanoporous Materials; Wiley-VCH: Weinheim, Germany, 2012. [Google Scholar]
- Kärger, J. Diffusionsuntersuchung von Wasser an 13X- sowie 4A- und 5A-Zeolithen mit Hilfe der Methode der gepulsten Feldgradienten. Z. Phys. Chem. 1971, 248, 27–41. [Google Scholar] [CrossRef]
- Kärger, J.; Vasenkov, S. Quantitation of diffusion in zeolite catalysts. Microporous Mesoporous Mater. 2005, 85, 195–206. [Google Scholar] [CrossRef]
- Valiullin, R. Diffusion NMR of Confined Systems; Royal Society of Chemistry: Cambridge, UK, 2016. [Google Scholar]
- Kärger, J.; Heink, W. The Propagator Representation of Molecular Transport in Microporous Crystallites. J. Magn. Reson. 1983, 51, 1–7. [Google Scholar] [CrossRef]
- Callaghan, P.T. Principles of NMR Microscopy; Clarendon Press: Oxford, UK, 1991. [Google Scholar]
- Price, W.S. NMR Studies of Translational Motion; Cambridge University Press: Cambridge, UK, 2009. [Google Scholar]
- Kärger, J. (Ed.) Leipzig, Einstein, Diffusion, 3rd ed.; Leipziger Universitätsverlag: Leipzig, Germany, 2014. [Google Scholar]
- Zhdanov, S.P.; Khvostchov, S.S.; Feoktistova, N.N. Synthetic Zeolites; Gordon and Breach: New York, NY, USA, 1990. [Google Scholar]
- Kärger, J. Measurement of diffusion in zeolites—A never ending challenge? Adsorption 2003, 9, 29–35. [Google Scholar] [CrossRef]
- Gaede, H.C.; Gawrisch, K. Multi-dimensional pulsed field gradient magic angle spinning NMR experiments on membranes. Magn. Reson. Chem. 2004, 42, 115–122. [Google Scholar] [CrossRef] [PubMed]
- Pampel, A.; Zick, K.; Glauner, H.; Engelke, F. Studying lateral diffusion in lipid bilayers by combining a magic angle spinning NMR probe with a microimaging gradient system. J. Am. Chem. Soc. 2004, 126, 9534–9535. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, M.; Kärger, J.; Freude, D.; Pampel, A.; van Baten, J.M.; Krishna, R. Mixture diffusion in zeolites studied by MAS PFG NMR and molecular simulation. Microporous Mesoporous Mater. 2007, 105, 124–131. [Google Scholar] [CrossRef]
- Romanova, E.E.; Grinberg, F.; Pampel, A.; Karger, J.; Freude, D. Diffusion studies in confined nematic liquid crystals by MAS PFG NMR. J. Magn. Reson. 2009, 196, 110–114. [Google Scholar] [CrossRef] [PubMed]
- Gratz, M.; Hertel, S.; Wehring, M.; Stallmach, F.; Galvosas, P. Mixture diffusion of adsorbed organic compounds in metal-organic frameworks as studied by magic-angle spinning pulsed-field gradient nuclear magnetic resonance. New J. Phys. 2011, 13, 045016. [Google Scholar] [CrossRef]
- Kimmich, R. NMR Tomography, Diffusometry, Relaxometry; Springer: Berlin, Germany, 1997. [Google Scholar]
- Valiullin, R.; Kärger, J. Confined Fluids: NMR Perspectives on Confinements and on Fluid Dynamics. In Diffusion NMR of Confined Systems; Valiullin, R., Ed.; Royal Society of Chemistry: Cambridge, UK, 2016; pp. 390–434. [Google Scholar]
- Harris, R.K.; Becker, E.D.; De Menezes, S.M.C.; Goodfellow, R.; Granger, P. NMR Nomenclature. Nuclear Spin Properties and Conventions for Chemical Shifts–(IUPAC Recommendations 2001). Pure Appl. Chem. 2001, 73, 1795–1818. [Google Scholar] [CrossRef]
- Callaghan, P.T.; Coy, A.; MacGowan, D.; Packer, K.J.; Zelaya, F.O. Diffraction-like effects in NMR diffusion studies of fluids in porous solids. Nature 1991, 351, 467–469. [Google Scholar] [CrossRef]
- Cussler, E.L. Diffusion: Mass Transfer in Fluid Systems, 3rd ed.; Cambridge University Press: Cambridge, UK, 2009. [Google Scholar]
- Rousselot-Pailley, P.; Maux, D.; Wieruszeski, J.M.; Aubagnac, J.L.; Martinez, J.; Lippens, G. Impurity detection in solid-phase organic chemistry: Scope and limits of HR MAS NMR. Tetrahedron 2000, 56, 5163–5167. [Google Scholar] [CrossRef]
- Schröder, H. High resolution magic angle spinning NMR for analyzing small molecules attached to solid support. J. Comb. Chem. 2003, 6, 741–753. [Google Scholar] [CrossRef]
- Pampel, A.; Fernandez, M.; Freude, D.; Kärger, J. New options for measuring molecular diffusion in zeolites by MAS PFG NMR. Chem. Phys. Lett. 2005, 407, 53–57. [Google Scholar] [CrossRef]
- Schlayer, S.; Stallmach, F.; Horch, C.; Splith, T.; Pusch, A.-K.; Pielenz, F.; Peksa, M. Konstruktion und Test eines Gradientensystems für NMR-Diffusionsuntersuchungen in Grenzflächensystemen. Chem. Ing. Tech. 2013, 85, 1755–1760. [Google Scholar] [CrossRef]
- Romanova, E.E.; Scheffler, F.; Freude, D. Crystallization of zeolite MFI under supergravity, studied in situ by 11B MAS NMR spectroscopy. Microporous Mesoporous Mater. 2009, 126, 268–271. [Google Scholar] [CrossRef]
- Dvoyashkina, N.; Freude, D.; Stepanov, A.G.; Böhlmann, W.; Krishna, R.; Kärger, J.; Haase, J. Alkane/alkene mixture diffusion in silicalite-1 studied by MAS PFG NMR. Microporous Mesoporous Mater. 2018, 257, 128–134. [Google Scholar] [CrossRef]
- Mildner, T.; Ernst, H.; Freude, D. 207Pb NMR detection of spinning-induced temperature gradients in MAS rotors. Solid State Nucl. Magn. Reson. 1995, 5, 269–271. [Google Scholar] [CrossRef]
- Ferguson, D.B.; Haw, J.F. Transient methods for in-situ NMR of reactions on solid catalysts using temperature jumps. Anal. Chem. 1995, 67, 3342–3348. [Google Scholar] [CrossRef]
- Neue, G.; Dybowski, C. Determining temperature in a magic-angle spinning probe using the temperature dependence of the isotropic chemical shift of lead nitrate. Solid State Nucl. Magn. Reson. 1997, 7, 333–336. [Google Scholar] [CrossRef]
- Chmelik, C.; Freude, D.; Bux, H.; Haase, J. Ethane/ethene mixture diffusion in the MOF sieve ZIF-8 studied by MAS PFG NMR diffusometry. Microporous Mesoporous Mater. 2012, 147, 135–141. [Google Scholar] [CrossRef]
- Chmelik, C. Characteristic features of molecular transport in MOF ZIF-8 as revealed by IR microimaging. Microporous Mesoporous Mater. 2015, 216, 138–145. [Google Scholar] [CrossRef]
- Park, K.S.; Ni, Z.; Cote, A.P.; Choi, J.Y.; Huang, R.D.; Uribe-Romo, F.J.; Chae, H.K.; O’Keeffe, M.; Yaghi, O.M. Exceptional chemical and thermal stability of zeolitic imidazolate frameworks. Proc. Natl. Acad. Sci. USA 2006, 103, 10186–10191. [Google Scholar] [CrossRef] [PubMed]
- Bux, H.; Liang, F.; Li, Y.; Cravillon, J.; Wiebcke, M.; Caro, J. Zeolitic imidazolate framework membrane with molecular sieving properties by microwave-assisted solvothermal synthesis. J. Am. Chem. Soc. 2009, 131, 16000–16001. [Google Scholar] [CrossRef] [PubMed]
- Lu, G.; Hupp, J.T. Metal-organic frameworks as sensors: A ZIF-8 based Fabry-Pérot device as a selective sensor for chemical vapors and gases. J. Am. Chem. Soc. 2010, 132, 7832–7833. [Google Scholar] [CrossRef] [PubMed]
- Kärger, J.; Binder, T.; Chmelik, C.; Hibbe, F.; Krautscheid, H.; Krishna, R.; Weitkamp, J. Microimaging of transient guest profiles to monitor mass transfer in nanoporous materials. Nat. Mater. 2014, 13, 333–343. [Google Scholar] [CrossRef] [PubMed]
- Gladstone, S.; Laidler, K.J.; Eyring, H. The Theory of Rate Processes; McGraw-Hill: New York, NY, USA, 1941. [Google Scholar]
- Ruthven, D.M.; Derrah, R.I. Transition state theory of zeolitic diffusion. J. Chem. Soc. Faraday Trans. I 1972, 68, 2332–2343. [Google Scholar] [CrossRef]
- Kärger, J.; Pfeifer, H.; Haberlandt, R. Application of absolute rate theory to intracrystalline diffusion in zeolites. J. Chem. Soc. Faraday Trans. I 1980, 76, 1569–1575. [Google Scholar] [CrossRef]
- Chmelik, C.; Kärger, J. The predictive power of classical transition state theory revealed in diffusion studies with MOF ZIF-8. Microporous Mesoporous Mater. 2016, 225, 128–132. [Google Scholar] [CrossRef]
- Caro, J.; Bülow, M.; Schirmer, W.; Kärger, J.; Heink, W.; Pfeifer, H.; Zhdanov, S.P. Microdynamics of methane, ethane and propane in ZSM-5 type zeolites. J. Chem. Soc. Faraday Trans. I 1985, 81, 2541–2550. [Google Scholar] [CrossRef]
- Heink, W.; Kärger, J.; Pfeifer, H.; Salverda, P.; Datema, K.P.; Nowak, A.K. High-temperature pulsed field gradient nuclear magnetic resonance self-diffusion measurements of n-Alkanes in MFI-type zeolites. J. Chem. Soc. Faraday Trans. 1992, 88, 3505–3509. [Google Scholar] [CrossRef]
- Jobic, H.; Schmidt, W.; Krause, C.; Kärger, J. PFG NMR and QENS diffusion studies of n-alkane homologues in MFI-type zeolites. Microporous Mesoporous Mater. 2006, 90, 299–306. [Google Scholar] [CrossRef]
- Tsai, H.L.; Sato, S.; Takahashi, R.; Sodesawa, T.; Takenaka, S. Liquid-phase hydrogenation of ketones in the mesopores of nickel catalysts. Phys. Chem. Chem. Phys. 2002, 4, 3537–3542. [Google Scholar] [CrossRef]
- Nguyen, N.Q.; McGann, M.R.; Lacks, D.J. Elastic stability limits of polyethylene and n-alkane crystals from molecular simulation. J. Phys. Chem. B 1999, 103, 10679–10683. [Google Scholar] [CrossRef]
- Kishore, K.; Bharat, S.; Kannan, S. Correlation of Kauzman temperature with odd-even effect in n-alkanes. J. Chem. Phys. 1996, 105, 11364–11365. [Google Scholar] [CrossRef]
- Takahashi, R.; Sato, S.; Sodesawa, T.; Ikeda, T. Diffusion coefficient of ketones in liquid media within mesopores. Phys. Chem. Chem. Phys. 2003, 5, 2476–2480. [Google Scholar] [CrossRef]
- Fernandez, M.; Pampel, A.; Takahashi, R.; Sato, S.; Freude, D.; Kärger, J. Revealing complex-formation in acetone-n-alkane mixtures by MAS PFG NMR diffusion measurement in nanoporous hosts. Phys. Chem. Chem. Phys. 2008, 10, 4165–4171. [Google Scholar] [CrossRef] [PubMed]
- Rincon Bonilla, M.; Bhatia, S.K. Diffusion in Pore Networks: Effective self-diffusivity and the concept of tortuosity. J. Phys. Chem. C 2013, 117, 3343–3357. [Google Scholar] [CrossRef]
- Rincon Bonilla, M.; Titze, T.; Schmidt, F.; Mehlhorn, D.; Chmelik, C.; Valiullin, R.; Bhatia, S.K.; Kaskel, S.; Ryoo, R.; Kärger, J. Diffusion study by IR micro-imaging of molecular uptake and release on mesoporous zeolites of structure type CHA and LTA. Materials 2013, 6, 2662–2688. [Google Scholar] [CrossRef] [PubMed]
- Kärger, J.; Valiullin, R. Mass transfer in mesoporous materials: The benefit of microscopic diffusion measurement. Chem. Soc. Rev. 2013, 42, 4172–4197. [Google Scholar] [CrossRef] [PubMed]
- Mehlhorn, D.; Kondrashova, D.; Küster, C.; Enke, D.; Emmerich, T.; Bunde, A.; Valiullin, R.; Kärger, J. Diffusion in complementary pore spaces. Adsorption 2016, 22, 879–890. [Google Scholar] [CrossRef]
- Stoeckel, D.; Kübel, C.; Hormann, K.; Höltzel, A.; Smarsly, B.M.; Tallarek, U. Morphological analysis of disordered macroporous-mesoporous solids based on physical reconstruction by nanoscale tomography. Langmuir 2014, 30, 9022–9027. [Google Scholar] [CrossRef] [PubMed]
- Kondrashova, D.; Lauerer, A.; Mehlhorn, D.; Jobic, H.; Feldhoff, A.; Thommes, M.; Chakraborty, D.; Gommes, C.; Zecevic, J.; de Jongh, P.; et al. Scale-dependent diffusion anisotropy in nanoporous silicon. Sci. Rep. 2017, 7, 40207. [Google Scholar] [CrossRef] [PubMed]
- Reich, S.-J.; Svidrytski, A.; Höltzel, A.; Florek, J.; Kleitz, F.; Wang, W.; Kübel, C.; Hlushkou, D.; Tallarek, U. Hindered diffusion in ordered mesoporous silicas: Insights from pore-scale simulations in physical reconstructions of SBA-15 and KIT-6 silica. J. Phys. Chem. C 2018, 122, 12350–12361. [Google Scholar] [CrossRef]
- Crawford, G.P.; Vilfan, M.; Doane, J.W.; Vilfan, I. Escaped-radial nematic configuration in submicrometer-size cylindrical cavities: Deuterium nuclear-magnetic-resonance study. Phys. Rev. A 1991, 43, 835–842. [Google Scholar] [CrossRef] [PubMed]
- Cramer, C.; Cramer, T.; Arndt, M.; Kremer, F.; Naji, L.; Stannarius, R. NMR and dielectric studies of nano-confined nematic liquid crystals. Mol. Cryst. Liq. Cryst. Sci. Technol. Sect. A Mol. Cryst. Liq. Cryst. 2006, 303, 209–217. [Google Scholar] [CrossRef]
- Gueudré, L.; Milina, M.; Mitchell, S.; Pérez-Ramírez, J. Superior mass transfer properties of technical zeolite bodies with hierarchical porosity. Adv. Funct. Mater. 2014, 24, 209–219. [Google Scholar] [CrossRef]
- Mitchell, S.; Pinar, A.B.; Kenvin, J.; Crivelli, P.; Kärger, J.; Pérez-Ramírez, J. Structural analysis of hierarchically organized zeolites. Nat. Commun. 2015, 6, 8633. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, M.; Schwieger, W. Hierarchically-structured porous materials: From basic understanding to applications. Chem. Soc. Rev. 2016, 45, 3311–3312. [Google Scholar] [CrossRef] [PubMed]
- Coasne, B. Multiscale adsorption and transport in hierarchical porous materials. New J. Chem. 2016, 40, 4078–4094. [Google Scholar] [CrossRef]
- Galarneau, A.; Guenneau, F.; Gedeon, A.; Mereib, D.; Rodriguez, J.; Fajula, F.; Coasne, B. Probing interconnectivity in hierarchical microporous/mesoporous materials using adsorption and nuclear magnetic resonance diffusion. J. Phys. Chem. C 2016, 120, 1562–1569. [Google Scholar] [CrossRef]
- Trogadas, P.; Nigra, M.M.; Coppens, M.-O. Nature-inspired optimization of hierarchical porous media for catalytic and separation processes. New J. Chem. 2016, 40, 4016–4026. [Google Scholar] [CrossRef]
- Coppens, M.O.; Ye, G. Nature-inspired optimization of transport in porous media. In Diffusive Spreading in Nature, Technology and Society; Bunde, A., Caro, J., Kärger, J., Vogl, G., Eds.; Springer International Publishing: Cham, Switzerland, 2018; pp. 203–232. [Google Scholar]
- Kärger, J. NMR Self-diffusion studies in heterogeneous systems. Adv. Colloid Interface Sci. 1985, 23, 129–148. [Google Scholar] [CrossRef]
- Reginald Waldeck, A.; Hossein Nouri-Sorkhabi, M.; Sullivan, D.R.; Kuchel, P.W. Effects of cholesterol on transmembrane water diffusion in human erythrocytes measured using pulsed field gradient NMR. Biophys. Chem. 1995, 55, 197–208. [Google Scholar] [CrossRef]
- Meier, C.; Dreher, W.; Leibfritz, D. Diffusion in compartmental systems. II. Diffusion weighted measurements of rat brain tissue in vivo and postmortem at very large b-values. Magn. Reson. Med. 2003, 50, 510–514. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, M.; Alerstam, E.; Wirestam, R.; Stahlberg, F.; Brockstedt, S.; Lätt, J. Evaluating the accuracy and precision of a two-compartment Kärger model using Monte Carlo simulations. J. Magn. Res. 2010, 206, 59–67. [Google Scholar] [CrossRef] [PubMed]
- Himmelein, S.; Sporenberg, N.; Schönhoff, M.; Ravoo, B.J. Size-selective permeation of water-soluble polymers through the bilayer membrane of cyclodextrin vesicles investigated by PFG-NMR. Langmuir 2014, 30, 3988–3995. [Google Scholar] [CrossRef] [PubMed]
- Melchior, J.-P.; Majer, G.; Kreuer, K.-D. Why do proton conducting polybenzimidazole phosphoric acid membranes perform well in high-temperature PEM fuel cells? Phys. Chem. Chem. Phys. 2016, 19, 601–612. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, A.S.D.; Barreiros, S.; Cabrita, E.J. Probing sol-gel matrices microenvironments by PGSE HR-MAS NMR. Magn. Reson. Chem. 2017, 55, 452–463. [Google Scholar] [CrossRef] [PubMed]
- Cabrita, E.J.; Berger, S.; Brauer, P.; Kärger, J. High-resolution DOSY NMR with spins in different chemical surroundings: Influence of particle exchange. J. Magn. Reson. 2002, 157, 124–131. [Google Scholar] [CrossRef] [PubMed]
- Kühl, G.H. Crystallization of low-silica faujasite. Zeolites 1987, 7, 451–457. [Google Scholar] [CrossRef]
- Schneider, D.; Toufar, H.; Samoson, A.; Freude, D. 17O DOR and other solid-state NMR studies concerning the basic properties of zeolites LSX. Solid State Nucl. Magn. Reson. 2009, 35, 87–92. [Google Scholar] [CrossRef] [PubMed]
- Freude, D.; Beckert, S.; Stallmach, F.; Kurzhals, R.; Täschner, D.; Toufar, H.; Kärger, J.; Haase, J. Ion and water mobility in hydrated Li-LSX zeolite studied by 1H, 6Li and 7Li NMR spectroscopy and diffusometry. Microporous Mesoporous Mater. 2013, 172, 174–181. [Google Scholar] [CrossRef]
- Beckert, S.; Stallmach, F.; Toufar, H.; Freude, D.; Kärger, J.; Haase, J. Tracing water and cation diffusion in hydrated zeolites of type Li-LSX by pulsed field gradient NMR. J. Phys. Chem. C 2013, 117, 24866–24872. [Google Scholar] [CrossRef]
- Lauerer, A.; Kurzhals, R.; Toufar, H.; Freude, D.; Kärger, J. Tracing compartment exchange by NMR diffusometry: Water in lithium-exchanged low-silica X zeolites. J. Magn. Reson. 2018, 289, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Laberty-Robert, C.; Vallé, K.; Pereira, F.; Sanchez, C. Design and properties of functional hybrid organic-inorganic membranes for fuel cells. Chem. Soc. Rev. 2011, 40, 961–1005. [Google Scholar] [CrossRef] [PubMed]
- Hickner, M.A.; Ghassemi, H.; Kim, Y.S.; Einsla, B.R.; McGrath, J.E. Alternative polymer systems for proton exchange membranes (PEMs). Chem. Rev. 2004, 104, 4587–4612. [Google Scholar] [CrossRef] [PubMed]
- Kreuer, K.-D.; Paddison, S.J.; Spohr, E.; Schuster, M. Transport in proton conductors for fuel-cell applications: simulations, elementary reactions, and phenomenology. Chem. Rev. 2004, 104, 4637–4678. [Google Scholar] [CrossRef] [PubMed]
- Sharifi, M.; Wark, M.; Freude, D.; Haase, J. Highly proton conducting sulfonic acid functionalized mesoporous materials studied by impedance spectroscopy, MAS NMR spectroscopy and MAS PFG NMR diffusometry. Microporous Mesoporous Mater. 2012, 156, 80–89. [Google Scholar] [CrossRef]
- Dvoyashkina, N.; Seidler, C.F.; Wark, M.; Freude, D.; Haase, J. Proton mobility in sulfonic acid functionalized mesoporous materials studied by MAS PFG NMR diffusometry and impedance spectroscopy. Microporous Mesoporous Mater. 2018, 255, 140–147. [Google Scholar] [CrossRef]
- D’Orazio, F.; Bhattacharja, S.; Halperin, W.P.; Gerhardt, R. Enhanced self-diffusion of water in restricted geometry. Phys. Rev. Lett. 1989, 63, 43–46. [Google Scholar] [CrossRef] [PubMed]
- Kärger, J.; Pfeifer, H.; Riedel, E.; Winkler, H. Self-diffusion measurements of water adsorbed in NaY zeolites by means of NMR pulsed field gradient techniques. J. Colloid Interface Sci. 1973, 44, 187–188. [Google Scholar] [CrossRef]
- Inayat, A.; Knoke, I.; Spieker, E.; Schwieger, W. Assemblies of mesoporous FAU-type zeolite nanosheets. Angew. Chem. Int. Ed. 2012, 51, 1962–1965. [Google Scholar] [CrossRef] [PubMed]
- Na, K.; Choi, M.; Ryoo, R. Recent advances in the synthesis of hierarchically nanoporous zeolites. Microporous Mesoporous Mater. 2013, 166, 3–19. [Google Scholar] [CrossRef]










© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kärger, J.; Freude, D.; Haase, J. Diffusion in Nanoporous Materials: Novel Insights by Combining MAS and PFG NMR. Processes 2018, 6, 147. https://doi.org/10.3390/pr6090147
Kärger J, Freude D, Haase J. Diffusion in Nanoporous Materials: Novel Insights by Combining MAS and PFG NMR. Processes. 2018; 6(9):147. https://doi.org/10.3390/pr6090147
Chicago/Turabian StyleKärger, Jörg, Dieter Freude, and Jürgen Haase. 2018. "Diffusion in Nanoporous Materials: Novel Insights by Combining MAS and PFG NMR" Processes 6, no. 9: 147. https://doi.org/10.3390/pr6090147
APA StyleKärger, J., Freude, D., & Haase, J. (2018). Diffusion in Nanoporous Materials: Novel Insights by Combining MAS and PFG NMR. Processes, 6(9), 147. https://doi.org/10.3390/pr6090147