3.1. Non-Ionized System
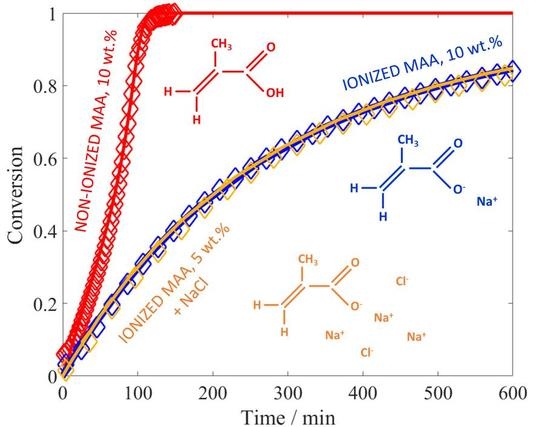
Reactions are carried out at 50 °C with 1, 5, and 10 wt % of initial monomer content (
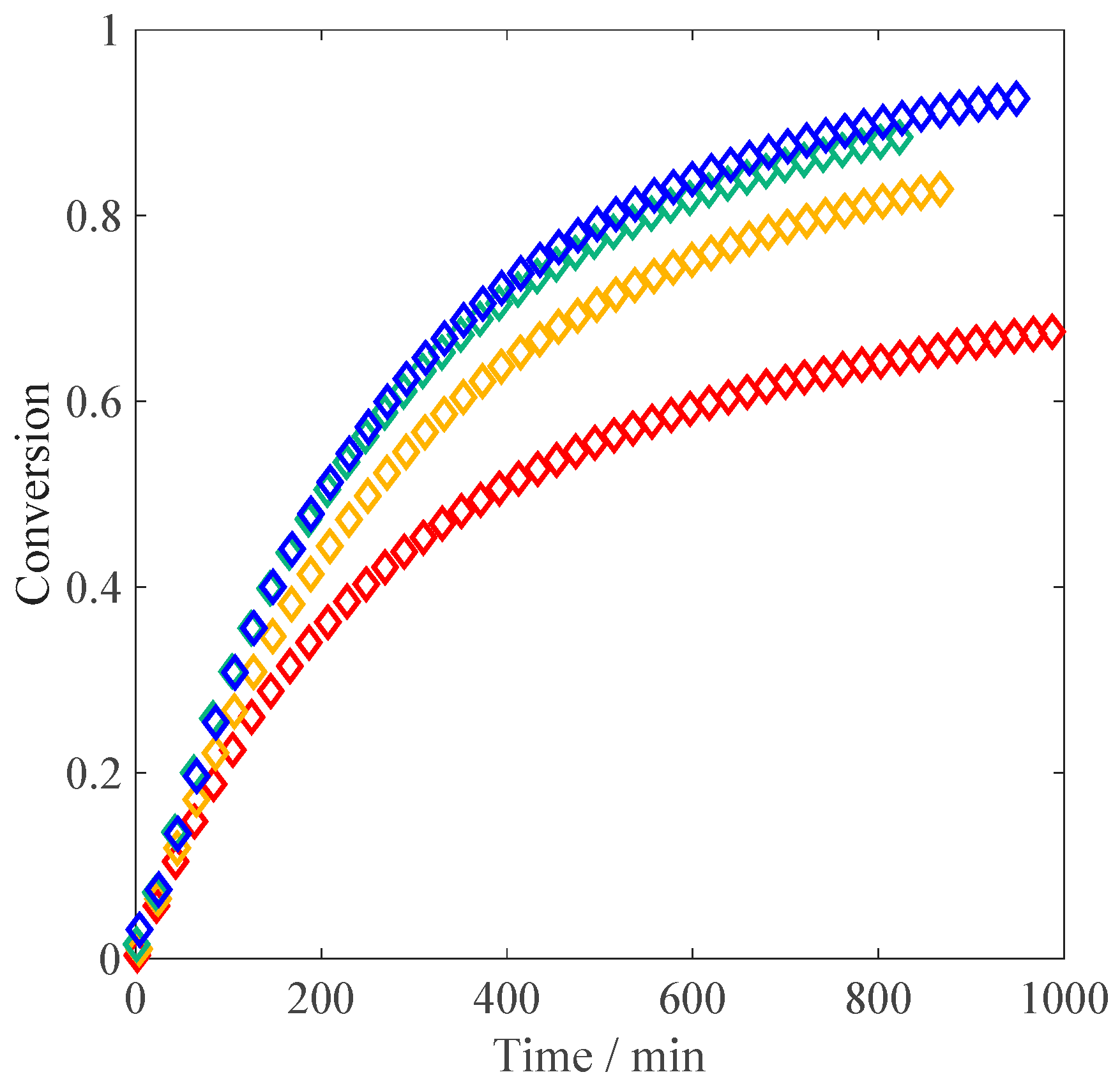
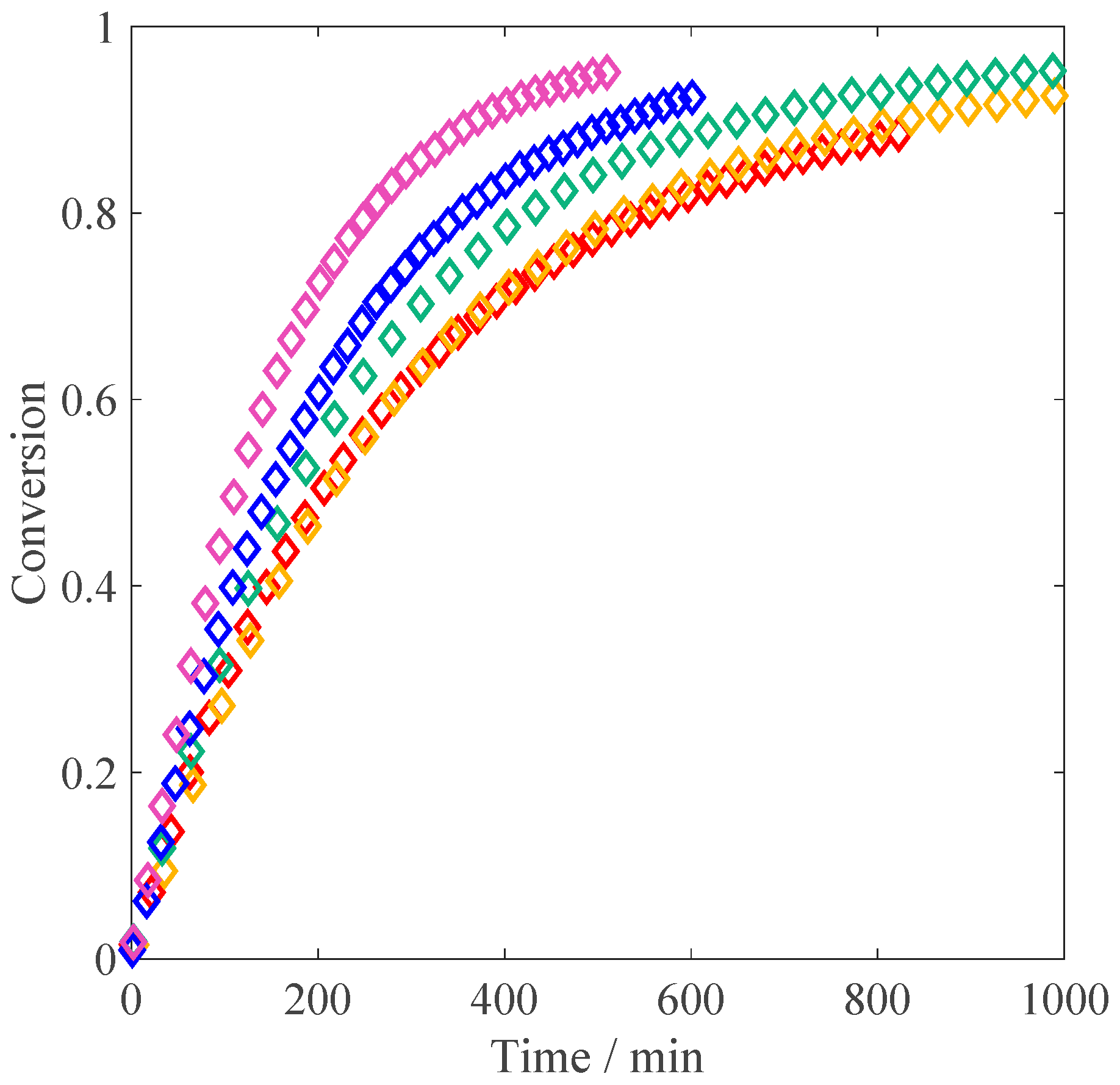
, weight fraction) and a constant amount of V-50 (0.02 wt %). With “non-ionized MAA” we mean the situation of natural dissociation of the monomer in the absence of any added base. The conversion versus time plots are shown in
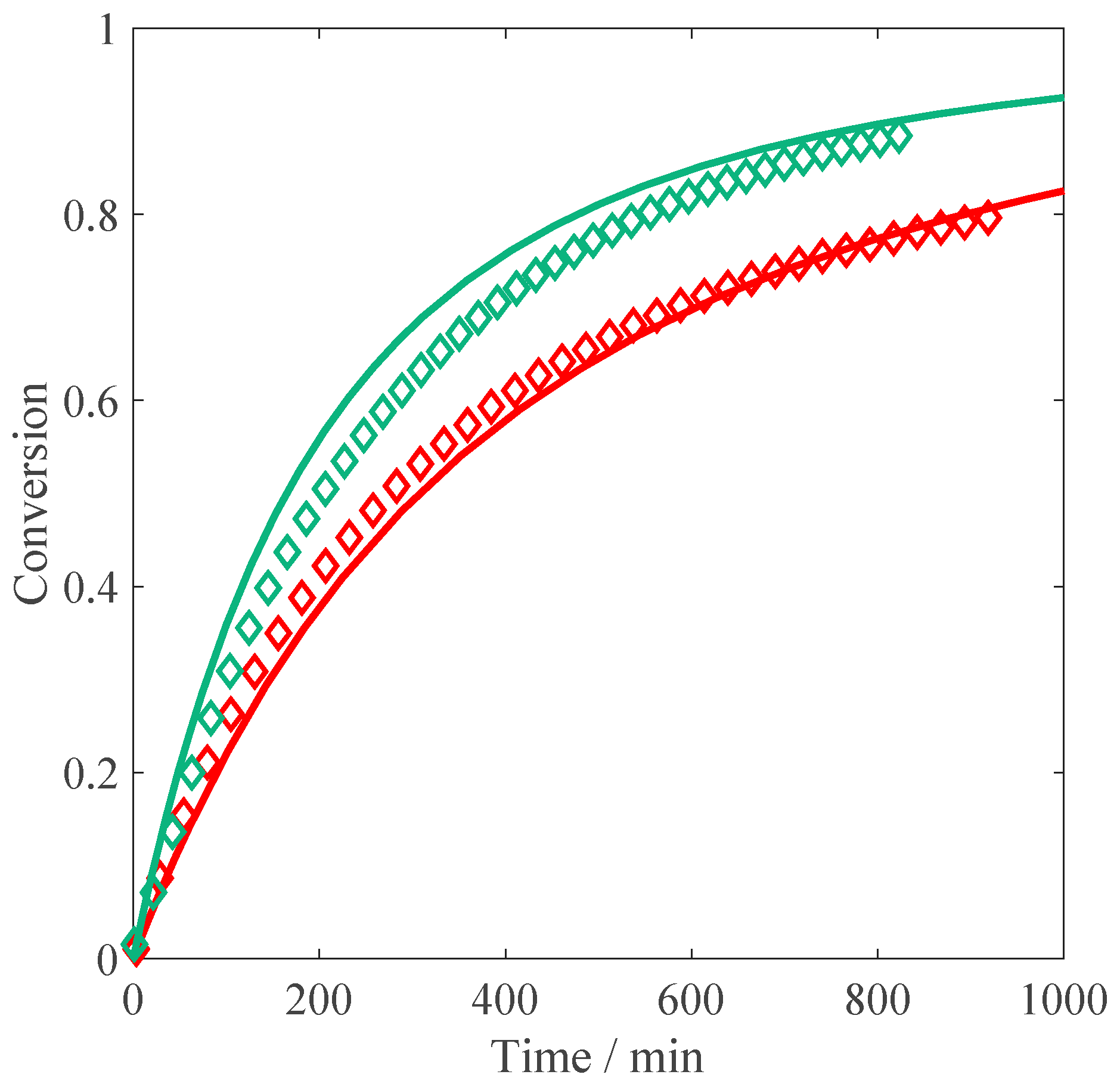
Figure 1. At 10 wt % of initial monomer, the slope of the conversion-time curve has reached its maximum value (i.e., the inhibitor is completely consumed) and remains constant until ca. 40% of conversion. After about 60 min, an increase of the slope is noticeable, which is a sign of diffusion limitations to termination (gel effect). This is fully consistent with the experimental results of Buback et al. [
23], who ran their reactions at larger initiator concentrations (5 mmol/L, equivalent to ca. 0.12 wt %). At lower monomer concentrations, the initial polymerization rate is faster than at 10 wt %, in agreement with the increase in the propagation rate coefficient as a function of
, revealed by PLP studies [
12]. It is also worth noting that at 5 wt % and 1 wt % of MAA, the autoacceleration of the reaction due to gel effect is less relevant: this behavior is consistent with the lower viscosity of the reacted solution observed in the NMR tube in the low monomer concentration cases compared to the situation at 10 wt % of initial MAA. Furthermore, it appears that at high conversion, the polymerization rate is slowing down: such an effect is particularly pronounced at 1 wt % of MAA, where full conversion is reached only hours later. The reproducibility of the obtained conversion versus time curves has been tested for the reaction at 1 wt % and 10 wt % of initial monomer. As shown in
Figure S3 in the
Supporting Information, at 1 wt %, the repeated experiment exhibits a slightly smoother behavior at conversions above 60%, i.e., showing a slightly less pronounced autoacceleration behavior. For the repetition of the reaction with 10 wt % MAA (
Figure S4), the difference in the slope above 40% conversion is a bit larger. Since this discrepancy (of similar size) at the same reaction conditions has already been reported in the literature [
23], we consider this accuracy to be sufficient.
A kinetic model suitable for evaluating the reaction rate and number average molecular weight of the active chains has been developed. The most conventional free-radical kinetic scheme is considered, with all the reactions listed in
Table 1. The initiator
decomposes to form two radicals
with rate coefficient
. Assuming that all terminations are accounted for by an initiator efficiency factor
, and applying the quasi-steady-state assumption for the radical fragments
, the rate of propagation of
is equal to the rate of initiation (i.e., the first term in the monomer balance equation in
Table 2). The radicals of any chain length
undergo propagation with a monomer unit
(rate coefficient
), and termination by combination (
) and by disproportionation (
). Chain transfer to the monomer is also considered (
). The population balance equations (PBEs) are given in
Table 2, where the definitions of the zero- and first-order moments of the chain length distribution of the active chains (
and
, respectively) and of their number average molecular weight (
) are as follows:
where
is the molar mass of MAA, thus the average chain length is defined in g mol
−1. The reaction rate coefficients are defined in
Table 3, where
is the conversion and
and
are set to 1.013 bar and 323 K, respectively. In the definition of the composite model for the termination rate coefficient, a critical average chain length
is considered, according to Wittenberg et al. [
25]. Since we do not include the population balance equations for the dead chains, we cannot compute the weight-average molecular weight required to calculate the viscosity parameter
as detailed by Wittenberg et al. [
25]; nevertheless, we obtained a satisfactory overlap of our model with the
versus conversion profiles at 30 wt % of MAA reported in the reference by setting
to 9. In the model equations, a constant density of 1.1 g mL
−1 is assumed for D
2O solutions.
Once all parameter values are set, the model is used to predict the kinetic profiles for our experiments at 1 wt %, 5 wt %, and 10 wt % of MAA. The resulting curves are shown in
Figure 1: a good agreement between the experimental data and the model predictions is found, with the initial slope perfectly reproduced at all the concentrations. At 10 wt % of initial monomer, both the simulated curve and the experimental data show an increase in polymerization rate due to the gel effect; moreover, they are nicely superimposed. On the other hand, at 1 wt % and 5 wt % the model predicts a slight increase in reaction rate, which is not observed experimentally. This discrepancy could reflect an increase in termination or a decrease in the propagation rate not accounted for by the model.
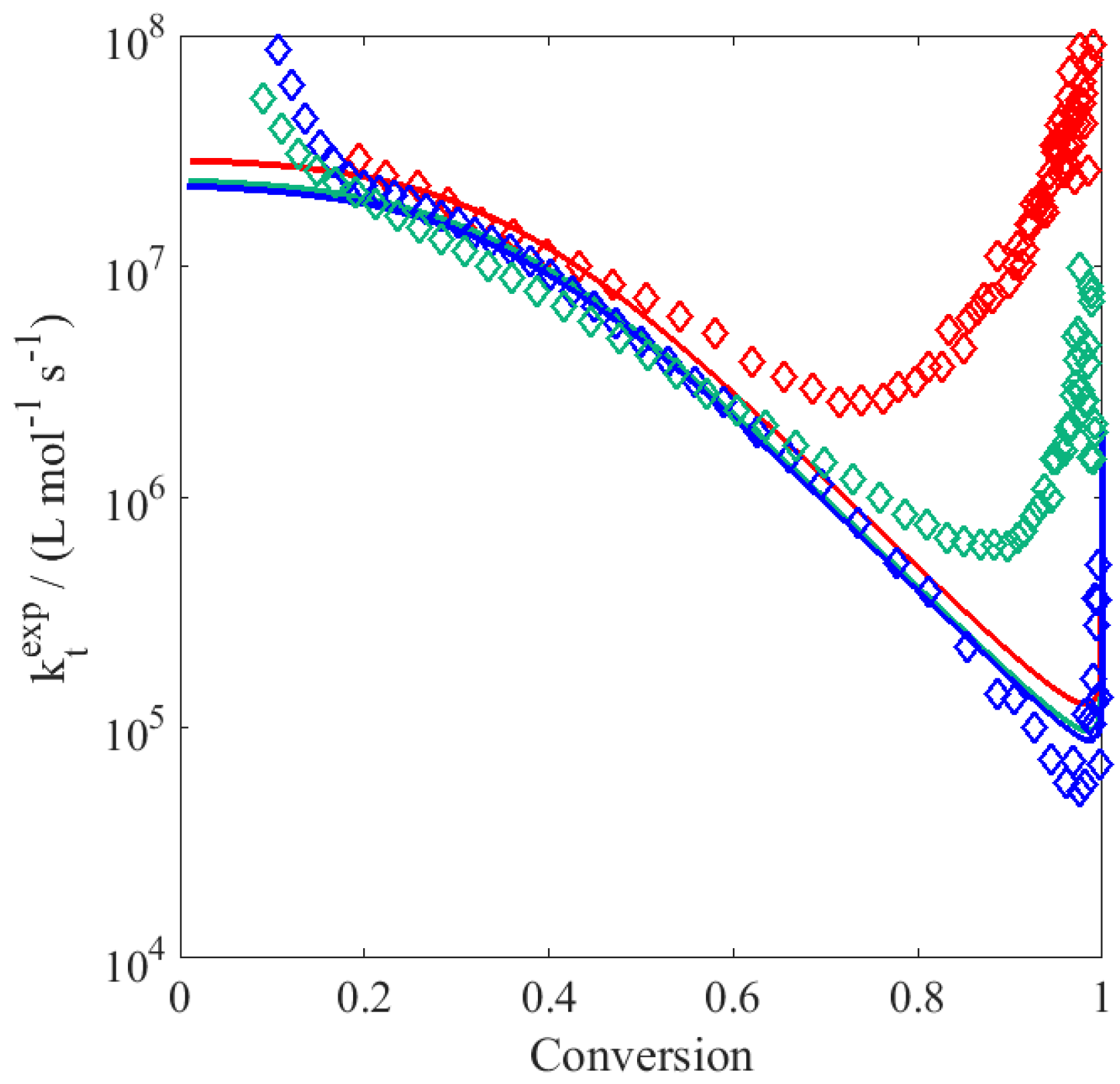
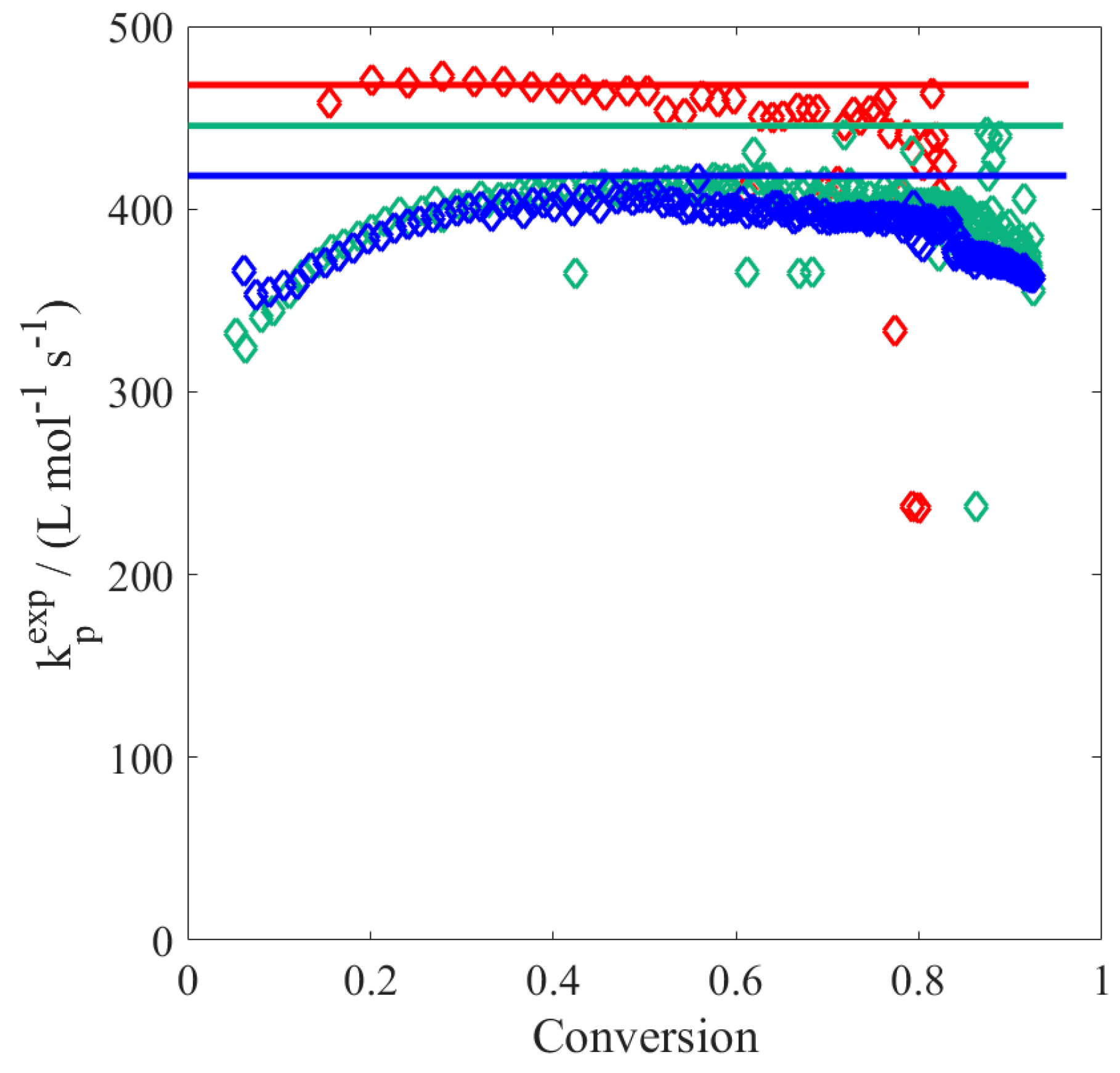
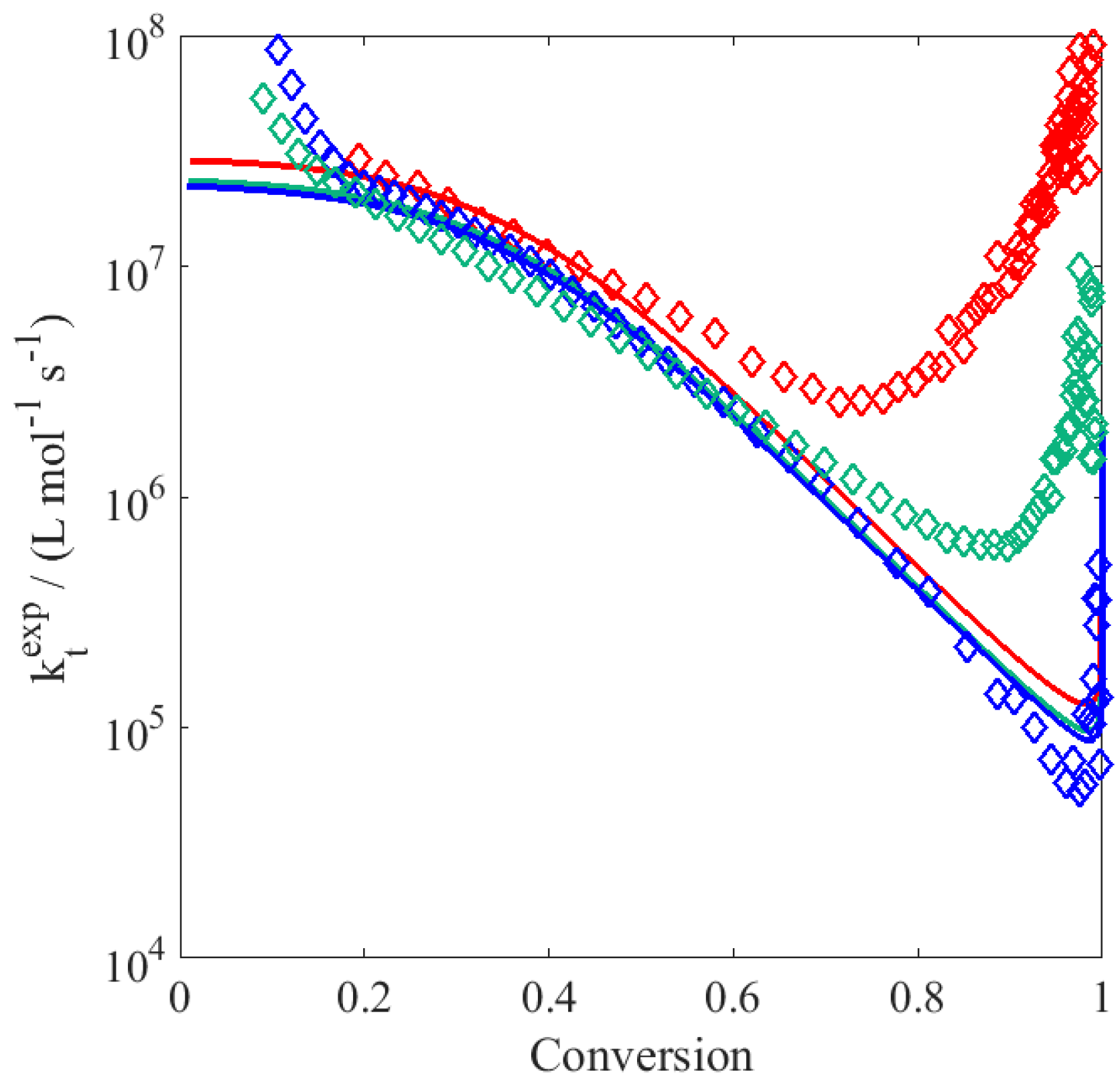
To better elucidate the possible reason for this disagreement, the experimental data have been processed as follows: (i) the local slope
is calculated at each time step; (ii) the product between the propagation rate coefficient and the actual radical concentration,
, is evaluated as:
(iii) using the
value defined as in
Table 3, the radical concentration is then obtained; and (iv) the apparent rate coefficient of termination is finally estimated as:
These resulting
values are depicted in
Figure 2: the increase in
reflecting the slowdown mentioned above becomes relevant at residual monomer concentrations below 0.5 wt %, i.e., at large polymer contents. Generally, a decrease (instead of an increase) in termination rate towards higher conversion is expected, thus the observed deceleration in the conversion versus time profiles should be imputed to a decrease in the propagation rate coefficient. Since
is chain-length dependent (cf.
Table 3), there is also the possibility that at high monomer conversion, the termination rate coefficient increases due to reduced monomer concentration leading to the production of shorter chains. However, from
Figure 2 such an increase in the literature
is observed only at very large conversion (i.e., above 95%): before that point, the termination rate coefficient decreases monotonously with conversion.
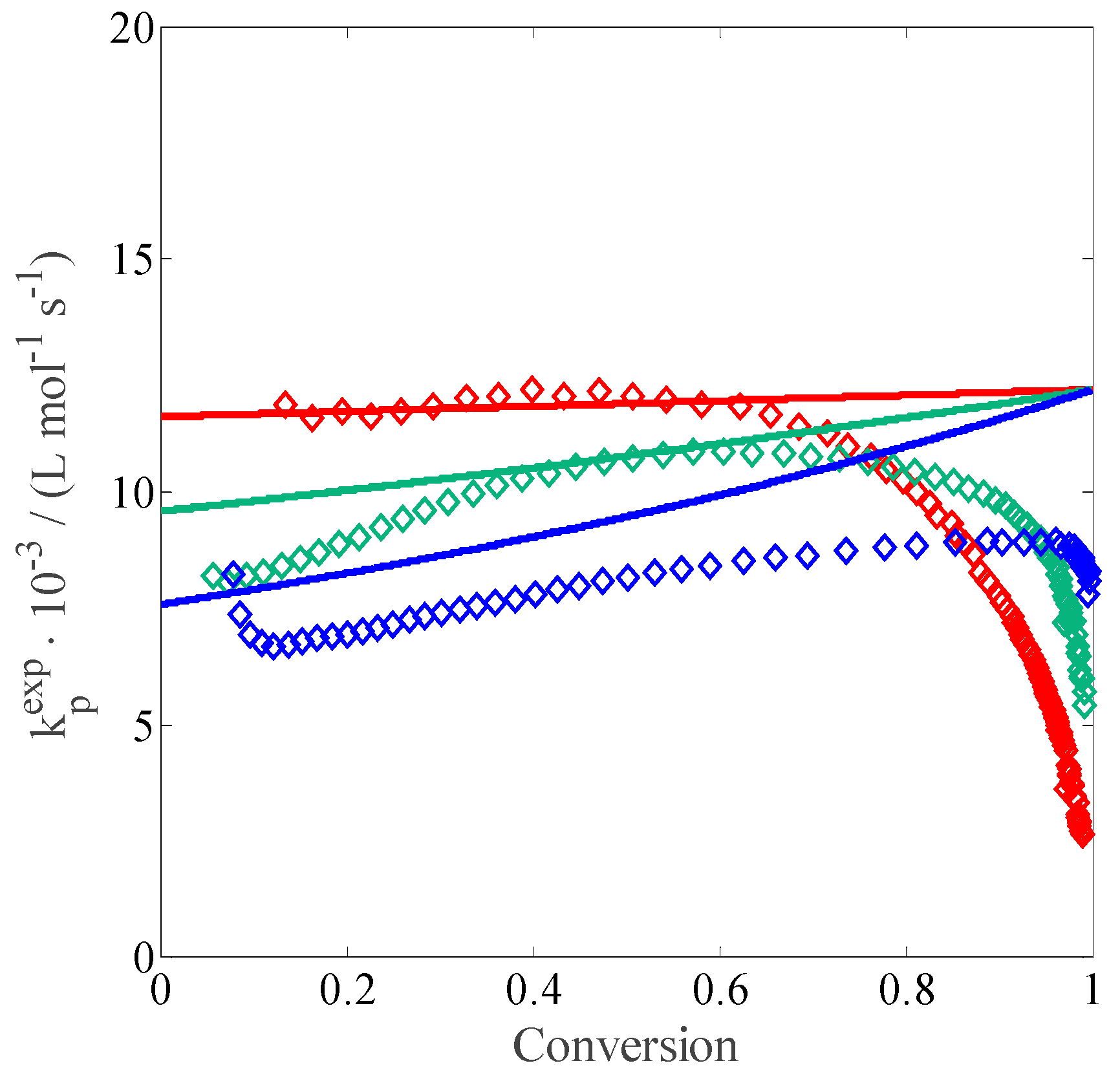
Therefore, a similar procedure as above has been applied to calculate the propagation rate coefficient deduced from experiments, this time by using the termination rate coefficient as in
Table 3. Since
is function of
, the procedure is not as straightforward: at each
step, the set of PBEs in
Table 2 is solved for different values of
while minimizing the error between the experimental and calculated conversion.
Figure 3 shows the resulting
curves as a function of conversion: all the curves exhibit a strong decrease at high conversion, more or less when the residual MAA concentration falls below 0.5 wt %. A similar behavior has been previously reported for the polymerization of acrylic acid (AA): a decrease of the apparent
at concentrations below 3 wt % has been noticed and imputed to differences in the local and overall monomer concentrations in solution, which may be a result of preferential solvation [
26]. This decrease explains the decreasing reaction rate at high conversion shown in
Figure 1. Apart from this discrepancy, the
predicted by the model equations provides a reasonable description of
as shown by
Figure 3. Therefore, we can conclude that the developed model is suitable for reproducing the experimental behavior of the system at medium-high monomer concentration and to qualitatively capture the trends of the propagation and termination rate coefficients as a function of conversion. The residual weakness in predicting the polymerization rate at lower monomer concentration is imputed to inaccurate
evaluation at high conversion.
3.2. Fully Ionized System
Three different sets of experiments are carried out for the polymerization of fully ionized MAA at 50 °C and 0.10 wt % V-50: the first set at different values of initial monomer concentration; the second set at different values of initial monomer concentration while keeping the ionic strength constant at an ionic strength equivalent to a monomer concentration of 10 wt %; and the third set at different values of ionic strength while keeping
constant at 5 wt %. In the second and third cases, the ionic strength is adjusted by the addition of NaCl. An overview over all these polymerization reactions is provided in
Table 4. A remarkably good reproducibility of the experiments in the fully ionized case has been observed for the reactions at 1 wt % and 10 wt % initial MAA, as reported in
Figures S5 and S6 of the
Supporting Information.
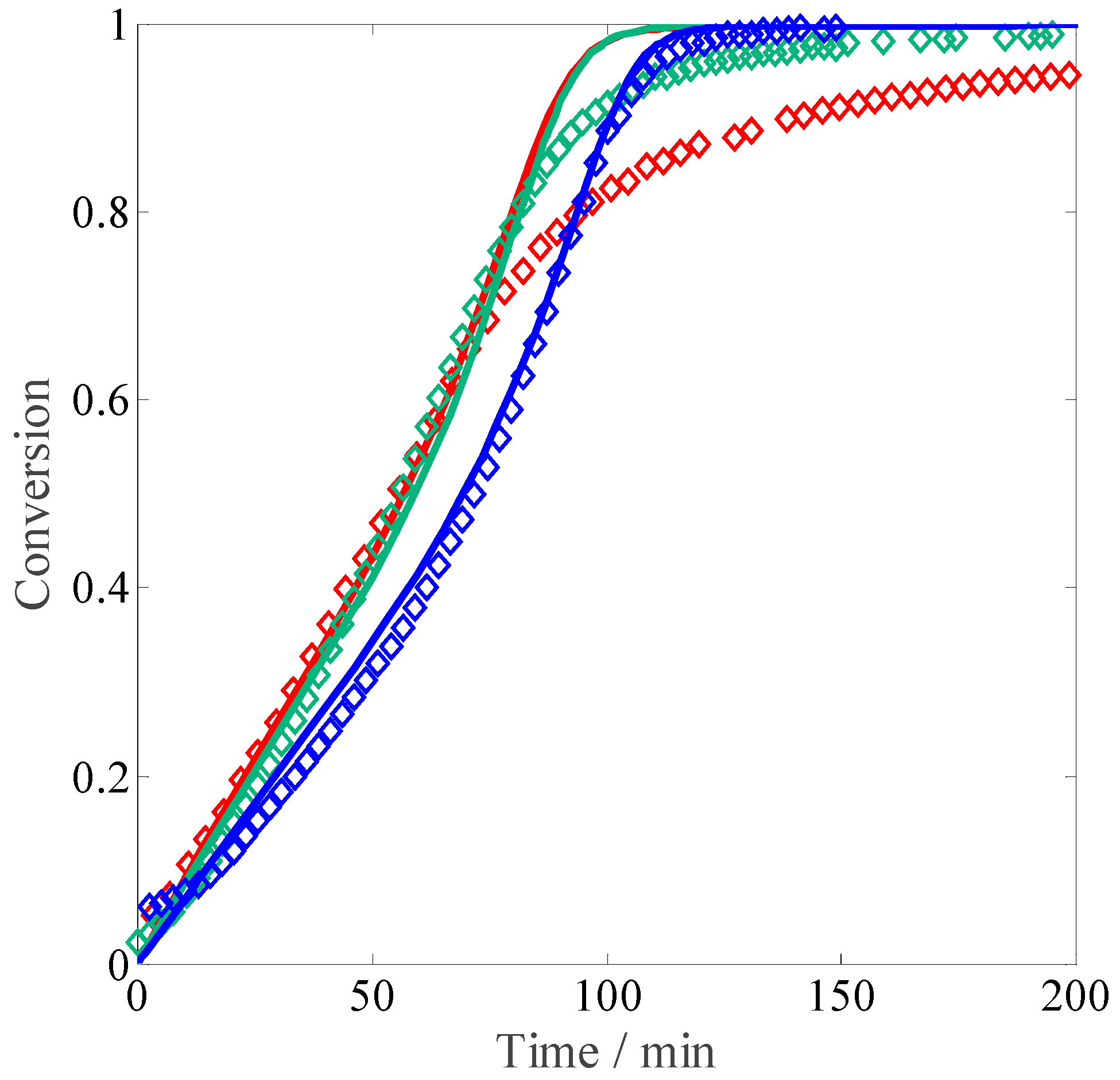
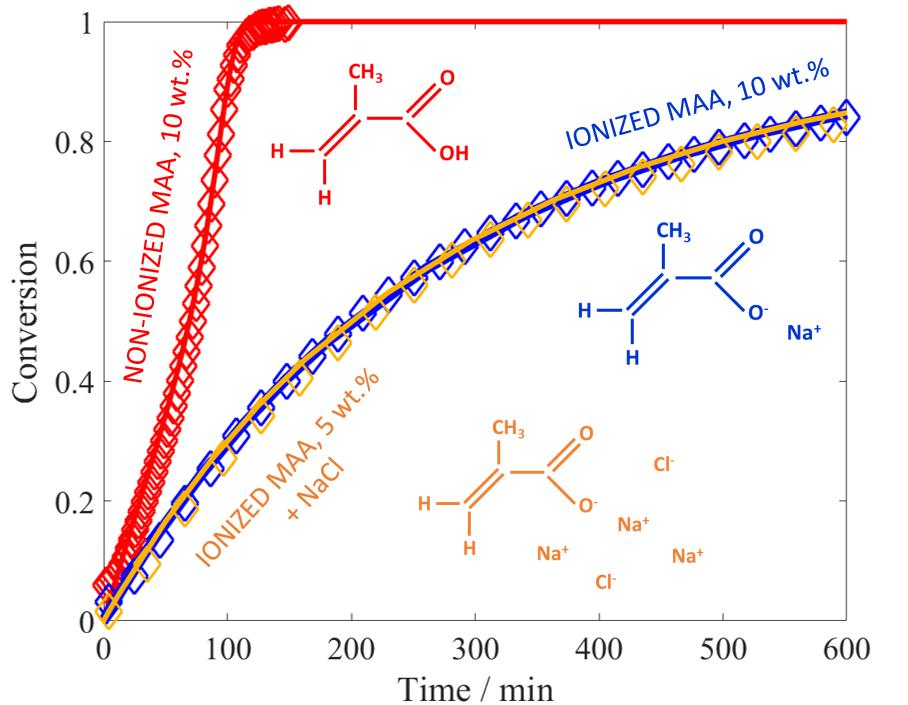
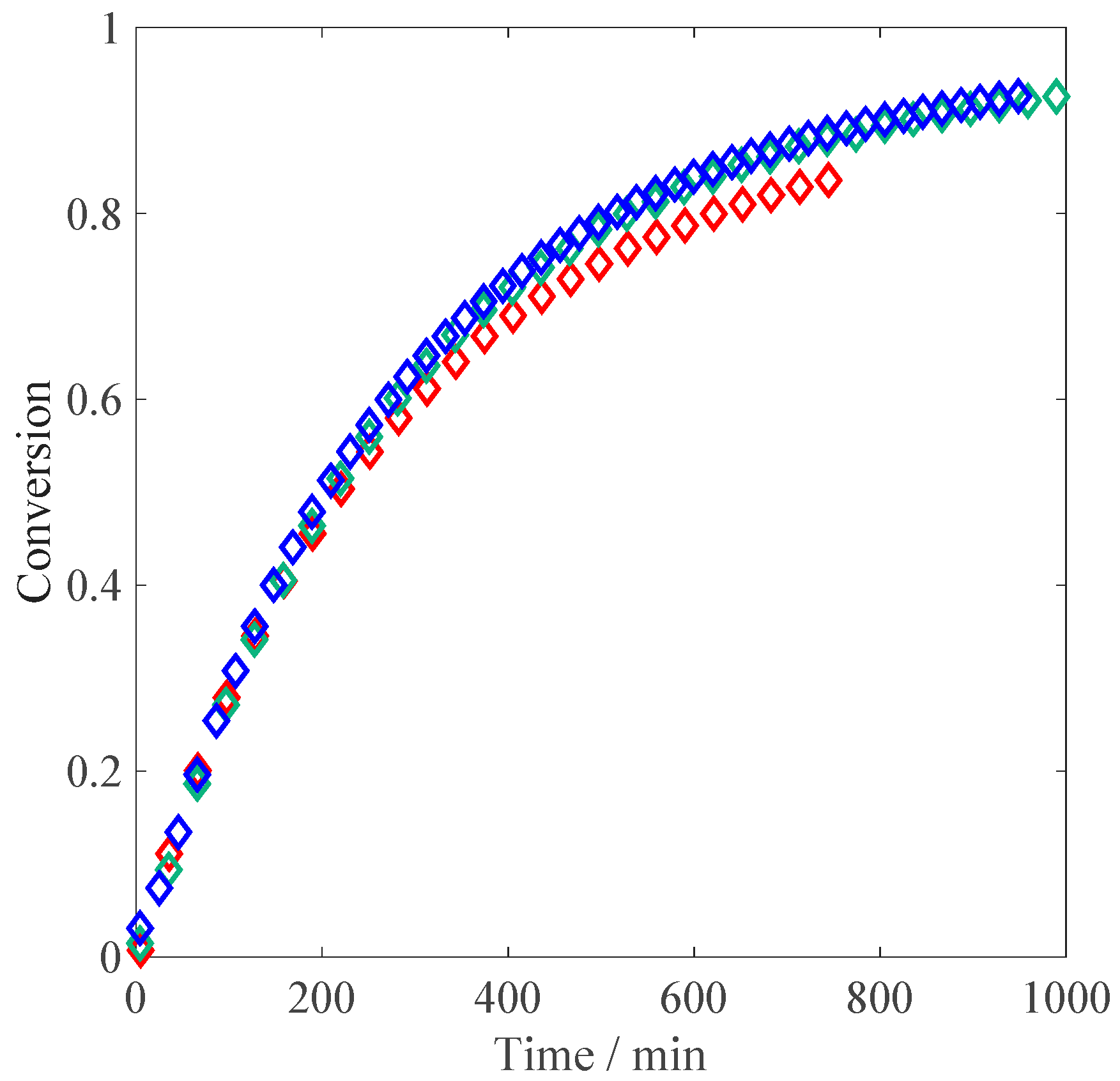
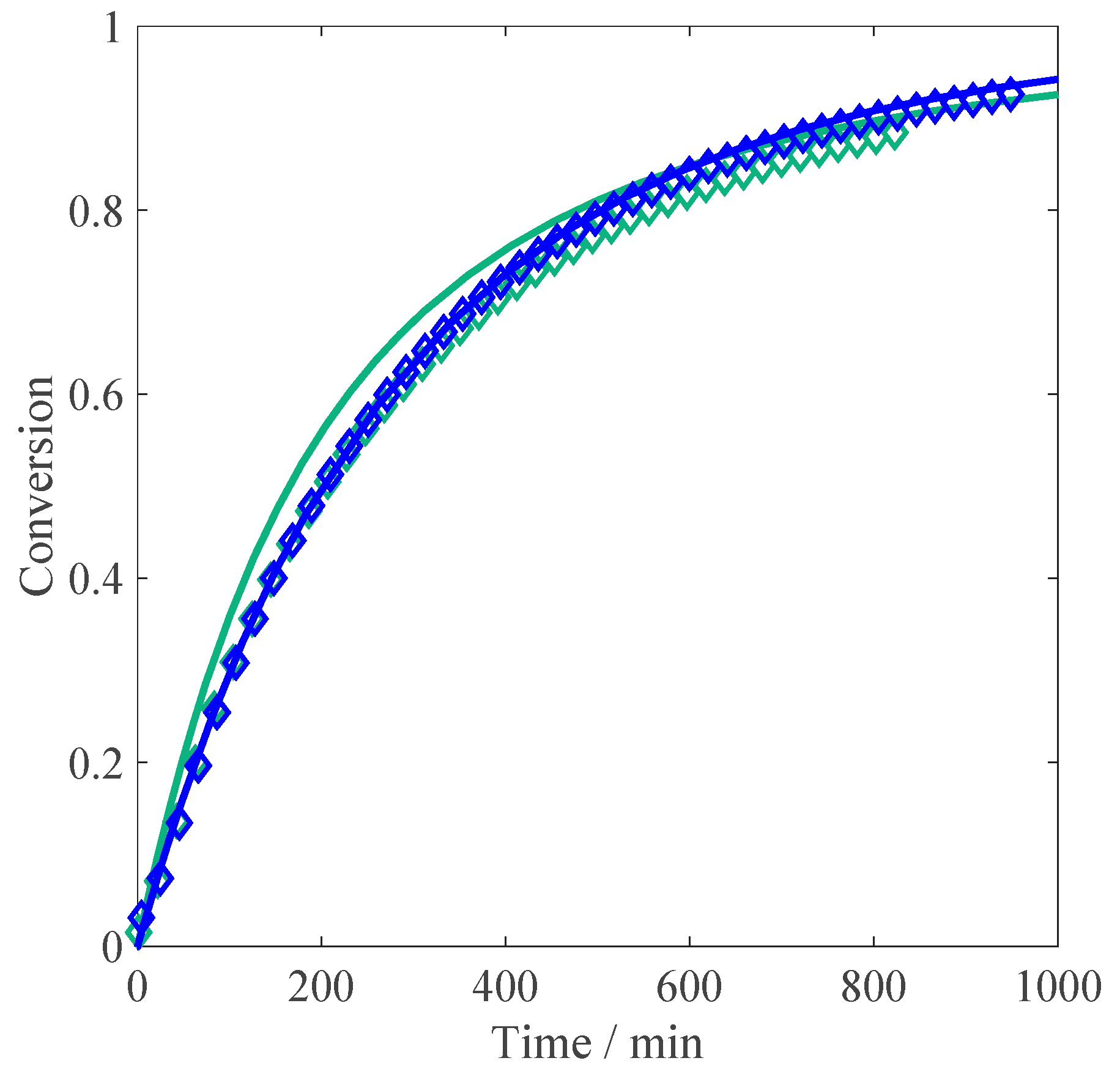
Let us consider the experiments at different monomer concentrations. When comparing the resulting conversion versus time curves shown in
Figure 4 to those reported above for the non-ionized case (
Figure 1), the initial reaction rates are much slower at full MAA ionization: this feature can be explained by the electrostatic repulsion between the charges of the ionized reactants in solution. An increase in the polymerization rate is observed when moving from 1 wt % of MAA to the higher concentrations, whereas between 2.5 wt % and 10 wt % the kinetics appears to be unaffected by the monomer concentration. The former effect corresponds to an increase in the propagation kinetics due to an increase in the solution ionic strength, in agreement with the so-called phenomenon of electrostatic screening: namely, higher initial concentration of the monomer corresponds to higher concentration of the electrolyte in the system [
6,
11]. The latter effect is more surprising, and reveals either a saturation of the electrostatic screening already at 2.5 wt % of MAA or a competition between electrostatic and non-electrostatic effects of monomer concentration on propagation kinetics. Another counterbalancing effect to the electrostatic-driven increase in
could be represented by the known dependence of termination kinetics of fully ionized MAA on monomer concentration [
27].
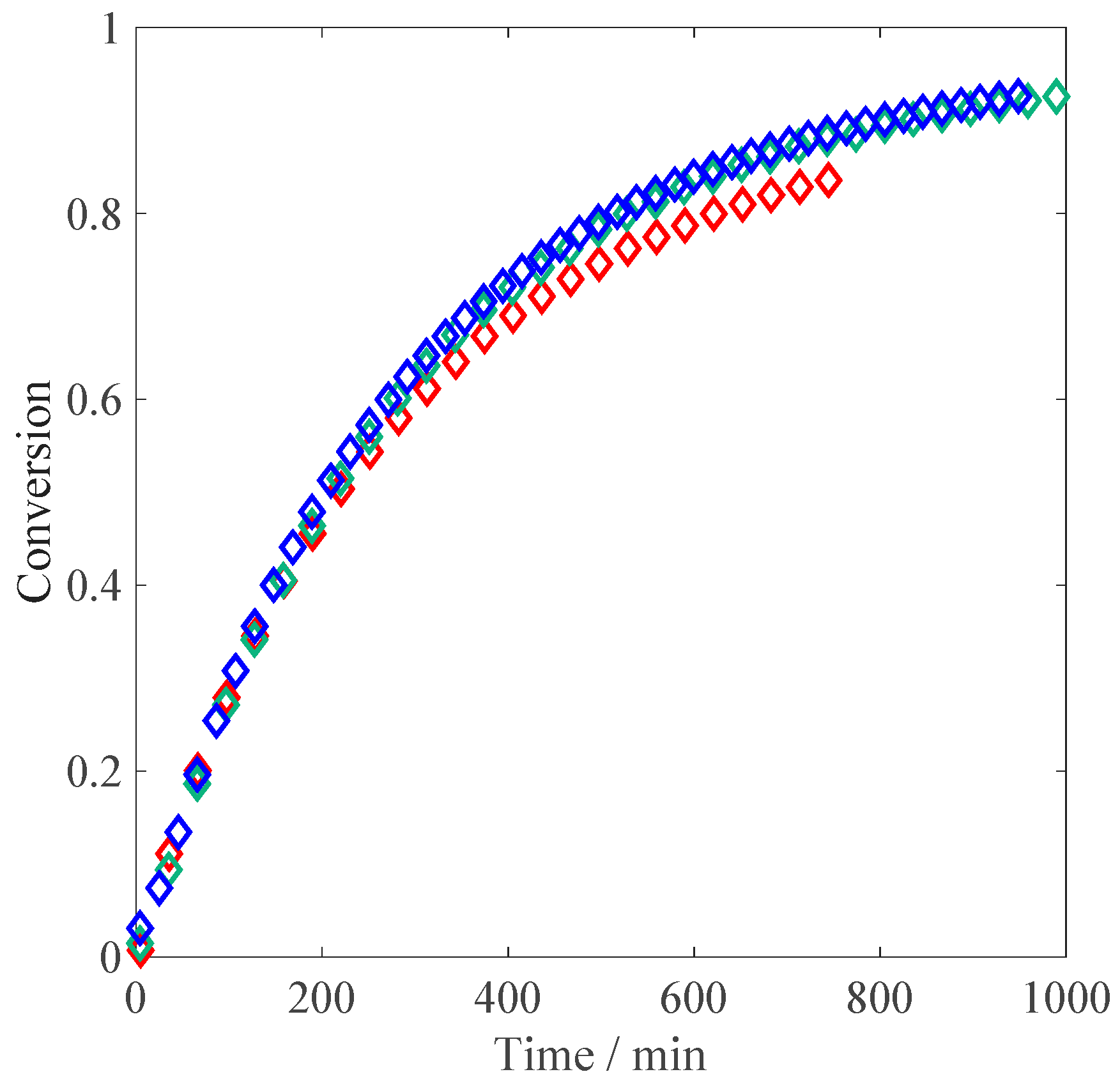
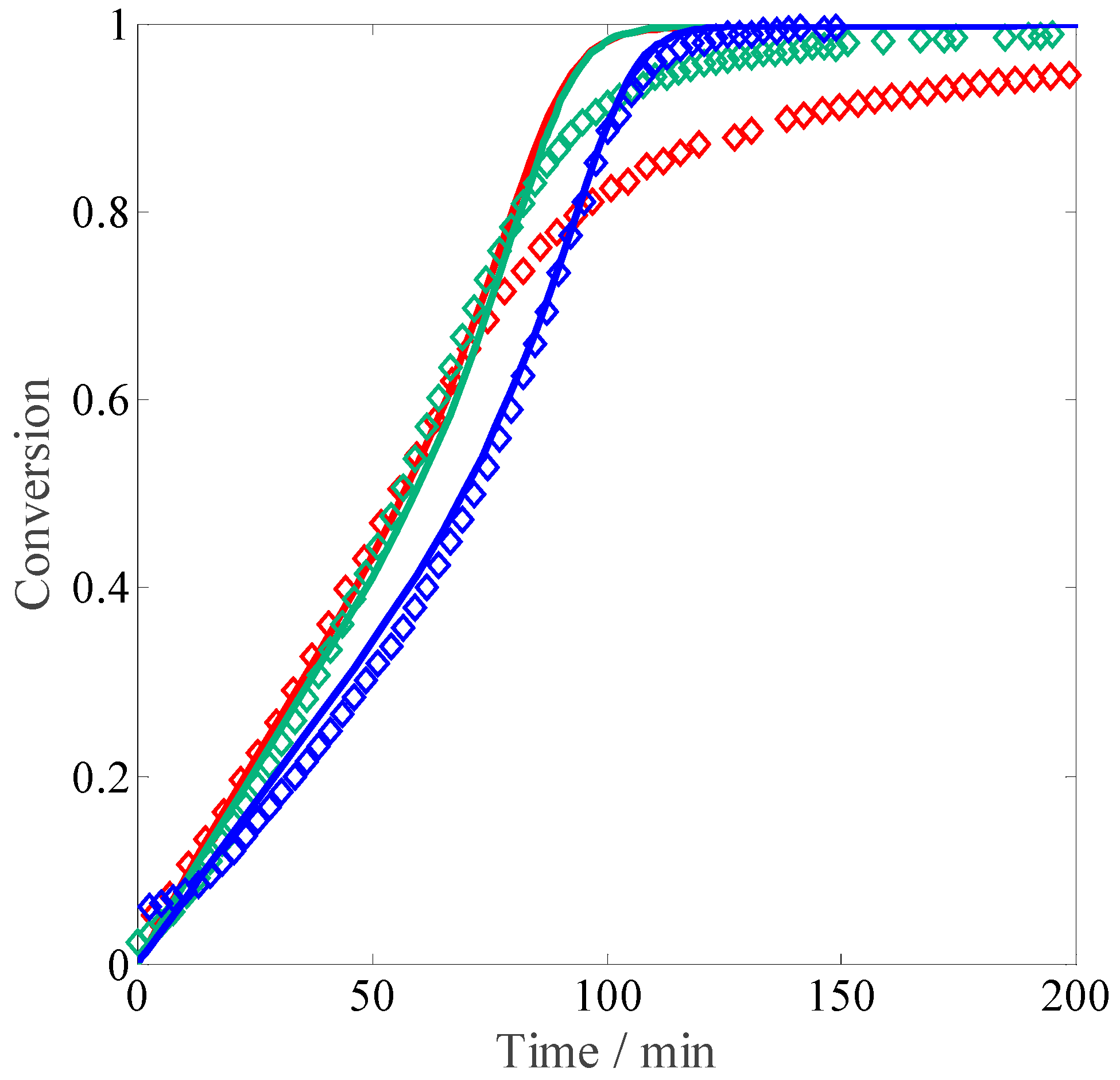
The impact of monomer concentration on the reacting system at constant ionic strength (equivalent to 10 wt % MAA) and 0.10 wt % of initiator is shown in
Figure 5. The idea behind this set of experiments is to reproduce the non-electrostatic influence of the monomer concentration, keeping the ionic strength constant. As shown in the figure, the initial reaction rate does not change with the monomer concentration. Only at a conversion higher than 50% does the curve at low monomer concentration seem to become slower than those at higher monomer concentration, where the propagation rate seems to be fully independent of the non-electrostatic (i.e., intrinsic) reactivity.
The impact of NaCl addition on the reacting system at constant monomer concentration is shown in
Figure 6. The aim is to reproduce the ionic strength of higher monomer concentrations while keeping constant the initial monomer concentration, in order to separate the effect of the charges from the mixed effects associated to an increase in monomer content (i.e., electrostatic and non-electrostatic ones). As shown in the figure, the initial reaction rate increases continuously with increasing amounts of added salt. Additionally, there is no visible effect of autoacceleration on the rate of polymerization in this case.
Since the propagation reaction seems to be affected by the electrolyte concentration in the entire explored range (i.e., up to 30 wt % of equivalent MAA), this same reaction is expected to never become fully kinetically-controlled: namely, the effects of electrostatic interactions and electrostatic screening on the propagation kinetics are likely to play a role at all explored values of initial monomer concentration, and some kind of “electrostatic saturation” at higher monomer content (i.e., above 2.5 wt % of MAA) cannot be assumed.
Aiming to develop a kinetic model for the polymerization of fully ionized MAA, the reaction scheme and population balances in
Table 1 and
Table 2 are again considered, whereas the validity of the reaction rate coefficients in
Table 3 is critically discussed hereinafter.
The propagation rate coefficient proposed by Beuermann et al. [
21] has been improved by Lacik et al. to take into account the effect of acid ionization [
20]. However, as this equation has been proposed for the specific range of initial monomer concentration
, it cannot be used for our experiments at 1 wt % and 2.5 wt % of initial monomer content, where the calculated
would be negative. Furthermore, this equation cannot be used to simulate the effect of ionic strength on the propagation kinetics coming from NaCl addition. For these reasons, we propose a rate law of propagation of fully ionized MAA composed of an intrinsic kinetics term and a diffusion term, the latter being driven by electrostatic interactions and thus a function of the electrolyte concentration, similar to the equation developed in a previous work [
11]:
where
,
,
, and
are fitting parameters, while
is the electrolyte concentration in mol kg
−1, which is a function of
:
where
and
are the initial weight fraction and molar mass of NaCl, respectively.
The first part of Equation (7) introduces the non-electrostatic dependence on the monomer concentration, similar to what has been done by Beuermann et al. for non-ionized MAA [
21]. However, it should be noted that such dependence can be in principle different for the fully ionized case: for this reason, the parameter
is estimated as well. For simplicity, the conversion-dependent term of the exponent
has been omitted. The parameter
identifies the intrinsic kinetics rate coefficient of fully ionized MAA, thus without any electrostatic interaction effect. The electrostatic and non-electrostatic dependence of
on
for the fully ionized case, defining respectively an increase and a decrease of the rate coefficient as the initial monomer concentration increases, are expected to compensate each other at higher monomer concentrations, thus reproducing the experimental behavior observed in
Figure 4.
In order to verify if the proposed rate law for
is reasonable, the propagation rate coefficient is evaluated as a function of conversion using the same procedure described for the non-ionized case, namely using the experimental data reported in
Figure 4 and
Figure 5. As for
, the rate coefficient from a recent publication by Kattner et al. [
27] has been adopted: the authors proposed an expression for the termination rate coefficient at monomer concentrations of 5 and 10 wt % as shown in
Table 5.
Similar to the propagation rate, it can be assumed that the termination reaction is also made of two contributions, one related to an intrinsic kinetics and the other driven by electrostatic interactions. Due to the limited amount of available data, we need to assume that only one of these two contributions majorly affects the termination rate. Since bimolecular termination of growing radicals is a reaction with a low activation energy barrier (mostly controlled by diffusion), we assume that the change in ionic strength upon change in monomer concentration is principally responsible for the
variation as a function of
. This is considered more realistic than an effect of monomer concentration on the fluidity of the transition state, as previously considered in the non-electrostatic effect of monomer concentration on the propagation kinetics of other water soluble compounds [
8]. Therefore, we will focus on those experiments with an ionic strength (obtained by a certain amount of monomer concentration or addition of NaCl) equivalent to a monomer concentration of 5 wt % or 10 wt % when considering the
expressions reported in
Table 5.
Equivalent to the procedure for the non-ionized case (cf.
Figure 3), the experimental propagation rates are shown in
Figure 7 and
Figure 8 for ionic strengths equal to 5 wt % and 10 wt %, respectively. The propagation rate coefficient of 5 wt % MAA without addition of NaCl can be compared with the experimental results of Lacik et al. [
20]: at our reaction temperature (50 °C), the Lacik equation predicts a propagation rate of about 770 L mol
−1 s
−1, whereas our experimentally deduced propagation rate is about five times smaller. At monomer concentrations of 5 wt % and 10 wt %, the propagation rate increases at low conversion, which is probably due to the competition between propagation and radical scavenging by the inhibitor. Above 50% conversion, a general decrease is visible. Apart from these minor variations with conversion, the results reveal that the reaction rate increases with ionic strength. In fact, for any monomer concentration, the propagation rate at an ionic strength of 10 wt % monomer concentration is 2–3 times faster than at 5 wt %. At constant ionic strength, a reduction of monomer concentration leads to a slight increase of the propagation rate coefficient. However, this effect is less relevant than the change in ionic strength. The observed competition between the influence of monomer concentration and ionic strength confirms the suitability of the propagation rate law in Equation (7) to describe at least qualitatively the kinetic behavior of the system.
The four parameters
,
,
, and
are estimated by fitting the model equations to the experimental data of conversion versus time for the fully ionized case. The ratio of the parameters
and
is expected to be close to unity: this way, the diffusion limitation to
due to the electrostatic interactions should be effective inside the entire range of ionic strength values explored, as deduced from the experimental results. It should be noted that
must be expressed in L mol
−1 s
−1 (kg mol
−1)
β in agreement with the definition of
in the model. The exponent
is dimensionless and it is expected to range between 0.1 and 10, while
is a measure of the influence of initial monomer concentration on the kinetics. As the value for non-ionized MAA has been determined as 5.3 [
21], a similar value (between 1 and 10) is expected.
The parameters are estimated by minimizing the square of the error between experimental and simulated conversion versus time data. This minimization is carried out by a genetic algorithm which is implemented using Matlab. An optimization run was conducted to evaluate the four mentioned adjustable parameters and leads to the results and parameter values reported in
Figure 9,
Figure 10 and
Figure 11 and
Table 6, respectively.
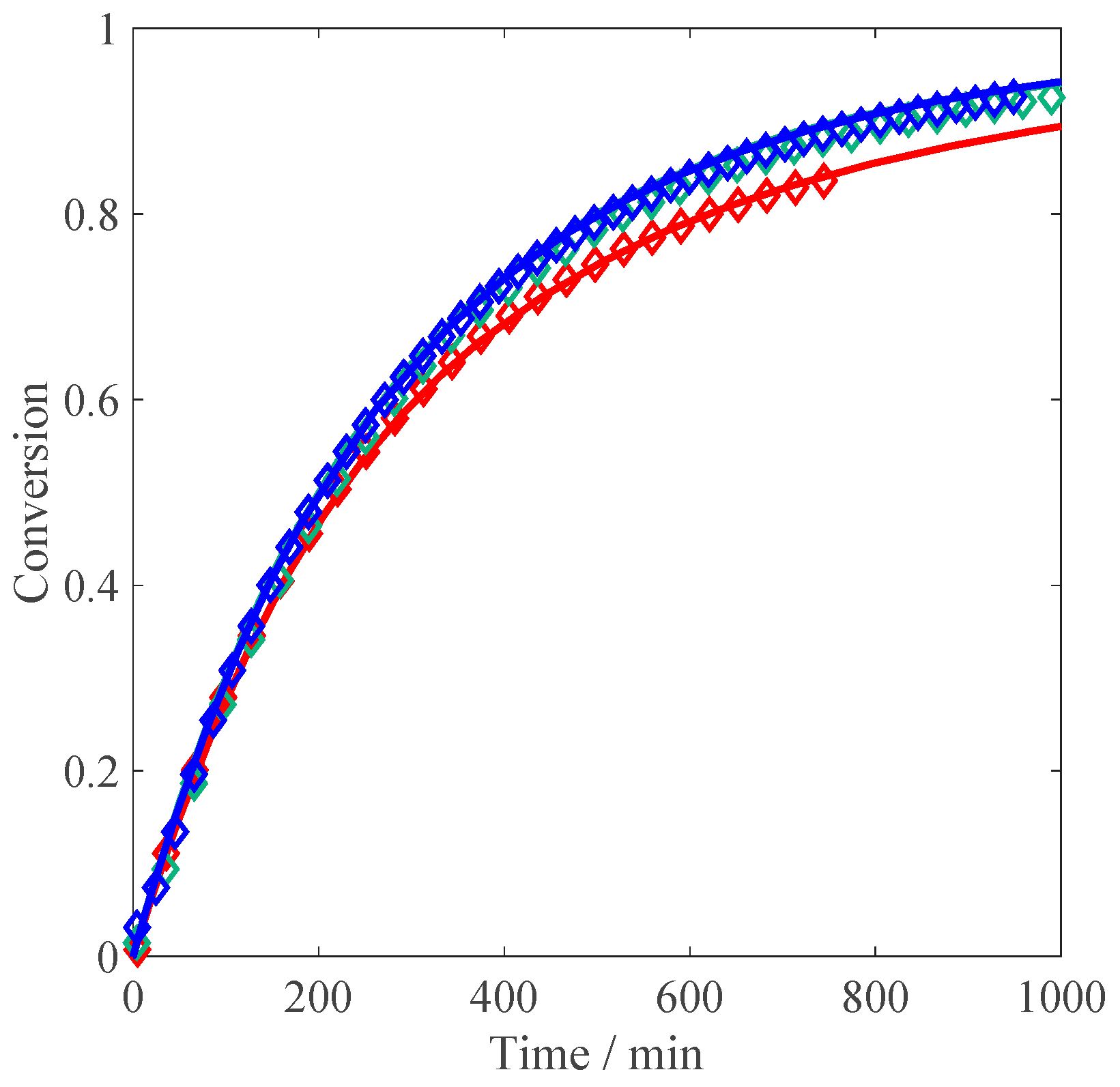
Figure 9 compares the influence of monomer concentration without salt on conversion: while the curve at 10 wt % is perfectly reproduced, at 5 wt % monomer, the model predicts a reaction rate which is slightly faster than that at 10 wt %, but while looking at the experimental data the rates should be exactly the same (i.e., the electrostatic and intrinsic kinetics effects on the propagation and termination rate coefficients should be perfectly balanced). Towards higher conversion, the model correctly predicts a decrease of the reaction rate at 5 wt %.
The influence of monomer concentration at an ionic strength equivalent to 5 wt % is shown in
Figure 10. Qualitatively, the initial slopes at varying amounts of MAA are well reproduced, although the impact of monomer concentration on the initial rate is slightly too strong. The decrease in the reaction rate at conversions above 50% is very well described for both monomer concentrations.
A similar comparison is shown in
Figure 11, where the ionic strength is set equivalent to 10 wt % MAA. As already mentioned in the discussion of
Figure 5, the initial slope is the same for all MAA concentrations, which is well reproduced by the model, as well as the behavior at higher conversion. Since the model is able to correctly simulate the reduction at 1 wt %, this decrease of the reaction rate cannot be imputed to smaller local concentrations around the polymer chain. It is rather due to a conversion-dependent increase of the termination rate coefficient (caused by a decrease of
). Note that an effect of local concentration can still be present when considering the reaction at 1 wt % without salt addition, but since we lack an appropriate description of the termination rate at this ionic strength, we are unable to predict this situation.
The parameter values are provided in
Table 6: the kinetic rates
and
are in the same order of magnitude and similar to the kinetic parameter obtained by Lacik et al. [
20], whereas
is slightly smaller than two. It is therefore clearly smaller than the 5.3 obtained by Beuermann et al. [
21], but since the reactivity of the ionized MAA is much smaller, it might be reasonable to assume that the effect of monomer concentration is also reduced. The parameter
is slightly larger than two, which is large enough to alter
in a way to qualitatively reproduce the experimental behavior at increasing ionic strength. The overall good balance between
and
leads to a good description of the system: at low ionic strength, the propagation reaction is diffusion limited, whereas at ionic strengths larger than 5 wt % (either by addition of NaCl or MAA), the propagation seems to become more reaction controlled, although never completely (as shown by the experimental results in
Figure 6). A comparison between the intrinsic kinetics and electrostatic contributions to the propagation rate coefficient for the considered reactions involving fully ionized MAA is given by
Figure S7 and
Table S2 in the
Supporting Information. Moreover, the sensitivity of the model performances upon limited variations of the model parameter values is reported in
Table S3 of the
Supporting Information, indicating that small variations of each parameter (i.e., within 10 wt %) induce a very large change in the total error between experiments and simulations.
Eventually, in order to better describe what happens at ionic strengths below 5 wt % and above 10 wt %, a more generalized description of the termination rate is required. This knowledge is especially needed to elucidate the reason for the drastically reduced reaction rate at 1 wt % MAA without salt addition.
As a final remark, the knowledge of molecular weights would allow us to improve the reliability of the developed kinetic models, especially for the fully ionized case. Nevertheless, our choice of focusing on time-conversion data only in the analysis and modeling of the system kinetics was motivated by the following reasons: (i) the estimation of accurate MWD for MAA and generally for aqueous polymers is not trivial and requires ad hoc experimental setup and procedures; (ii) the accuracy and reliability of the kinetic data obtained by in-situ NMR is much higher compared to our current capability of measuring MWD; (iii) the availability of independent values of the termination rate coefficients from PLP of non-ionized MAA and, for specific reaction conditions, also for fully ionized MAA makes our approach reasonable for determining the propagation kinetics by focusing only on the measured polymerization rates.










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}