


Ni Supported on Pr-Doped Ceria as Catalysts for Dry Reforming of Methane

 , , , ,
, , , ,
Abstract

1. Introduction
2. Materials and Methods
2.1. Catalyst Preparation
2.2. XRD
2.3. TEM
2.4. SEM
2.5. TPR
2.6. DRM Reaction
2.7. TPO
3. Results and Discussion
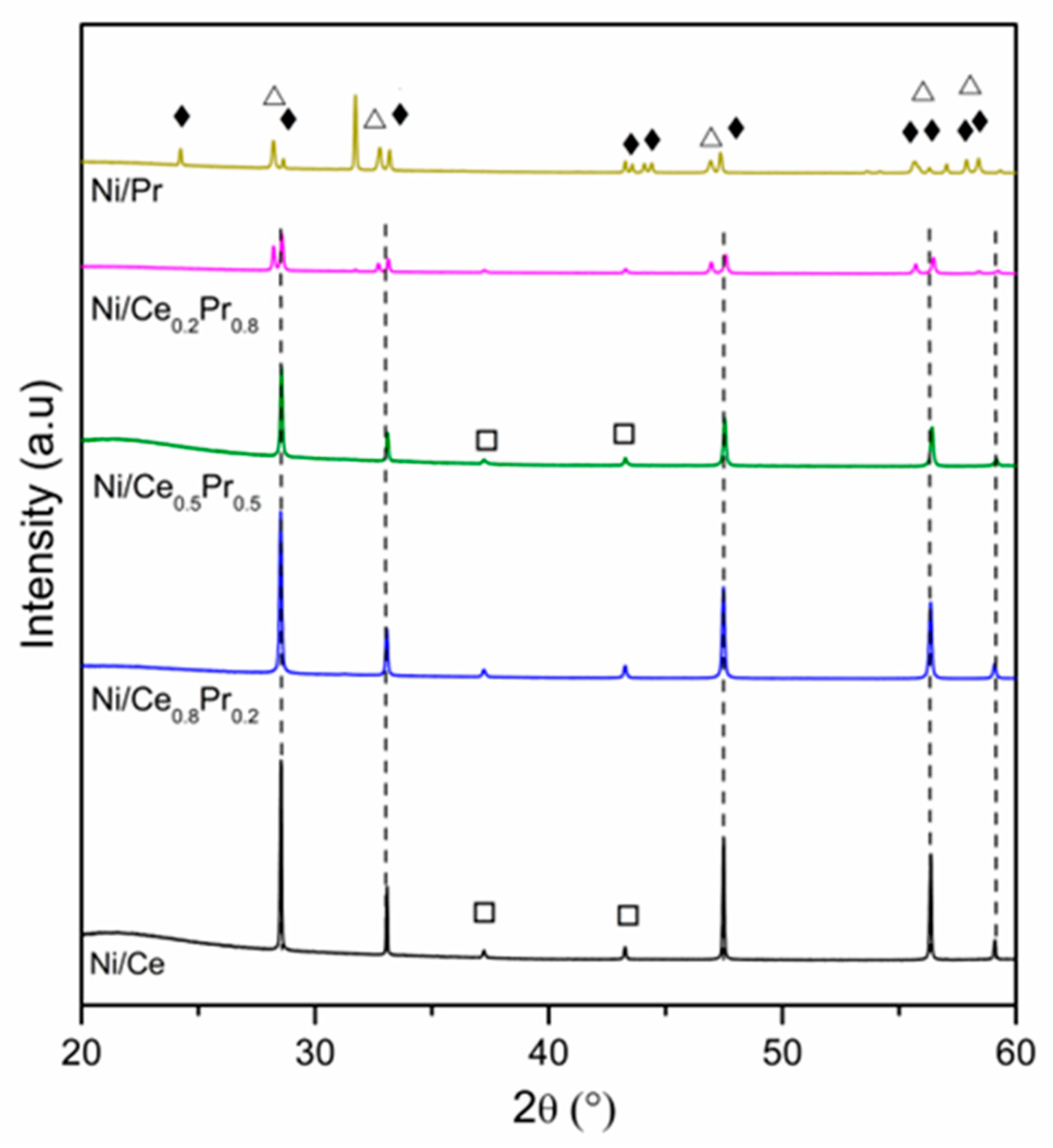
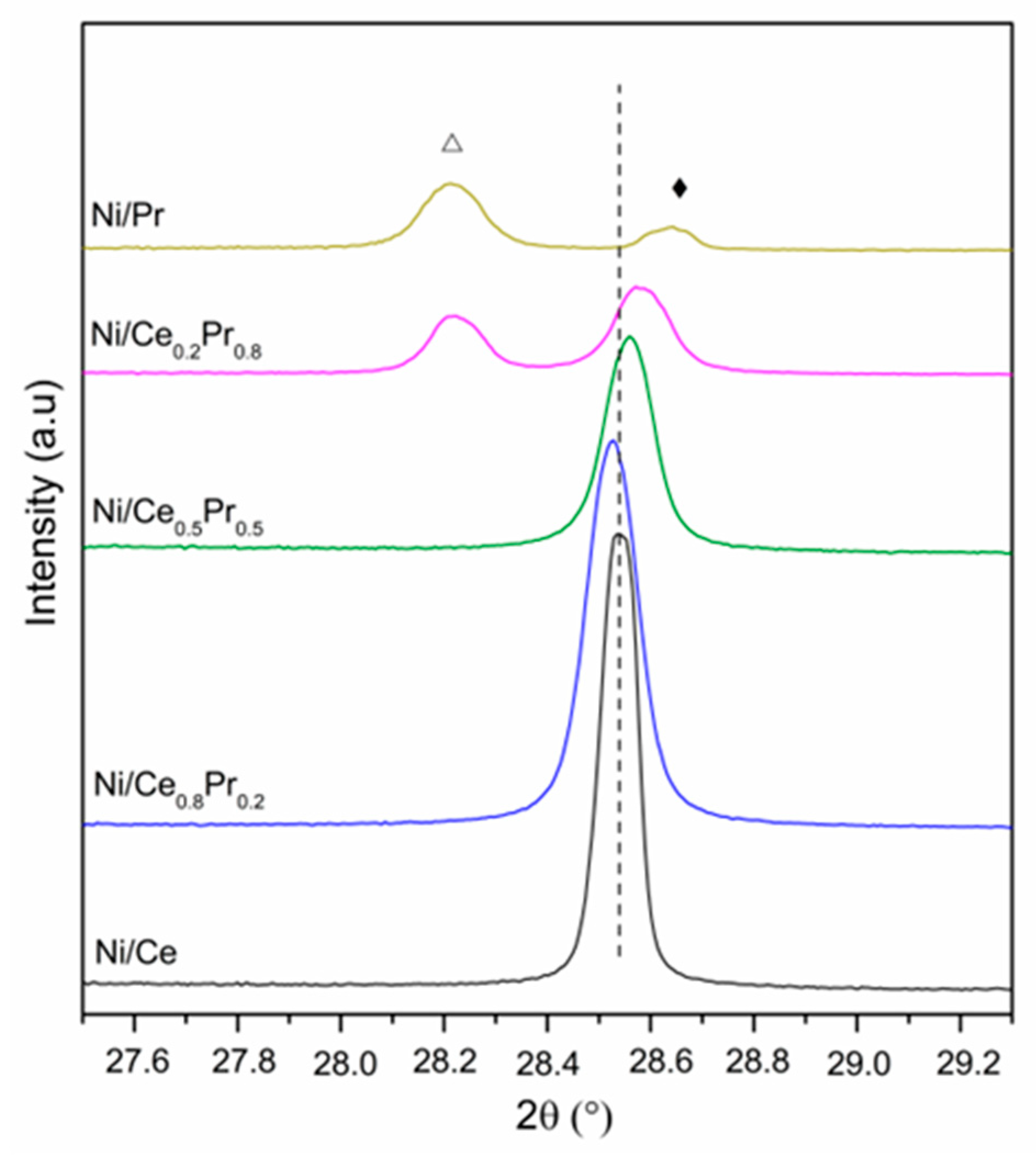
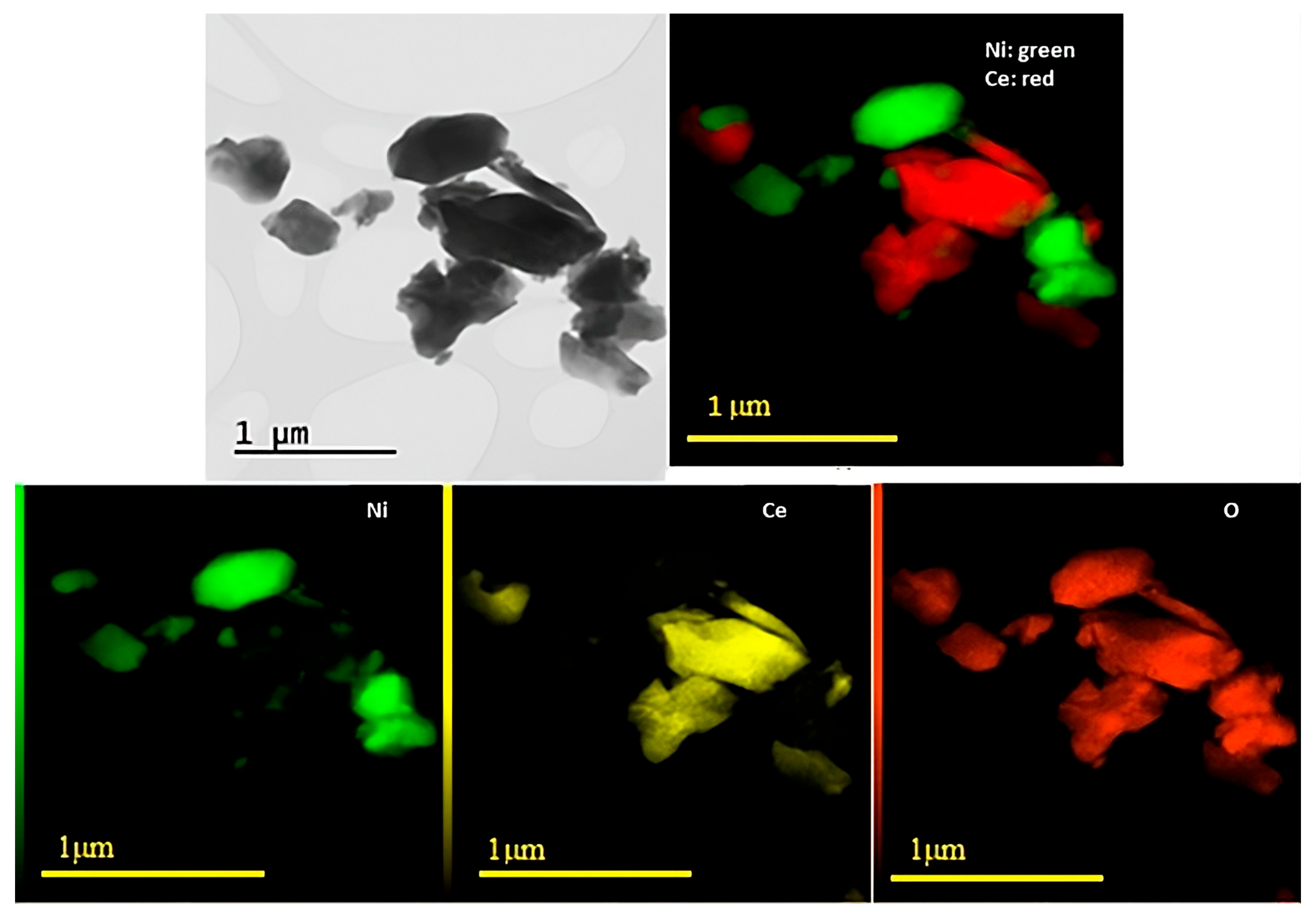
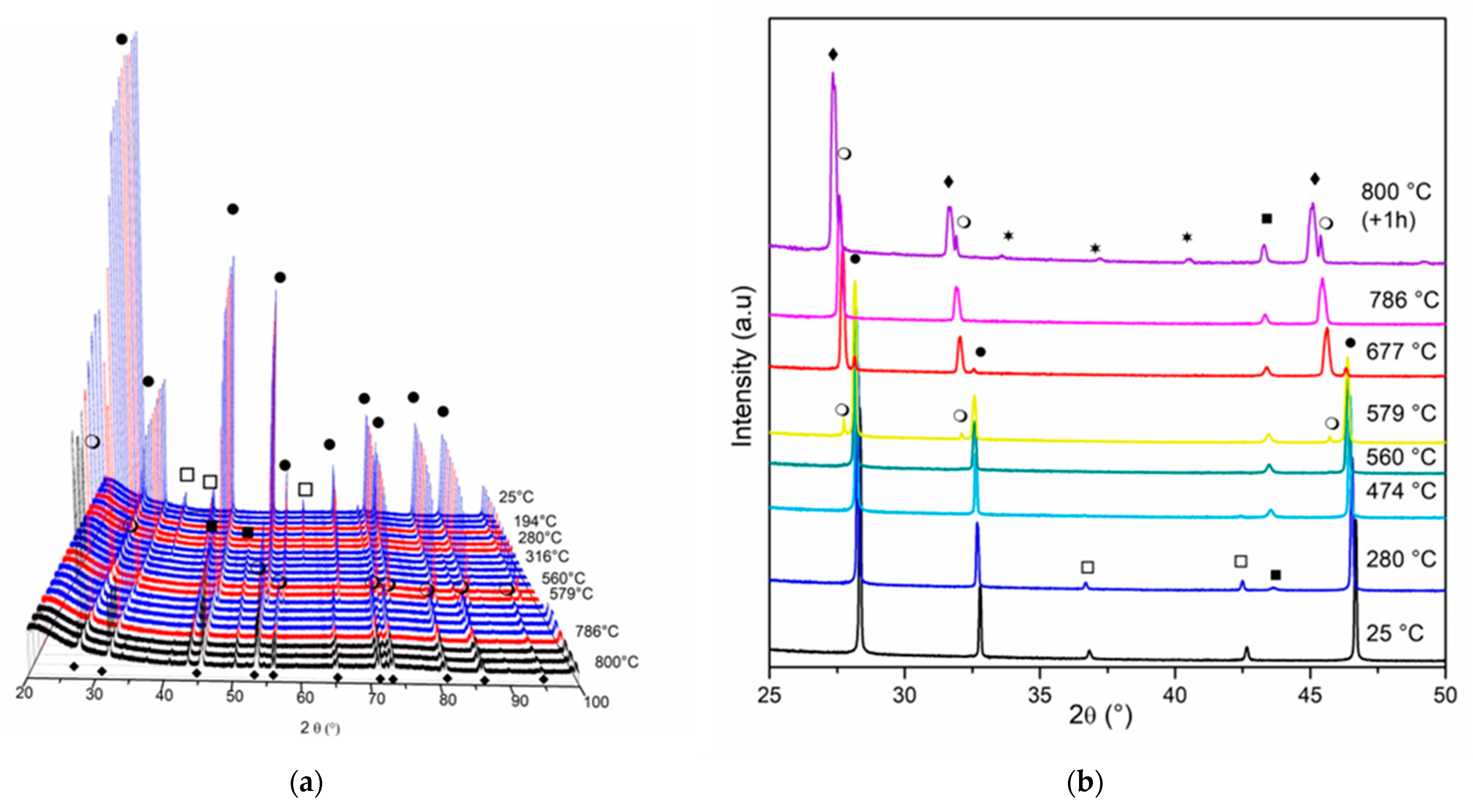
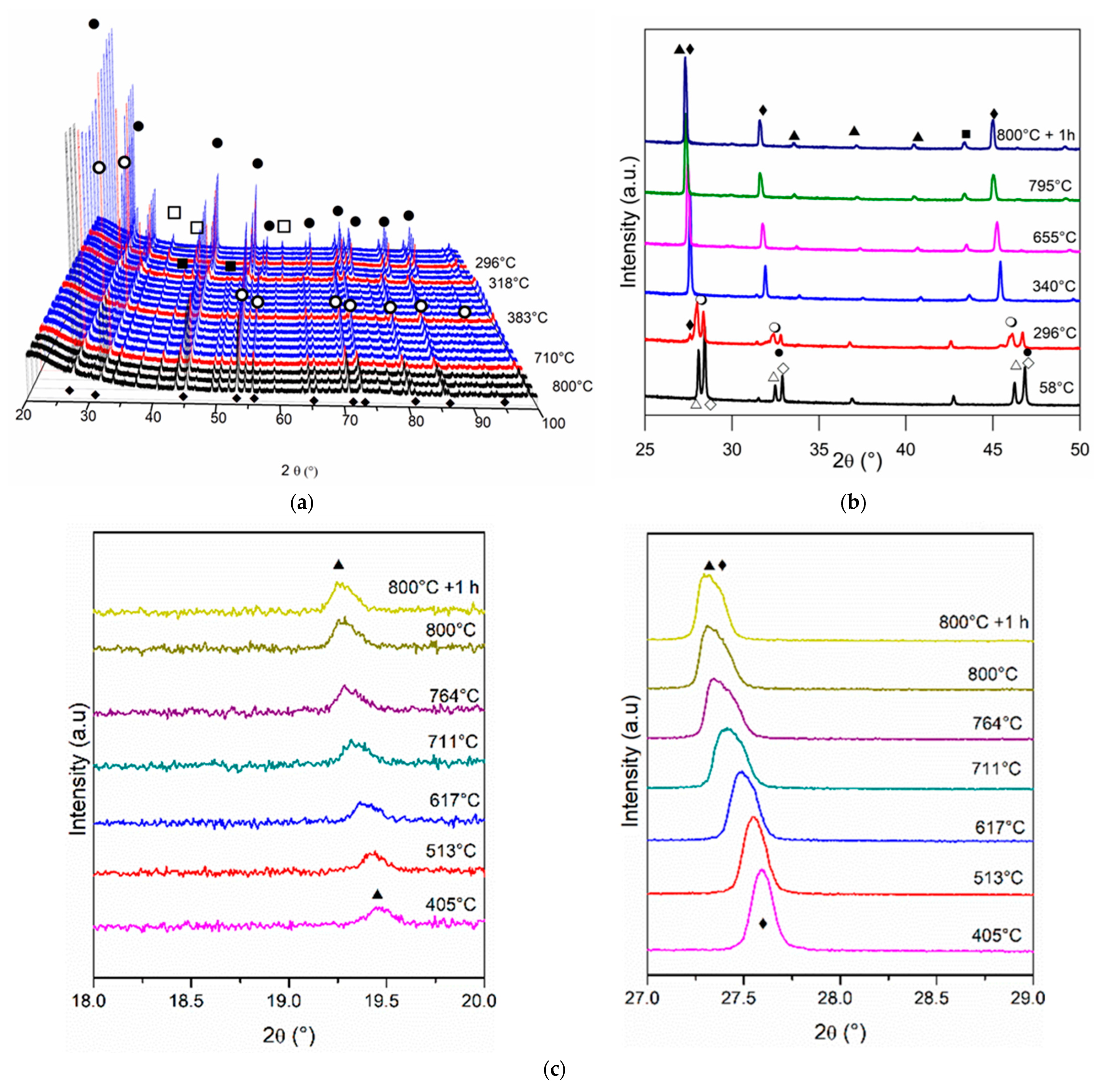
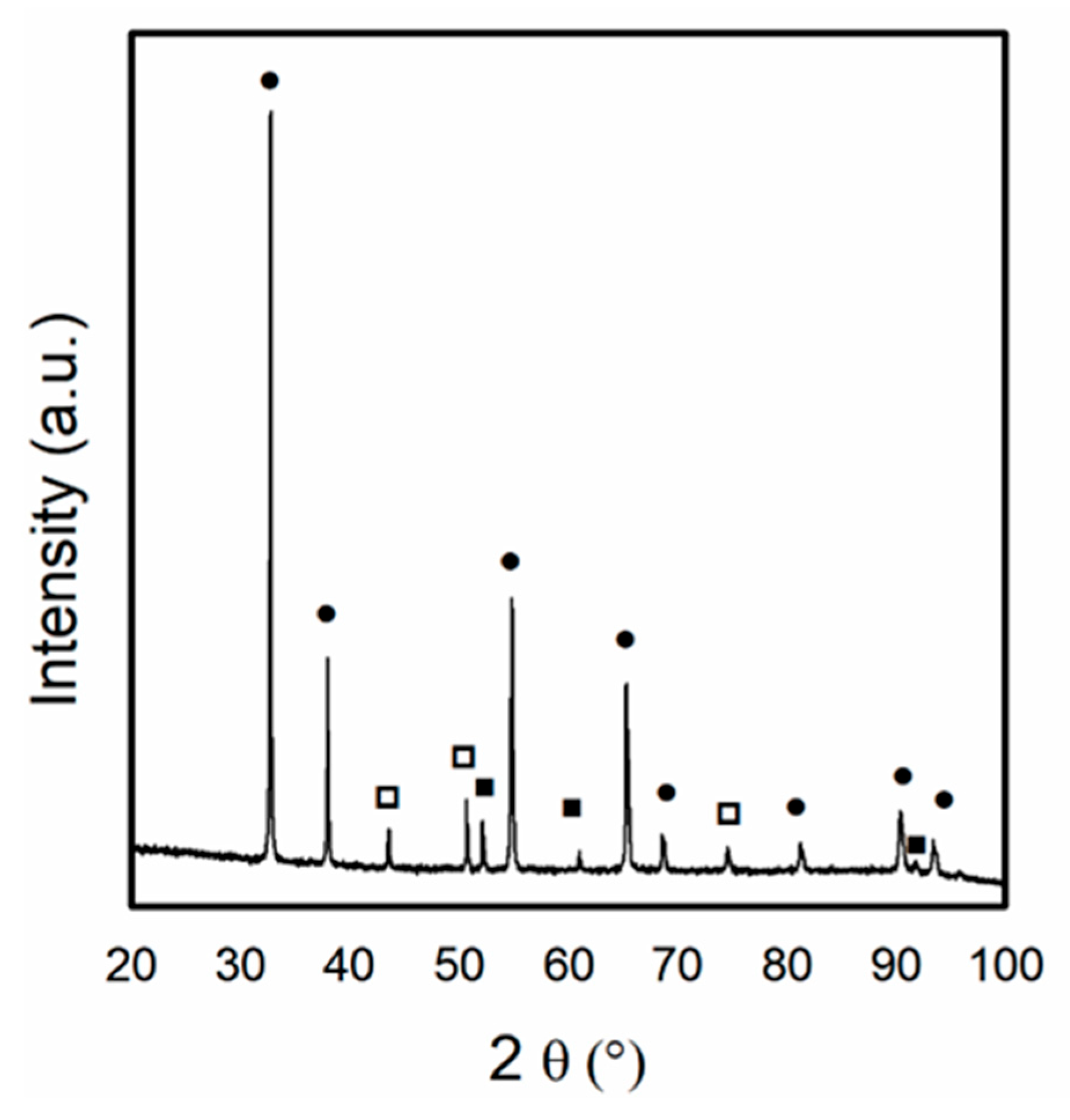
3.1. Catalyst Characterization
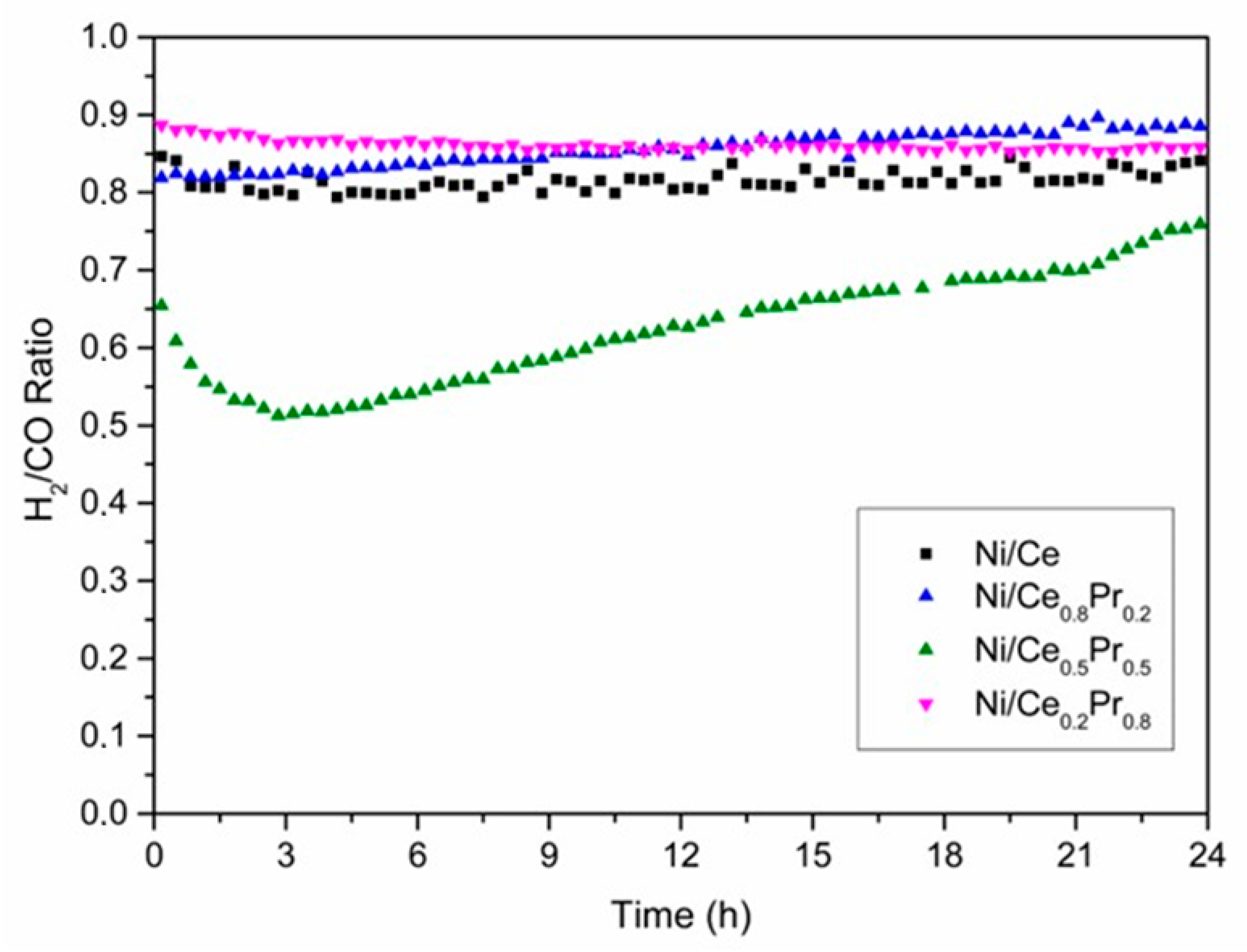
3.2. CO2 Reforming of Methane
3.3. Characterization of Used Catalysts
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| DIR-SOFC | Direct Internal Reforming Solid Oxide Fuel Cell |
| DRM | Dry Reforming of Methane |
| XRD | X-ray diffraction |
| SEM | Scanning Electron Microscopy |
| TPR | Temperature-programmed Reduction |
| TEM | Transmission Electron Microscopy |
| TPO | Temperature-programmed Oxidation |
References
- Veiga, S.; Romero, M.; Faccio, R.; Segobia, D.; Apesteguía, C.; Laura Pérez, A.; Dante Brondino, C.; Bussi, J. Biogas Dry Reforming over Ni-La-Ti Catalysts for Synthesis Gas Production: Effects of Preparation Method and Biogas Composition. Fuel 2023, 346, 128300. [Google Scholar] [CrossRef]
- Escudero, M.J.; Valero, C.; Cauqui, M.Á.; Goma, D.; Yeste, M.P. Ni-Ce-ZrO2 System as Anode Material for Direct Internal Reforming Biogas Solid Oxide Fuel Cells. Fuel 2022, 322, 124247. [Google Scholar] [CrossRef]
- Rayner, A.J.; Briggs, J.; Tremback, R.; Clemmer, R.M.C. Design of an Organic Waste Power Plant Coupling Anaerobic Digestion and Solid Oxide Fuel Cell Technologies. Renew. Sustain. Energy Rev. 2017, 71, 563–571. [Google Scholar] [CrossRef]
- Reeping, K.W.; Bohn, J.A.; Walker, R.A. Chlorine-Induced Degradation in SOFCs Operating with Biogas. Sustain. Energy Fuels 2017, 1, 1320–1328. [Google Scholar] [CrossRef]
- Saadabadi, S.A.; Thallam Thattai, A.; Fan, L.; Lindeboom, R.E.F.; Spanjers, H.; Aravind, P.V. Solid Oxide Fuel Cells Fuelled with Biogas: Potential and Constraints. Renew. Energy 2019, 134, 194–214. [Google Scholar] [CrossRef]
- Papurello, D.; Silvestri, S.; Modena, S. Biogas Trace Compounds Impact on High-Temperature Fuel Cells Short Stack Performance. Int. J. Hydrogen Energy 2021, 46, 8792–8801. [Google Scholar] [CrossRef]
- Bochentyn, B.; Chlipała, M.; Gazda, M.; Wang, S.-F.; Jasiński, P. Copper and Cobalt Co-Doped Ceria as an Anode Catalyst for DIR-SOFCs Fueled by Biogas. Solid State Ion. 2019, 330, 47–53. [Google Scholar] [CrossRef]
- Tsipis, E.V.; Matveev, D.V.; Sharafutdinov, A.U.; Yalovenko, D.V.; Samoilov, A.V.; Fedotov, Y.S.; Dyakina, M.S.; Zhigacheva, D.V.; Agarkov, D.A.; Bredikhin, S.I.; et al. Performance of SOFCs Using Model Waste Gases: A Case Study. Fuel 2024, 358, 130129. [Google Scholar] [CrossRef]
- Illathukandy, B.; Saadabadi, S.A.; Kuo, P.-C.; Wasajja, H.; Lindeboom, R.E.F.; Vijay, V.K.; Aravind, P.V. Solid Oxide Fuel Cells (SOFCs) Fed with Biogas Containing Hydrogen Chloride Traces: Impact on Direct Internal Reforming and Electrochemical Performance. Electrochim. Acta 2022, 433, 141198. [Google Scholar] [CrossRef]
- Singh, M.; Zappa, D.; Comini, E. Solid Oxide Fuel Cell: Decade of Progress, Future Perspectives and Challenges. Int. J. Hydrogen Energy 2021, 46, 27643–27674. [Google Scholar] [CrossRef]
- da Silva, F.S.; de Souza, T.M. Novel Materials for Solid Oxide Fuel Cell Technologies: A Literature Review. Int. J. Hydrogen Energy 2017, 42, 26020–26036. [Google Scholar] [CrossRef]
- Piroonlerkgul, P.; Laosiripojana, N.; Adesina, A.A.; Assabumrungrat, S. Performance of Biogas-Fed Solid Oxide Fuel Cell Systems Integrated with Membrane Module for CO2 Removal. Chem. Eng. Process. Process Intensif. 2009, 48, 672–682. [Google Scholar] [CrossRef]
- Chen, T.; Wang, W.G.; Miao, H.; Li, T.; Xu, C. Evaluation of Carbon Deposition Behavior on the Nickel/Yttrium-Stabilized Zirconia Anode-Supported Fuel Cell Fueled with Simulated Syngas. J. Power Sources 2011, 196, 2461–2468. [Google Scholar] [CrossRef]
- Li, Y.; Pang, Y.; Tu, H.; Torrigino, F.; Biollaz, S.M.A.; Li, Z.; Huang, Y.; Yin, X.; Grimm, F.; Karl, J. Impact of Syngas from Biomass Gasification on Solid Oxide Fuel Cells: A Review Study for the Energy Transition. Energy Convers. Manag. 2021, 250, 114894. [Google Scholar] [CrossRef]
- da Silva, A.A.A.; Steil, M.C.; Tabuti, F.N.; Rabelo-Neto, R.C.; Noronha, F.B.; Mattos, L.V.; Fonseca, F.C. The Role of the Ceria Dopant on Ni / Doped-Ceria Anodic Layer Cermets for Direct Ethanol Solid Oxide Fuel Cell. Int. J. Hydrogen Energy 2021, 46, 4309–4328. [Google Scholar] [CrossRef]
- Faria, E.C.; Neto, R.C.R.; Colman, R.C.; Noronha, F.B. Hydrogen Production through CO2 Reforming of Methane over Ni/CeZrO2/Al2O3 Catalysts. Catal. Today 2014, 228, 138–144. [Google Scholar] [CrossRef]
- Rabelo-Neto, R.C.; Sales, H.B.E.; Inocêncio, C.V.M.; Varga, E.; Oszko, A.; Erdohelyi, A.; Noronha, F.B.; Mattos, L.V. CO2 Reforming of Methane over Supported LaNiO3 Perovskite-Type Oxides. Appl. Catal. B 2018, 221, 349–361. [Google Scholar] [CrossRef]
- da Silva, A.A.A.; Bion, N.; Epron, F.; Baraka, S.; Fonseca, F.C.; Rabelo-Neto, R.C.; Mattos, L.V.; Noronha, F.B. Effect of the Type of Ceria Dopant on the Performance of Ni/CeO2 SOFC Anode for Ethanol Internal Reforming. Appl. Catal. B 2017, 206, 626–641. [Google Scholar] [CrossRef]
- Adjah-Tetteh, C.; Wang, Y.; Niaz, A.K.; Jia, Z.; Zhou, X.-D. Understanding the Physical Properties of Pr-Gd Co-Doped Ceria and Its Role as the Interlayer in Solid Oxide Cells. ECS Meet. Abstr. 2023, MA2023-01, 252. [Google Scholar] [CrossRef]
- Wang, H.; Chroneos, A.; Schwingenschlögl, U. Impact of Doping on the Ionic Conductivity of Ceria: A Comprehensive Model. J. Chem. Phys. 2013, 138, 224705. [Google Scholar] [CrossRef]
- Grope, B.O.H.; Zacherle, T.; Nakayama, M.; Martin, M. Oxygen Ion Conductivity of Doped Ceria: A Kinetic Monte Carlo Study. Solid State Ion. 2012, 225, 476–483. [Google Scholar] [CrossRef]
- Lyu, Z.; Wang, Y.; Zhang, Y.; Han, M. Solid Oxide Fuel Cells Fueled by Simulated Biogas: Comparison of Anode Modification by Infiltration and Reforming Catalytic Layer. Chem. Eng. J. 2020, 393, 124755. [Google Scholar] [CrossRef]
- Błaszczak, P.; Łapiński, M.; Wang, S.-F.; Jasiński, P.; Bochentyn, B. Exsolution of Ni Nanoparticles on the Surface of Cerium and Nickel Co-Doped Lanthanum Strontium Titanate as a New Anodic Layer for DIR-SOFC. Anti-Coking Potential and H2S Poisoning Resistance of the Prepared Material. Int. J. Hydrogen Energy 2020, 45, 29186–29200. [Google Scholar] [CrossRef]
- da Fonseca, R.O.; da Silva, A.A.A.; Signorelli, M.R.M.; Rabelo-Neto, R.C.; Noronha, F.B.; Simões, R.C.C.; Mattos, L.V. Nickel/Doped Ceria Solid Oxide Fuel Cell Anodes for Dry Reforming of Methane. J. Braz. Chem. Soc. 2014, 25, 235–2363. [Google Scholar] [CrossRef]
- Modafferi, V.; Panzera, G.; Baglio, V.; Frusteri, F.; Antonucci, P.L. Propane Reforming on Ni–Ru/GDC Catalyst: H2 Production for IT-SOFCs under SR and ATR Conditions. Appl. Catal. A Gen. 2008, 334, 1–9. [Google Scholar] [CrossRef]
- Vasiliades, M.A.; Makri, M.M.; Djinović, P.; Erjavec, B.; Pintar, A.; Efstathiou, A.M. Dry Reforming of Methane over 5 wt% Ni/Ce1–XPrxO2–δ Catalysts: Performance and Characterisation of Active and Inactive Carbon by Transient Isotopic Techniques. Appl. Catal. B 2016, 197, 168–183. [Google Scholar] [CrossRef]
- Dogdibegovic, E.; Wang, Y.; Wright, C.J.; Zhou, X.-D. Origin for Retained Activity in Pr2NiO4 While Undergoing Substantial Phase Transformation in a Long-Term Solid Oxide Cell Operation. Nano Energy 2022, 103, 107684. [Google Scholar] [CrossRef]
- Krishna, K.; Bueno-López, A.; Makkee, M.; Moulijn, J.A. Potential Rare-Earth Modified CeO2 Catalysts for Soot Oxidation. Appl. Catal. B 2007, 75, 210–220. [Google Scholar] [CrossRef]
- Krishna, K.; Bueno-López, A.; Makkee, M.; Moulijn, J.A. Potential Rare-Earth Modified CeO2 Catalysts for Soot Oxidation Part II: Characterisation and Catalytic Activity with NO + O2. Appl. Catal. B 2007, 75, 201–209. [Google Scholar] [CrossRef]
- Mahato, N.; Banerjee, A.; Gupta, A.; Omar, S.; Balani, K. Progress in Material Selection for Solid Oxide Fuel Cell Technology: A Review. Prog. Mater. Sci. 2015, 72, 141–337. [Google Scholar]
- Stagg-Williams, S.M.; Noronha, F.B.; Fendley, G.; Resasco, D.E. CO2 Reforming of CH4 over Pt/ZrO2 Catalysts Promoted with La and Ce Oxides. J. Catal. 2000, 194, 240–249. [Google Scholar] [CrossRef]
- Ahn, K.; Yoo, D.S.; Prasad, D.H.; Lee, H.-W.; Chung, Y.-C.; Lee, J.-H. Role of Multivalent Pr in the Formation and Migration of Oxygen Vacancy in Pr-Doped Ceria: Experimental and First-Principles Investigations. Chem. Mater. 2012, 24, 4261–4267. [Google Scholar] [CrossRef]
- Chiba, R.; Taguchi, H.; Komatsu, T.; Orui, H.; Nozawa, K.; Arai, H. High Temperature Properties of Ce1–XPrxO2–δ as an Active Layer Material for SOFC Cathodes. Solid State Ion. 2011, 197, 42–48. [Google Scholar] [CrossRef]
- Fan, M.S.; Abdullah, A.Z.; Bhatia, S. Catalytic Technology for Carbon Dioxide Reforming of Methane to Synthesis Gas. ChemCatChem 2009, 1, 192–208. [Google Scholar] [CrossRef]
- Wang, S.; Kato, T.; Nagata, S.; Honda, T.; Kaneko, T.; Iwashita, N.; Dokiya, M. Ni/Ceria Cermet as Anode of Reduced-Temperature Solid Oxide Fuel Cells. J. Electrochem. Soc. 2002, 149, A927. [Google Scholar] [CrossRef]
- Bekheet, M.F.; Grünbacher, M.; Schlicker, L.; Gili, A.; Doran, A.; Epping, J.D.; Gurlo, A.; Klötzer, B.; Penner, S. On the Structural Stability of Crystalline Ceria Phases in Undoped and Acceptor-Doped Ceria Materials under in Situ Reduction Conditions. CrystEngComm 2019, 21, 145–154. [Google Scholar] [CrossRef] [PubMed]
- Kümmerle, E.A.; Heger, G. The Structures of C–Ce2O3+δ, Ce7O12, and Ce11O20. J. Solid State Chem. 1999, 147, 485–500. [Google Scholar] [CrossRef]
- da Fonseca, R.O.; Ponseggi, A.R.; Rabelo-Neto, R.C.; Simões, R.C.C.; Mattos, L.V.; Noronha, F.B. Controlling Carbon Formation over Ni/CeO2 Catalyst for Dry Reforming of CH4 by Tuning Ni Crystallite Size and Oxygen Vacancies of the Support. J. CO2 Util. 2022, 57, 101880. [Google Scholar] [CrossRef]
- Tarutin, A.; Lyagaeva, J.; Farlenkov, A.; Plaksin, S.; Vdovin, G.; Demin, A.; Medvedev, D. A Reversible Protonic Ceramic Cell with Symmetrically Designed Pr2NiO4+δ-Based Electrodes: Fabrication and Electrochemical Features. Materials 2018, 12, 118. [Google Scholar] [CrossRef]
- Osinkin, D.A. Precursor of Pr2NiO4+δ as a Highly Effective Catalyst for the Simultaneous Promotion of Oxygen Reduction and Hydrogen Oxidation Reactions in Solid Oxide Electrochemical Devices. Int. J. Hydrogen Energy 2021, 46, 24546–24554. [Google Scholar] [CrossRef]
- Dong, W.S.; Roh, H.S.; Jun, K.W.; Park, S.E.; Oh, Y.S. Methane Reforming over Ni/Ce-ZrO2 Catalysts: Effect of Nickel Content. Appl. Catal. A Gen. 2002, 226, 63–72. [Google Scholar] [CrossRef]
- Augusto, B.L.; Costa, L.O.O.; Noronha, F.B.; Colman, R.C.; Mattos, L.V. Ethanol Reforming over Ni/CeGd Catalysts with Low Ni Content. Int. J. Hydrogen Energy 2012, 37, 12258–12270. [Google Scholar] [CrossRef]
- Yao, H.C.; Yao, Y.F.Y. Ceria in Automotive Exhaust Catalysts. I. Oxygen Storage. J. Catal. 1984, 86, 254–265. [Google Scholar] [CrossRef]
- Ahn, K.; He, H.; Vohs, J.M.; Gorte, R.J. Enhanced Thermal Stability of SOFC Anodes Made with CeO2-ZrO2 Solutions. Electrochem. Solid-State Lett. 2005, 8, A414. [Google Scholar] [CrossRef]
- Zoellner, M.H.; Niu, G.; Jhang, J.-H.; Schaefer, A.; Zaumseil, P.; Bäumer, M.; Schroeder, T. Temperature-Dependent Reduction of Epitaxial Ce1–xPrxO2–δ (x = 0–1) Thin Films on Si(111): A Combined Temperature-Programmed Desorption, X-Ray Diffraction, X-Ray Photoelectron Spectroscopy, and Raman Study. J. Phys. Chem. C 2013, 117, 24851–24857. [Google Scholar] [CrossRef]
- Muñoz, M.A.; Calvino, J.J.; Rodríguez-Izquierdo, J.M.; Blanco, G.; Arias, D.C.; Pérez-Omil, J.A.; Hernández-Garrido, J.C.; González-Leal, J.M.; Cauqui, M.A.; Yeste, M.P. Highly Stable Ceria-Zirconia-Yttria Supported Ni Catalysts for Syngas Production by CO2 Reforming of Methane. Appl. Surf. Sci. 2017, 426, 864–873. [Google Scholar] [CrossRef]
- Marinho, A.L.A.; Toniolo, F.S.; Noronha, F.B.; Epron, F.; Duprez, D.; Bion, N. Highly Active and Stable Ni Dispersed on Mesoporous CeO2-Al2O3 Catalysts for Production of Syngas by Dry Reforming of Methane. Appl. Catal. B 2021, 281, 119459. [Google Scholar] [CrossRef]
- Marinho, A.L.A.; Rabelo-Neto, R.C.; Epron, F.; Bion, N.; Toniolo, F.S.; Noronha, F.B. Embedded Ni Nanoparticles in CeZrO2 as Stable Catalyst for Dry Reforming of Methane. Appl. Catal. B 2020, 268, 118387. [Google Scholar] [CrossRef]
- Luisetto, I.; Tuti, S.; Romano, C.; Boaro, M.; Di Bartolomeo, E.; Kesavan, J.K.; Kumar, S.S.; Selvakumar, K. Dry Reforming of Methane over Ni Supported on Doped CeO2: New Insight on the Role of Dopants for CO2 Activation. J. CO2 Util. 2019, 30, 63–78. [Google Scholar] [CrossRef]
- Luisetto, I.; Tuti, S.; Battocchio, C.; Lo Mastro, S.; Sodo, A. Ni/CeO2–Al2O3 Catalysts for the Dry Reforming of Methane: The Effect of CeAlO3 Content and Nickel Crystallite Size on Catalytic Activity and Coke Resistance. Appl. Catal. A Gen. 2015, 500, 12–22. [Google Scholar] [CrossRef]
- Pu, Z.; Liu, Q.; Chen, C.; Wang, F. Promoting Surface Lattice Oxygen and Oxygen Vacancy of CeO2 for Photothermal Methane Dry Reforming over Ni/CeO2 Catalysts. Chem. Eng. J. 2024, 497, 154861. [Google Scholar] [CrossRef]
- Gonzalez-Delacruz, V.M.; Ternero, F.; Pereñíguez, R.; Caballero, A.; Holgado, J.P. Study of Nanostructured Ni/CeO2 Catalysts Prepared by Combustion Synthesis in Dry Reforming of Methane. Appl. Catal. A Gen. 2010, 384, 1–9. [Google Scholar] [CrossRef]
- Cozendey da Silva, H.N.; Prata, D.M.; Alves Lima, G.B.; Zotes, L.P.; Mattos, L.V. A Techno-Economic Evaluation of the Energy Generation by Proton Exchange Membrane Fuel Cell Using Biogas Reforming. J. Clean. Prod. 2018, 200, 598–608. [Google Scholar] [CrossRef]
- Takanabe, K.; Nagaoka, K.; Nariai, K.; Aika, K. Influence of Reduction Temperature on the Catalytic Behavior of Co/TiO2 Catalysts for CH4/CO2 Reforming and Its Relation with Titania Bulk Crystal Structure. J. Catal. 2005, 230, 75–85. [Google Scholar] [CrossRef]
- Zhdanov, V.P.; Kasemo, B. Cabrera–Mott Kinetics of Oxidation of Nm-Sized Metal Particles. Chem. Phys. Lett. 2008, 452, 285–288. [Google Scholar] [CrossRef]
- Chernavskii, P.A.; Pankina, G.V.; Chernavskii, A.P.; Peskov, N.V.; Afanasiev, P.; Perov, N.S.; Tennov, V.A. In Situ Magnetic Study of the Low-Temperature Oxidation of Carbon-Supported Cobalt Nanoparticles. J. Phys. Chem. C 2007, 111, 5576–5581. [Google Scholar] [CrossRef]
- Wang, F.; Xu, L.; Zhang, J.; Zhao, Y.; Li, H.; Li, H.X.; Wu, K.; Xu, G.Q.; Chen, W. Tuning the Metal-Support Interaction in Catalysts for Highly Efficient Methane Dry Reforming Reaction. Appl. Catal. B 2016, 180, 511–520. [Google Scholar] [CrossRef]
- Rezaei, M.; Alavi, S.M.; Sahebdelfar, S.; Yan, Z.-F. Syngas Production by Methane Reforming with Carbon Dioxide on Noble Metal Catalysts. J. Nat. Gas Chem. 2006, 15, 327–334. [Google Scholar] [CrossRef]
- Damaskinos, C.M.; Zavašnik, J.; Djinović, P.; Efstathiou, A.M. Dry Reforming of Methane over Ni/Ce0.8Ti0.2O2–δ: The Effect of Ni Particle Size on the Carbon Pathways Studied by Transient and Isotopic Techniques. Appl. Catal. B 2021, 296, 120321. [Google Scholar] [CrossRef]
- Liang, W.; Yan, H.; Chen, C.; Lin, D.; Tan, K.; Feng, X.; Liu, Y.; Chen, X.; Yang, C.; Shan, H. Revealing the Effect of Nickel Particle Size on Carbon Formation Type in the Methane Decomposition Reaction. Catalysts 2020, 10, 890. [Google Scholar] [CrossRef]
- Rostrup-Nielsen, J.; Trimm, D.L. Mechanisms of Carbon Formation on Nickel-Containing Catalysts. J. Catal. 1977, 48, 155–165. [Google Scholar] [CrossRef]
- Trimm, D.L. Catalysts for the Control of Coking during Steam Reforming. Catal. Today 1999, 49, 3–10. [Google Scholar] [CrossRef]
- Bengaard, H.S.; Nørskov, J.K.; Sehested, J.; Clausen, B.S.; Nielsen, L.P.; Molenbroek, A.M.; Rostrup-Nielsen, J.R. Steam Reforming and Graphite Formation on Ni Catalysts. J. Catal. 2002, 209, 365–384. [Google Scholar] [CrossRef]
- Li, D.; Nakagawa, Y.; Tomishige, K. Methane Reforming to Synthesis Gas over Ni Catalysts Modified with Noble Metals. Appl. Catal. A Gen. 2011, 408, 1–24. [Google Scholar] [CrossRef]
- Chen, D.; Christensen, K.; Ochoafernandez, E.; Yu, Z.; Totdal, B.; Latorre, N.; Monzon, A.; Holmen, A. Synthesis of Carbon Nanofibers: Effects of Ni Crystal Size during Methane Decomposition. J. Catal. 2005, 229, 82–96. [Google Scholar] [CrossRef]
- Toebes, M. Impact of the Structure and Reactivity of Nickel Particles on the Catalytic Growth of Carbon Nanofibers. Catal. Today 2002, 76, 33–42. [Google Scholar] [CrossRef]
- Minh, N.Q. Ceramic Fuel Cells. J. Am. Ceram. Soc. 1993, 76, 563–588. [Google Scholar] [CrossRef]
- Vogt, C.; Kranenborg, J.; Monai, M.; Weckhuysen, B.M. Structure Sensitivity in Steam and Dry Methane Reforming over Nickel: Activity and Carbon Formation. ACS Catal. 2020, 10, 1428–1438. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst | Lattice Parameter of Calcined Samples (Å) 1 | CeO2 Crystallite Sizes (nm) 2 | NiO Crystallite Sizes (nm) 2 | Ni0 Crystallite Sizes (nm) 3 |
|---|---|---|---|---|
| Ni/Ce | 5.4137 | 95 | 81 | 116 |
| Ni/Ce0.8Pr0.2 | 5.4155 | 81 | 90 | 98 |
| Ni/Ce0.5Pr0.5 | 5.4095 | 116 | 78 | 73 |
| Ni/Ce0.2Pr0.8 | 5.4050 | 107 | 88 | 63 |
| Catalyst | Reduction Degree at Low Temperature Region (T < 750 °C) (%) | Reduction Degree at Low Temperature Region (T < 750 °C) (%) | Total H2 Consumption (mol/g Catalyst) |
|---|---|---|---|
| Ni/Ce | 99 | 32 | 2.36 × 10−3 |
| Ni/Ce0.8Pr0.2 | 131 | 12 | 2.37 × 10−3 |
| Ni/Ce0.5Pr0.5 | 162 | 13 | 2.91 × 10−3 |
| Ni/Ce0.2Pr0.8 | 231 | 12 | 2.77 × 10−3 |
| Catalyst | Tcalc. (°C) a | Treaction (°C) b | Ni0 Crystallite Size (nm) | Initial XCH4 (%) c | mgC/ (gcat·h) | Reference |
|---|---|---|---|---|---|---|
| 5%Ni/CeO2 | 400 | 800 | 10 | ~66 | 1.6 | [38] |
| 10%Ni/CeO2 | 400 | 800 | 20 | ~63 | 20.9 | [38] |
| 10%Ni/CeO2 | 600 | 800 | 28 | ~71 | 10.6 | [38] |
| 10%Ni/CeO2 | 800 | 800 | 46 | ~61 | 5.9 | [38] |
| 5%Ni/CeO2/YSZ | 500 | 750 | 15 | ~100 | 2.9 | [46] |
| 10%Ni-CeAl | 800 | 800 | 11 | ~70 | 2.0 | [47] |
| 10%Ni@CeO2 | 400 | 800 | 19 | ~60 | 1.6 | [48] |
| 10%Ni/CeO2 | 400 | 800 | 33 | ~60 | 9.7 | [48] |
| 10%Ni/Ce0.2Pr0.8 | 1200 | 800 | 63 | 72 | 0.36 | this work |
| 10%Ni/Ce0.5Pr0.5 | 1200 | 800 | 73 | 46 | 0.20 | this work |
| 10%Ni/Ce0.8Pr0.2 | 1200 | 800 | 98 | 56 | 0.29 | this work |
| 10%Ni/Ce | 1200 | 800 | 116 | 58 | 0.33 | this work |
| NiO | 1200 | 800 | 132 | 37 | 0.0 | [38] |
| 5%Ni/CeO2 | 750 | 750 | 31 | 80 | 14.8 | [26] |
| 5%Ni/Ce0.8Pr0.2O2 | 750 | 750 | 34 | 75 | 12.0 | [26] |
| 5%Ni/Ce0.5Pr0.52 | 750 | 750 | 30 | 70 | 0.4 | [26] |
| 5&Ni/Ce0.2Pr0.8O2 | 750 | 750 | 36 | 50 | 0.06 | [26] |
| 18%Ni/CeNb | 800 | 800 | 32 | 66 | 6.5 | [24] |
| 18%Ni/CeZr | 800 | 800 | 24 | 74 | 9.68 | [24] |
| 18%Ni/CePr | 800 | 800 | 33 | 66 | 0.42 | [24] |
| 5%Ni/CeO2 | 750 | 800 | 24 | ~55 | 4.8 | [49] |
| 5%Ni/Zr-DC (Zr-doped ceria) | 750 | 800 | 9.2 | ~55 | 3.2 | [49] |
| 5%Ni/Sm-DC (Sm-doped ceria) | 750 | 800 | 19 | ~38 | 0.6 | [49] |
| 5%Ni/La-DC (La-doped ceria) | 750 | 800 | 21 | ~45 | 0.4 | [49] |
| 10%Ni/Ce/Al | 800 | 800 | 4.6 | ~40 | n.d. | [16] |
| 10%Ni/Ce0.75Zr0.25O2/Al | 800 | 800 | 6.6 | ~70 | n.d. | [16] |
| 10%Ni/Ce0.50Zr0.50O2/Al | 800 | 800 | 5.2 | ~70 | [16] | |
| 10%Ni/20%CeO2-Al2O3 Co-precipitation method | 600 | 800 | 22.6 | ~75 | 200 | [50] |
| 10%Ni/20%CeO2-Al2O3 Wet impregnation method | 600 | 800 | 11.1 | ~55 | 138 | [50] |
| 10%Ni/20%CeO2-Al2O3 Sol-gel method | 600 | 800 | 9.5 | ~55 | 50 | [50] |
| 10%Ni/20%CeO2-Al2O3 Citric acid method | 600 | 800 | 5.8 | ~70 | 12.5 | [50] |
| 20%Ni/CeO2-NR (nanorods morphology) | 500 | 700 | 20.5 | ~45 | 0.54 | [51] |
| 5%Ni/CeO2-NR (nanorods morphology) | 500 | 700 | 6.8 | ~70 | 0.21 | [51] |
| 3%Ni/CeO2-NR (nanorods morphology) | 500 | 700 | 6.2 | ~55 | 0.50 | [51] |
| 26%Ni/CeO2-ac25 Combustion method (red./oxi. ratio = 2.5) | 750 | 750 | 28 | ~50 | n.d. | [52] |
| 13%Ni/CeO2-ac25 Combustion method (red./oxid. ratio = 2.5) | 750 | 750 | 15 | ~41 | n.d. | [52] |
| 7%Ni/CeO2-ac25 Combustion method (red./oxid. ratio = 2.5) | 750 | 750 | 16 | ~35 | n.d. | [52] |
| 13%Ni/CeO2-ac10 Combustion method (red./oxid. ratio = 1.0) | 750 | 750 | 18 | ~65 | n.d. | [52] |
| 13%Ni/CeO2-ac15 Combustion method (red./oxid. ratio = 1.5) | 750 | 750 | 12 | ~90 | n.d. | [52] |
| Catalyst | Rate of Carbon Formation mgC/(gcat·h) | Rate of Carbon Formation (mgC·gcat−1·h−1·molCH4conv−1) |
|---|---|---|
| Ni/Ce | 0.33 | 0.23 |
| Ni/Ce0.8Pr0.2 | 0.29 | 0.19 |
| Ni/Ce0.5Pr0.5 | 0.20 | 0.15 |
| Ni/Ce0.2Pr0.8 | 0.36 | 0.19 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ponseggi, A.R.; Guimarães, A.d.C.P.; da Fonseca, R.O.; Rabelo-Neto, R.C.; Xing, Y.; Silva, A.A.A.; Noronha, F.B.; Mattos, L.V. Ni Supported on Pr-Doped Ceria as Catalysts for Dry Reforming of Methane. Processes 2025, 13, 2119. https://doi.org/10.3390/pr13072119
Ponseggi AR, Guimarães AdCP, da Fonseca RO, Rabelo-Neto RC, Xing Y, Silva AAA, Noronha FB, Mattos LV. Ni Supported on Pr-Doped Ceria as Catalysts for Dry Reforming of Methane. Processes. 2025; 13(7):2119. https://doi.org/10.3390/pr13072119
Chicago/Turabian StylePonseggi, Antonella R., Amanda de C. P. Guimarães, Renata O. da Fonseca, Raimundo C. Rabelo-Neto, Yutao Xing, Andressa A. A. Silva, Fábio B. Noronha, and Lisiane V. Mattos. 2025. "Ni Supported on Pr-Doped Ceria as Catalysts for Dry Reforming of Methane" Processes 13, no. 7: 2119. https://doi.org/10.3390/pr13072119
APA StylePonseggi, A. R., Guimarães, A. d. C. P., da Fonseca, R. O., Rabelo-Neto, R. C., Xing, Y., Silva, A. A. A., Noronha, F. B., & Mattos, L. V. (2025). Ni Supported on Pr-Doped Ceria as Catalysts for Dry Reforming of Methane. Processes, 13(7), 2119. https://doi.org/10.3390/pr13072119