Abstract
The combustion of biomass fuels releases alkali metals, which induce severe catalyst deactivation due to alkali metal (K) poisoning in low-temperature ammonia selective catalytic reduction (NH3-SCR) systems. To address this issue, this study developed a series of heteropoly acid (HPA)-modified Ce/AC catalysts prepared via incipient wetness impregnation. The low-temperature NH3-SCR performance (80–200 °C) of these catalysts was systematically evaluated, with particular emphasis on their denitrification activity and K-poisoning resistance. The silicotungstic-acid (TSiA)-modified Ce/Ac (TSiA-Ce/AC) catalyst showed an improvement (>20%) in NO conversion activity under the K poisoning condition. The superior K-poisoning resistance of the TSiA-Ce/AC catalyst was attributed to the high density of Brønsted acidic sites and the strong K binding affinity of TSiA, which together protected active sites and preserved the standard SCR reaction pathway under K contaminations. This study proposes a novel strategy for enhancing catalyst K resistance in low-temperature NH3-SCR systems.
1. Introduction
As a sustainable energy alternative, biomass offers multiple environmental advantages, including minimal pollution emissions and reduced reliance on conventional fossil fuels [1], aligning with global strategic objectives for sustainable energy development. Although biomass combustion predominantly yields CO2 and H2O under complete combustion conditions, the growing deployment of biomass-fueled power plants still releases atmospheric pollutants, such as NOx; consequently, considerable attention has been directed toward flue gas treatment and NOx emission reduction technologies. Among them, selective catalytic reduction (SCR) represents the predominant technology for NOx abatement in industrial applications [2].
The primary reaction in low-temperature NH3-SCR involves the reduction of NO by NH3 as the reductant, yielding harmless N2 and H2O, thereby enabling NOx removal. Macroscopically, under a stoichiometric NH3:NO = 1:1 condition, Equation (1) represents the standard NH3-SCR reaction, while Equation (2) constitutes a competing pathway arising from NH3 oxidation and non-selective catalytic reduction (NSCR) [3]:
However, practical flue gas streams exhibit greater compositional complexity. Biomass contains elevated levels of alkaline substances (such as K, Na, Ca, Cl, etc.) [4]. During the combustion of substantial quantities of biomass, chlorinated alkali is released via vaporization; this gaseous substance may subsequently participate in chemical reactions or physical transformations and can condense to form fly ash or aerosols in the flue gas [5]. Furthermore, alkali metal aerosols may interact with one another or with other ash components and ultimately exist as solid-phase oxides or salts [6]. Alkali metals present in exhaust gases adversely affect the catalytic activity of SCR denitrification catalysts [7].
Currently, vanadium–titanium-based catalysts (e.g., V2O5-WO3/TiO2 and V2O5-MoO3/TiO2) are the most widely adopted commercial SCR catalysts. However, their application is limited by inherent disadvantages, including the requirement for high operating temperatures and high susceptibility to deactivation by alkali/alkaline earth metals present in flue gas [8]. The flue gas temperature following dust removal and desulfurization processes typically falls below 200 °C. However, the optimal operating temperature window for vanadium–titanium denitrification catalysts ranges from 300–400 °C. This temperature mismatch requires the integration of a preheating system upstream of the SCR unit to maintain catalytic conversion efficiency, thereby leading to increased operational costs. Consequently, substantial research efforts have been devoted to developing low-temperature SCR catalysts (operating range: 100–200 °C) suitable for the downstream dust removal and desulfurization units [9].
Alkali metals represent the most detrimental class of poisoning elements for denitrification catalysts, including severe deactivation [10] through similar poisoning mechanisms, though with varying intensities. Potassium (K) demonstrates the most severe deleterious effects among these elements [11]. The progressive accumulation of K on the catalyst surface leads to gradual degradation of both surface acidity and redox properties, resulting in significant deterioration of catalytic activity [12]. Therefore, enhancing the potassium resistance of low-temperature NH3-SCR catalysts is key to addressing the rapid deactivation in practical flue gas conditions.
Heteropoly acids (HPAs), as environmentally friendly solid acids with metal–oxygen bridged structures, exhibit unique characteristics, including strong acidity and pseudo-liquid phase behavior [13]. As a result of their strong redox properties and high stability, HPAs show excellent catalytic activity. Therefore, HPA catalysts have been the subject of extensive basic and applied catalytic research [14,15] and exhibit superior Brønsted acidity compared to conventional solid acids, such as metal oxides and zeolites [16]. Putluru et al. [17] demonstrated that the SCR activity and alkali deactivation resistance of HPA-promoted V2O5/TiO2 catalysts was found to be much higher than for WO3-promoted catalysts.
Among Keggin-type HPAs, the most prevalent and stable structures include silicotungstic acid (H4[SiW12O40], TSiA), phosphotungstic acid (H3[PW12O40], TPA), and phosphomolybdic acid (H3[PMo12O40], MPA) [18,19]. Dong et al. [20] incorporated MPA uniformly and dispersively to enhance both the N2 selectivity and low-temperature SCR activity of Mn-based oxide catalysts concurrently, thereby regulating the oxidation capability of the catalysts and improving the NH3 adsorption-storage capacity. Geng et al. [21] investigated the catalytic performance of a series of heteropoly acid-grafted Fe2O3 catalysts (HPA/Fe2O3) in NH3-SCR and found that TSiA/Fe2O3 and TPA/Fe2O3 exhibited superior SCR activity below 250 °C compared to MPA/Fe2O3. Xue et al. [22] demonstrated that incorporating TPA markedly enhanced the alkali metal resistance of MnOx catalysts at temperatures below 180 °C. However, current research predominantly focuses on the enhancement effect of coexisting TPA and metal oxides on catalytic activity [23,24], whereas investigations specifically addressing the low-temperature activity and alkali metal resistance of TSiA-modified NH3-SCR catalysts remain scarce.
In this study, we developed an acidic-site-enriched (ASE) strategy by co-impregnating heteropoly acids (HPAs) with cerium onto an AC support using the incipient wetness method. We tested three HPAs (MPA, TPA, and TSiA) and systematically investigated the role of these HPAs in enhancing the K resistance and catalytic activity of Ce-loaded AC catalysts in low-temperature NH3-SCR (80–200 °C). Subsequently, the optimal TSiA-modified catalyst was selected for further investigation of potassium resistance performance. The TSiA-modified Ce/AC catalyst showed an enhancement of 20% NO conversion efficiency under the K poisoning conditions (maximum 100 μmol/g). The K resistance mechanism was systematically analyzed through catalyst characterizations and kinetic studies.
2. Experimental Catalyst Preparation
2.1. AC Pretreatment
The AC was ground using an agate mortar, sieved to a particle size fraction of 10–20 mesh, treated in boiling water with continuous stirring for 1 h, thoroughly rinsed with deionized water, and subsequently dried in an oven at 105 °C to obtain AC0 support. The purified support material was treated with a 30% nitric acid (HNO3) solution in a temperature-controlled water bath at 60 °C for 1 h. After the acid treatment, the material was repeatedly rinsed with water until neutral pH was reached, followed by drying in an oven at 105 °C to obtain the pretreated AC support.
2.2. Synthesis of Ce/AC and HPA-Ce/AC Catalysts
Catalyst preparation was performed via the incipient wetness impregnation method. Three heteropoly acid modifiers, H3PW12O40 (TPA), H4SiW12O40 (TSiA), and H3PMo12O40, (MPA) were employed, with Ce(NO3)3 serving as the metal precursor for active component loading. The pretreated support was impregnated with an aqueous solution of Ce(NO3)3 to achieve a 5 wt% Ce loading. The impregnated sample was evaporated to dryness under constant stirring in a temperature-controlled water bath at 60 °C. The resulting material was then dried at 105 °C, followed by calcination in a muffle furnace at 400 °C for 2 h to obtain the final Ce/AC catalyst. The pretreated support was impregnated with TSiA solution to achieve loadings ranging from 1 to 10 wt%. The impregnated sample was then evaporated to dryness under continuous stirring in a temperature-controlled water bath at 60 °C. After oven-drying at 105 °C for 12 h, the material was calcined in a muffle furnace at 200–700 °C for 2 h to obtain the final TSiA-modified denitrification catalyst (TSiA-Ce/AC). The MPA-Ce/AC and TPA-Ce/AC catalysts were prepared following an identical synthesis protocol to that described for the TSiA-Ce/AC catalyst.
2.3. Simulation of K Poisoning on Catalysts
To simulate potassium poisoning, the catalysts were impregnated with a potassium nitrate (KNO3) as the K precursor and introduced during the cerium loading step. Following identical drying and calcination procedures, the thermally decomposed KNO3 yielded K2O, which subsequently interacted with the catalyst’s active surface components. This approach effectively simulated K poisoning of the catalyst system. The K-modified catalysts were prepared by introducing 100 μmol/g of K catalyst to each HPA-modified sample, designated as MPA-K-Ce/AC, TPA-K-Ce/AC, and TSiA-K-Ce/AC to reflect their respective compositions. Different amounts of K (0, 30, 60, 100 μmol/g) were added to Ce/AC and TSiA-Ce/AC catalysts, and the corresponding samples after the K poisoning simulation were numbered as xK-Ce/AC and TSiA-xK-Ce/AC, and x was the amount of potassium added. The obtained catalysts are shown in Table 1, and the catalysts’ preparation process are shown in Figure 1.

Table 1.
Name and content of HPA-Ce/AC catalysts.
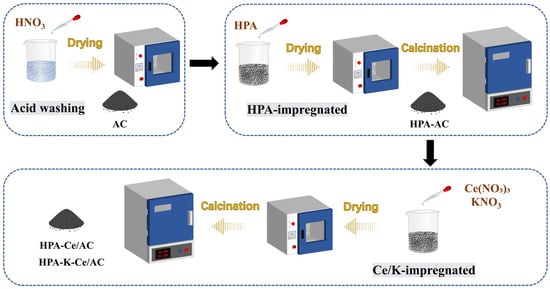
Figure 1.
Chart of catalyst preparation process.
2.4. Catalytic Activity Measurements
The catalysts’ performance was evaluated in a fixed-bed reactor system under atmospheric pressure conditions, employing NH3 as the reducing agent for the standard SCR reaction. The denitrification activity was assessed by monitoring NO conversion efficiency across the temperature range of interest. The fixed-bed reactor (20 mm inner diameter) was loaded with approximately 3 g of catalyst, corresponding to a bed height of 2.5 cm under standard packing conditions. The flow rate of the simulated flue gas was 800 mL min−1, generating gaseous hourly space velocity (GHSV) = 10,000 h−1/20,000 h−1. The simulated flue gas composition consisted of approximately 500 ppm nitric oxide (NO), an equimolar NH3 concentration (NH3/NO = 1:1), and 4% O2, with N2 as the balance gas.
The catalytic performance was evaluated across a temperature range of 80–200 °C under continuous flow conditions.
The low-temperature denitrification performance of the catalysts was evaluated based on NO conversion efficiency, calculated according to the following equation:
Kinetic measurements were conducted under identical experimental conditions to the activity tests, with the exception that NO conversion was maintained below 20%. Previous studies have demonstrated that the standard NH3-SCR reaction follows apparent first-order kinetics under low-temperature conditions (<200 °C) [14,25]. Accordingly, the reaction rate was normalized using a first-order kinetic model, expressed as follows:
where k represents the normalized reaction rate (μmol·g−1·s−1), V is the total gas flow rate (cm3·s−1), W is the catalyst mass (g), and x is the fractional NO conversion. The apparent activation energy (Ea) was determined by Arrhenius analysis through linear regression of the temperature-dependent reaction rates, according to the following equation:
where A is the pre-exponential factor (cm3∙g−1∙s−1) and the apparent activation energy Ea (J∙mol−1), R is the universal gas constant (8.314 J·mol−1·K−1), and T is the thermodynamic temperature (K).
2.5. Catalyst Characterization
The catalysts’ phase structures were analyzed by X-ray diffraction (XRD; Rigaku MiniFlex, Tokyo, Japan). The surface functional groups were characterized by Fourier transform infrared spectroscopy (FTIR) using a Nicolet 6700 spectrometer (Thermo Scientific, Waltham, MA, USA). Ammonia temperature-programmed desorption (NH3-TPD) analysis was conducted to characterize the surface acid strength distribution using a PreeRight TP5000 chemisorption analyzer. The surface topography of the materials, microscale elemental composition, and quantitative evaluation of the constituent elements analyses were performed using a JEOL JSM-7500F field emission scanning electron microscope (SEM) equipped with an energy-dispersive X-ray spectroscopy (EDS) system (JEOL Ltd., Tokyo, Japan).
3. Results and Discussion
3.1. Effect of HPA Modification on Denitrification Performance
3.1.1. Catalytic Activities of Catalysts
Figure 2 illustrates the denitrification performance of the heteropoly acids (MPA, TPA, and TSiA) supported on Ce/AC catalysts under their respective optimal loading conditions. As observed, within the temperature range of 80–200 °C, the NH3-SCR activity follows the sequence of TSiA-Ce/AC > TPA-Ce/AC > MPA-Ce/AC > Ce/AC, indicating that the impregnation of HPAs could greatly enhance the SCR performance of Ce/AC. The results demonstrate that TSiA exhibited the most obvious modification effect, significantly enhancing the denitrification performance. Specifically, TSiA-modified Ce/AC achieved a nearly 15% increase in NO removal efficiency at 140 °C compared to the unmodified catalyst. TPA exhibited a relatively lower denitrification activity comparable to that of TSiA (12% increase at 140 °C). In contrast, MPA showed a comparatively weaker improvement, increasing the denitrification activity by only 6% under the same conditions. In conclusion, all three heteropoly acids (MPA, TPA, and TSiA) demonstrate the potential for enhancing the low-temperature catalytic activity of Ce/AC catalysts. The TSiA-Ce/AC catalyst, synthesized with a 3% TSiA loading, followed by calcination at 500 °C, demonstrated optimal low-temperature denitrification performance among the tested materials.
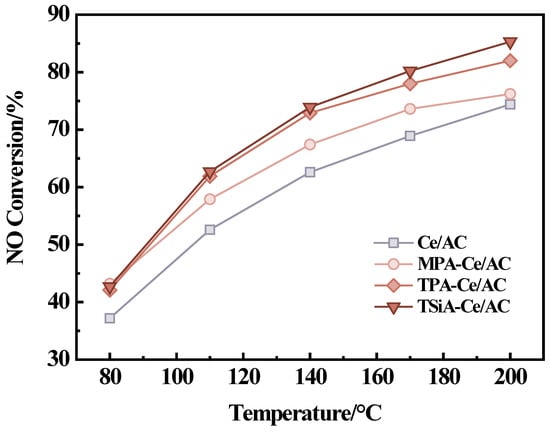
Figure 2.
NH3-SCR activity of TSiA-Ce/AC, TPA-Ce/AC, MPA-Ce/AC, and Ce/AC. Reaction conditions: [NO] = [NH3] = 500 ppm, 4 vol% [O2], N2 balance, and GHSV = 20,000 h−1.
Figure 3 presents the selection of the highest-activity TSiA-modified catalyst alongside the optimized synthesis parameters: 500 °C calcination temperature and 3% TSiA loading. In Figure 3a, the TSiA-Ce/AC catalyst calcined at 500 °C exhibits a NO removal rate of 72% at 80 °C, markedly superior to the samples treated at other temperatures. However, above 140 °C, the catalysts calcined at 400 °C, 500 °C, and 600 °C demonstrated comparable activities, with NO removal rates exceeding 85%. Collectively, the catalysts calcined at 500 °C demonstrated optimal NO removal activity across the 80–200 °C temperature range. Figure 3b reveals comparable NO removal rates across the 80–200 °C range for the TSiA-Ce/AC catalysts with varying TSiA loadings. The denitration activity decreased in the following sequence: 3%-TSiA-Ce/AC > 1%-TSiA-Ce/AC > 5%-TSiA-Ce/AC > 8%-TSiA-Ce/AC.
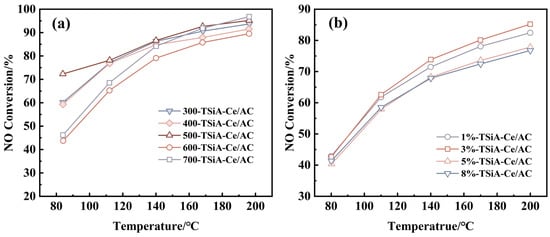
Figure 3.
Low-temperature denitration activity of TSiA-modified catalysts as a function of (a) calcination temperature and (b) TSiA loading. Reaction conditions: [NO] = [NH3] = 500 ppm, 4 vol% [O2], N2 balance, and GHSV = 20,000 h−1.
3.1.2. Acidity Analysis of Catalysts
Surface acidity plays a crucial role in determining the denitrification activity of catalysts. To investigate this relationship, we conducted NH3 temperature-programmed desorption (NH3-TPD) characterization to evaluate the surface acidity of HPA-Ce/AC catalysts modified with three heteropoly acids (MPA, TPA, and TSiA). The resulting NH3-TPD profiles, presented in Figure 4, reveal distinct acid site distributions and strengths among the modified catalysts. The NH3-TPD profiles of the heteropoly acid-modified catalysts revealed two distinct ammonia desorption peaks in the temperature ranges of 100–250 °C and 300–500 °C, corresponding to weak and medium–strong acid sites, respectively. This bimodal distribution indicated the presence of acid sites with different strengths on the catalyst surface. Previous studies have suggested that NH3 coordinated to Lewis acid sites (L acid sites) exhibits greater thermal stability than NH3 bound to Brønsted acid sites (B acid sites) [26]. Accordingly, the low-temperature desorption peak (<300 °C) in our NH3-TPD profiles can be assigned to the following: (1) physically adsorbed NH3 and (2) NH4+ species associated with B acid sites [27]. The high-temperature desorption peak (>300 °C) corresponds to strongly coordinated NH3 on L acid sites [28].
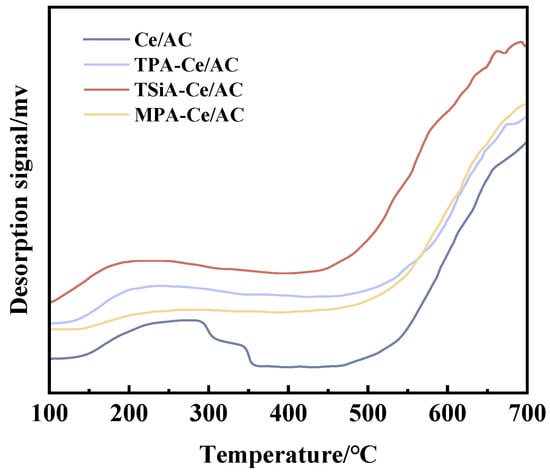
Figure 4.
NH3-TPD profiles of different catalysts.
The NH3-TPD analysis revealed a substantially larger desorption peak area for HPA-Ce/AC compared to the unmodified Ce/AC catalyst, demonstrating that heteropoly acid (HPA) modification significantly increases the total number of acid sites. The increased acid site density facilitates enhanced adsorption and activation of reactant gases. This preferential adsorption promoted greater NH3 availability for selective catalytic reduction (SCR) rather than non-selective oxidation pathways, thereby significantly improving SCR activity [29].
Notably, persistent desorption peaks above 600 °C were observed in all the catalyst spectra. This high-temperature desorption may originate from either (1) ammonia molecules adsorbed on non-acidic sites of the AC support or (2) thermal decomposition of oxygen-containing functional groups on the carbon carrier surface. Fu et al. [30] reported analogous high-temperature desorption behavior (>600 °C) in NH3-TPD studies of Al2O3-TiO2-SiO2-supported denitrification catalysts, attributing these features to NH3 desorption from non-acidic support sites. This consistency with our observations suggested such high-temperature interactions represent a general phenomenon in supported catalyst systems.
Quantitative analysis of the NH3-TPD profiles reveals that HPAs modification significantly enhances the surface acidity of the Ce/AC catalysts, with the degree of improvement following the order TSiA-Ce/AC > TPA-Ce/AC > MPA-Ce/AC. This trend, determined by integrating the desorption peak areas, demonstrates that TSiA induces the most substantial acid site formation, followed by TPA and then MPA. The denitrification activity of the modified catalysts exhibited a strong positive correlation with their surface acidity (TSiA-Ce/AC > TPA-Ce/AC > MPA-Ce/AC), demonstrating that HPA modification enhanced catalytic performance through two key mechanisms: (1) increasing the concentration of surface acid sites and (2) modulating the strength distribution of acid centers. This structure–activity relationship confirmed that optimized surface acidity facilitated both NH3 adsorption and activation, thereby improving the overall SCR efficiency.
To investigate alterations in the surface functional groups of activated carbon following the nitric acid modification, FT-IR was employed to characterize both pre- and post-modification samples. As shown in Figure 5, the FT-IR spectrum of acid-treated AC exhibits a significantly intensified absorption peak at 3400 cm−1. This enhancement is primarily attributed to O-H stretching vibrations on the carbon surface, indicating the presence of carboxyl groups, hydroxyl groups, and chemically adsorbed water [31]. The distinctive absorption band observed at approximately 1600 cm−1 in the FT-IR spectrum of nitric-acid-modified AC corresponds to C=O stretching vibrations, characteristic of carbonyl functional groups, such as carboxyl groups, esters, or other carbonyl-containing moieties [32]. The absorption peak at 1434 cm−1 in the untreated activated carbon (AC0) corresponds to aliphatic C-H bending vibrations [33]. Following nitric acid modification, this vibrational feature diminishes in the spectrum of AC due to oxidative degradation of aliphatic structures under strongly oxidizing conditions. Both AC and AC0 exhibit strong absorption bands at 1092 cm−1, characteristic of C-O stretching vibrations. Comparative analysis reveals a significant attenuation of this band in AC relative to AC0, indicating partial oxidative conversion of surface C-O groups into acidic oxygen- and nitrogen-containing functional groups—including carboxyl, lactone, hydroxyl, and nitro groups—under nitric acid treatment [34]. These functional groups exhibit enhanced reactivity toward NO.
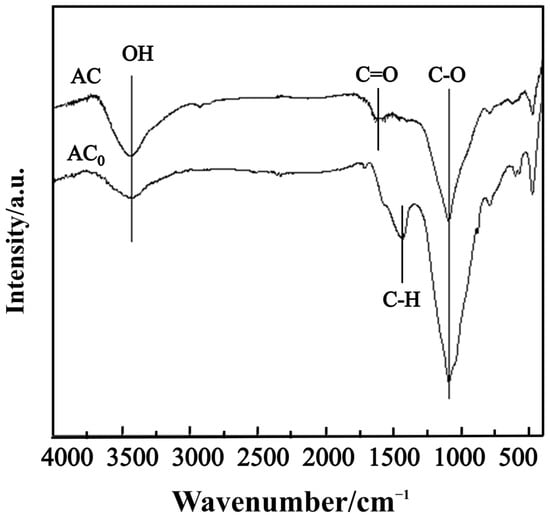
Figure 5.
FT-IR spectra of AC0 and AC (HNO3 treatment).
To elucidate the structural stability of the most active catalyst after HPA modification, we conducted comparative FT-IR analyses of fresh TSiA-Ce/AC and post-reaction TSiA-Ce/AC, which are shown in Figure 6. All the catalysts exhibit an absorption band at 1071 cm−1, corresponding to C-O stretching vibrations. The Keggin-type heteropoly acid structure is primarily characterized by vibrational modes in the range of 650–1150 cm−1, with distinct peaks corresponding to the metal–oxygen stretching mode [17].
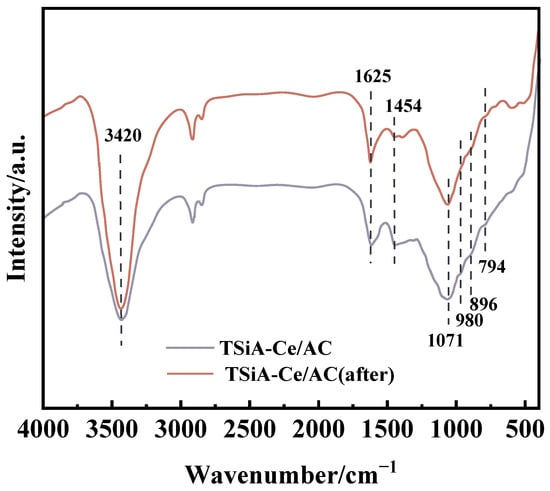
Figure 6.
FT-IR spectra of TSiA-Ce/AC catalysts in fresh and post-reaction states.
As shown in Figure 6, the infrared spectra of the catalyst before and after evaluation exhibited characteristic structural peaks of TSiA, including 980 cm−1 (asymmetric W═O stretching), 896 cm−1 (W-O-W bridging vibration in TSiA structures), and 794 cm−1 (symmetric Si-O stretching) [35]. The intensities of the TSiA characteristic peaks remained unchanged before and after evaluation, confirming that the TSiA structure in the catalyst remained intact during both catalyst preparation and activity testing. This stability further indicated that the Keggin-structured TSiA was successfully immobilized on the surface and pores of the activated carbon. Furthermore, the characteristic Keggin peaks of TSiA in the activated-carbon-supported catalyst exhibited a redshift compared to those of pure TSiA. This shift can be attributed to hydrogen bonding and weak interactions between TSiA and Ce/AC [36], which stabilize the Keggin structure and ensure uniform dispersion of TSiA within Ce/AC. Compared to pure TSiA, the characteristic Keggin peak attenuates in the activated-carbon-supported catalyst, indicating partial conversion of TSiA to WO3 during 500 °C calcination.
In the post-reaction FTIR spectrum, a new absorption band emerged at 1454 cm−1, corresponding to the N-H bending vibration of NH4+ species adsorbed on B acid sites of the catalyst surface [37]. The intensified absorption band at 3420 cm−1 corresponded to the O-H stretching vibration on the catalyst surface [38]. This enhancement occurred because NH3 adsorption at B acid sites generated NH4+ species, simultaneously producing additional hydroxyl groups. These results demonstrate that TSiA modification substantially increased the number of B acid sites in the catalyst. This enhancement facilitated NH3 adsorption and activation, thereby improving the catalyst’s low-temperature SCR activity. These findings are consistent with the NH3-TPD results.
3.2. Effect of HPAs on Anti-K Performance
3.2.1. HPAs’ Effect on K-Poisoning Resistance in Low-Temperature NH3-SCR Activity
Figure 7 compares the catalytic performance of three different HPA-modified Ce/AC catalysts, prepared with identical loading amounts and calcined at their respective optimal temperatures, to evaluate their modification effects. The line chart revealed that within the reaction temperature range of 80–200 °C, all the HPA-modified catalysts exhibited progressively increasing NOx conversion efficiency, and the NH3-SCR activity sequence follows the order of 5%-TSiA-Ce/AC > 5%-TPA-Ce/AC > 5%-MPA-Ce/AC. The catalytic evaluation revealed that silicon-containing heteropoly acids exhibited superior denitrification performance compared to phosphorus-containing analogues. Notably, all three modified catalysts achieved high NOx conversion efficiencies exceeding 90% at 200 °C, demonstrating their excellent low-temperature activity.
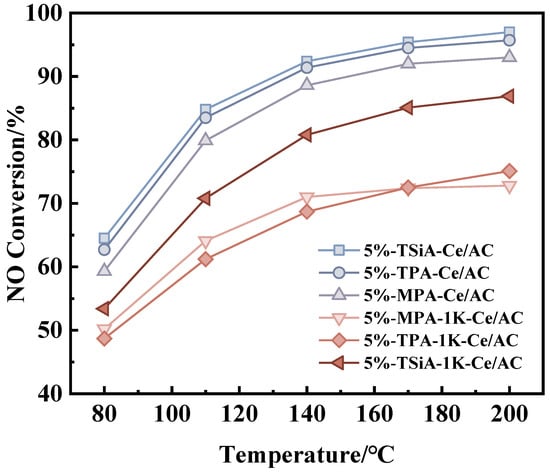
Figure 7.
NH3-SCR activity of different K+-poisoned catalysts. Reaction conditions: [NO] = [NH3] = 500 ppm, 4 vol% [O2], N2 balance, and GHSV = 10,000 h−1.
Additionally, Figure 7 shows the catalytic activity of the catalysts after K poisoning with 100 μmol/g. The comparison of catalytic performance revealed that among the three K-poisoning HPA-Ce/AC catalysts (5%-MPA-1K-Ce/AC, 5%-TPA-1K-Ce/AC, and 5%-TSiA-1K-Ce/AC), the TSiA-modified variant demonstrates superior NO removal efficiency throughout the 80–200 °C temperature range. At the temperature of 170 °C, 5%-TSiA-1K-Ce/AC exhibited approximately 10% higher denitrification activity compared to both 5%-MPA-1K-Ce/AC and 5%-TPA-1K-Ce/AC. The results demonstrate that among the three heteropoly acids evaluated, TSiA modification effectively enhanced both the denitrification efficiency and K resistance of the Ce/AC catalyst. Compared to TPA and MPA modifications, the TSiA-modified catalyst exhibits superior performance in maintaining catalytic activity under K poisoning conditions. Consequently, the effect of K tolerance on the denitrification activity of the Ce/AC catalysts was systematically investigated using TSiA-Ce/AC as a representative model catalyst.
3.2.2. Effect of K Tolerance on TSiA-Ce/AC
Figure 8 presents a comparative analysis of the denitrification activities of the TSiA-xK-Ce/AC and xK-Ce/AC catalysts (x = 0.3, 0.6, 1) with varying K-poisoning levels. The activity profiles in the panels demonstrate that TSiA modification significantly enhances the catalytic performance of Ce/AC under K-poisoned conditions across the entire test temperature range (80–200 °C). The K resistance of the TSiA-modified Ce/AC catalysts exhibited temperature-dependent enhancement, becoming more significant at elevated temperatures. Notably, TSiA modification obviously improved the catalyst’s K tolerance, particularly under high K level conditions, demonstrating superior stability compared to unmodified Ce/AC. At a K loading of 100 μmol/g, TSiA modification boosted the denitrification efficiency of the Ce/AC catalysts by over 20%, highlighting its role in mitigating K poisoning effects.
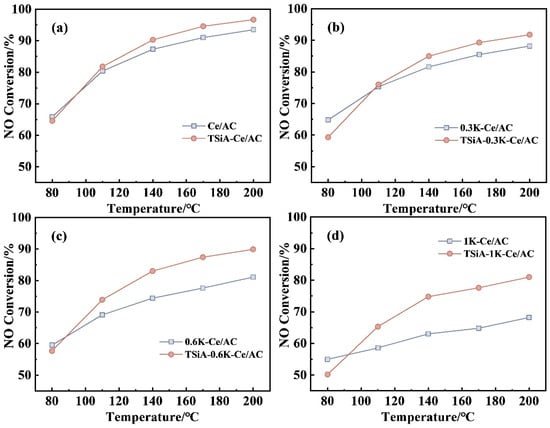
Figure 8.
(a–d) Comparison of NH3-SCR activity between TSiA-xK-Ce/AC and xK-Ce/AC catalysts (x = 0.3, 0.6, 1) with varying K loadings. Reaction conditions: [NO] = [NH3] = 500 ppm, 4 vol% [O2], N2 balance, and GHSV = 10,000 h−1.
Figure 9 presents the EDS analyses conducted on both the Ce/AC and TSiA-Ce/AC catalysts with varying K poisoning levels. Figure 10 presents the SEM images and EDS elemental mapping for the Ce/AC, 1K-Ce/AC, TSiA-Ce/AC, and TSiA-1K-Ce/AC catalysts, with the corresponding mass ratios of the C, O, Si, K, Ce, and W elements for each catalyst provided in Table 2. It is evident from Figure 9a,c that the fresh catalysts exhibit well-developed pore structures, which facilitate the reaction with and adsorption of NH3-SCR gases. Figure 8 clearly demonstrated the presence of K peaks in all the K-poisoned catalyst samples, confirming successful K deposition on the catalyst surfaces. Figure 9b reveals that the pore structures on the K-poisoned catalyst surface are nearly obscured, whereas TSiA doping effectively preserves the pore architecture of the Ce/AC catalyst (Figure 9c,d). Notably, the abundant pore structure of the TSiA-1K-Ce/AC catalyst reemerges, approximating that of the non-poisoned catalysts. BET analysis of the Ce-based catalyst series [39,40,41] revealed that K addition induces moderate modifications in a specific surface area. However, this structural parameter does not constitute the primary determinant of catalytic activity.
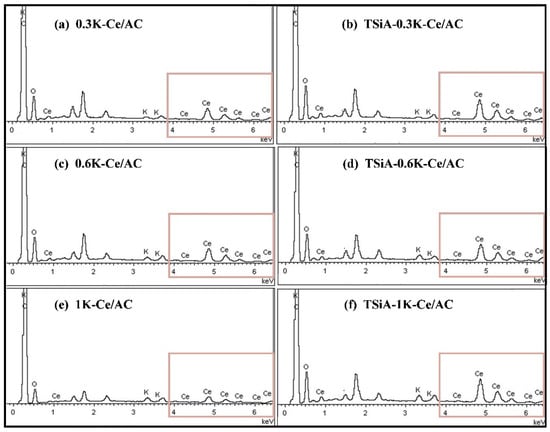
Figure 9.
EDS spectra of catalysts with varying potassium loadings.
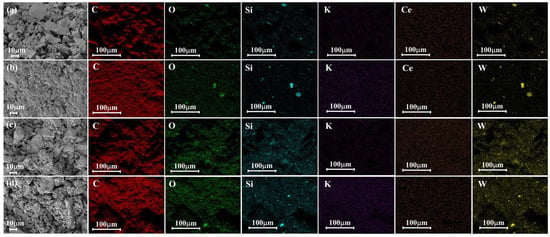
Figure 10.
SEM and elements mapping images of (a) Ce/AC, (b) 1K-Ce/AC, (c) TSiA-Ce/AC, and (d) TSiA—1K-Ce/AC catalysts.
Table 2.
Surface elemental composition of the catalysts.
Figure 10 displays the EDS elemental mapping and demonstrates uniform distribution of the active components across all the catalyst surfaces. For the 1K-Ce/AC catalyst in Figure 9c, K species are dispersed across the catalyst surface, while the detected Ce content is reduced by more than half—a principal factor contributing to catalyst deactivation. Notably, Ce signals became virtually undetectable in the 1K-Ce/AC sample, indicating complete K-induced coverage or leaching of active Ce species. However, following TSiA loading (Figure 8 and Figure 9d), the masking effect of K on surface Ce is substantially mitigated, with Ce becoming uniformly distributed across the catalyst surface. The EDS-derived Ce content of 7.45% closely approaches the 8.40% observed in the undoped Ce/AC catalysts (Table 2). Du et al. [42] demonstrated that K obscured the underlying Ce species, preventing their detection by surface spectroscopic techniques. The 1K-Ce/AC catalyst exhibited extensive K coverage of Ce active sites, and this obvious K-induced masking effect significantly compromised the catalytic performance of the Ce/AC system. These results demonstrate that TSiA modification effectively shielded active sites from K poisoning, thereby significantly mitigating its detrimental impact on catalyst denitrification performance.
The XRD patterns of the Ce/AC and TSiA-Ce/AC catalysts before and after potassium poisoning are displayed in Figure 11. The diffraction peaks observed at 20–30° and 40–45° across all the catalysts correspond to disordered graphitic crystallites within the activated carbon matrix [31]. SiO2 signatures at 20.9°, 26.7°, 36.6°, and 50.2° originate from crystalline silica in AC ash (JCPDS: 79-1906) [43]. Weak characteristic peaks of CeO2 (JCPDS: 44-0398) are observed at 2θ = 35.2°. These peaks exhibit further attenuation in the catalyst modified with TSiA, suggesting that TSiA enhances the dispersion of Ce species on the AC surface. Meanwhile, weak diffraction peaks corresponding to WO3 (JCPDS: 32-1395) are observed at 2θ = 22.6°, 23.3°, 24.0°, 33.0°, and 33.9°. The emergence of these WO3 peaks is attributed to the partial thermal decomposition of TSiA during calcination [44]. In both the 1K-Ce/AC and TSiA-1K-Ce/AC catalysts, no characteristic diffraction peak attributable to potassium oxide species was discernible. This absence is attributable primarily to the low K loading and the limited crystallite size [45]. Furthermore, the diffraction peak positions and characteristic peak profiles of the catalyst following K poisoning align with those of the unpoisoned catalysts. This indicates that K poisoning exerts minimal influence on the catalyst’s crystal structure.
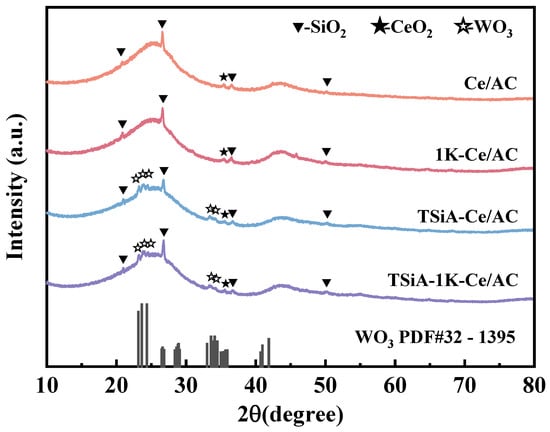
Figure 11.
XRD patterns of Ce/AC, 1K-Ce/AC, TSiA-Ce/AC, and TSiA—1K-Ce/AC catalysts.
Figure 12 presents the FT-IR spectra of the Ce/AC and TSiA-Ce/AC catalysts before and after K poisoning, showing a distinct broad absorption band in the 3130–3635 cm−1 range, with peak intensity at 3439 cm−1. The elastic vibration of the -OH group in adsorbed water [46], associated with carboxyl, hydroxyl, or AC surfaces, remained largely unaffected by the K-toxicity process. Additionally, several absorption bands were observed on other AC surfaces in the FT-IR spectra of all the samples. These bands correspond to the C=C vibration (1560–1570 cm−1) of the aromatic carbon ring skeleton and the in-plane C–H bending vibration (700–900 cm−1) in the aromatic group [47]. The infrared sample pretreatment was mixed with potassium bromide, so the infrared characterization cannot detect the relevant functional groups of K. However, it can be seen from the comparison of the spectra in the figure that the absorption peaks at 2921 cm−1 and 2847 cm−1 of the infrared spectra of Ce/AC and TSiA-Ce/AC disappear after the addition of K. These two peaks are likely attributed to the aliphatic C–H stretching vibrations of CH2 and CH3 groups [47], suggesting that K-induced poisoning results in the coverage or degradation of organic functional groups on the Ce/AC surface.
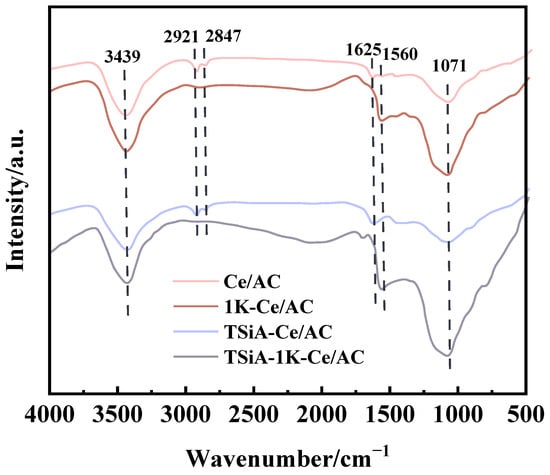
Figure 12.
FT-IR spectra of Ce/AC and TSiA-Ce/AC catalysts before and after K poisoning.
For K2O poisoning, K+ combines with Brønsted acid sites (M−OH) to form M−O−K species. This reaction blocks NH3 adsorption at active sites, consequently reducing catalytic activity [48,49]. This suggests that K+ substitution for H+ in surface M−OH groups deactivates these Brønsted acid sites, eliminating their capacity to adsorb NO and NH3. Following K impregnation, the 1625 cm−1 absorption band in 1K-Ce/AC corresponds to modified C=O stretching vibrations in quinone or carboxyl groups. NO adsorption occurs preferentially at the C=O of carboxyl functionalities. Surface oxygen functional groups can enhance active phase dispersion and serve as acid sites for NH3 adsorption [46]. Consequently, the depletion of both oxygen functionalities and Brønsted acid sites following K-doping accounts for the diminished SCR activity of the Ce/AC catalysts.
The absorption bands observed in the 1000–1400 cm−1 range are attributed to C–O stretching and O–H bending vibrations characteristic of ethers, alcohols, and carboxylic acids [50]. The peak at 1071 cm−1, corresponding to the C-O vibrational band, exhibited significant enhancement following K poisoning, with this effect being more pronounced after TSiA modification. This suggests that the primary K+-binding sites on Ce/AC were predominantly oxygen-containing functional groups of the AC support. However, when functionalized with TSiA, the TSiA-Ce/AC catalyst possesses abundant base-trapping sites via Si-O [51] and W-O groups [52]; notably, the W-O groups provide additional acidic sites for enhanced K+ bonding stability. This enhanced binding stability could explain the observed intensification of vibrational signals. Notably, the TSiA-induced increase in acidic sites and oxygen-containing functional groups appeared to mitigate the deleterious effects of potassium on the Ce/AC catalyst system.
The NH3-SCR denitrification reaction was originally proposed to commence through chemisorption and activation of ammonia at the surface acid sites. Consequently, the catalyst surface acidity constitutes a critical determinant of catalytic performance [53]. Surface acidity of the catalyst was characterized via NH3-TPD both pre- and post-TSiA modification, with the results presented in Figure 13. For the K-poisoned catalysts, the NH3-TPD profiles of 1K-Ce/AC and TSiA-1K-Ce/AC exhibit a narrow low-temperature peak (<300 °C, indicative of Brønsted acid sites) and absence of the broad high-temperature peak (300–500 °C, characteristic of Lewis acid sites) observed in the unpoisoned analogues. Extensive evidence confirms that NH3 adsorption constitutes the initial step in the NH3-SCR reaction pathway, with its extent governed predominantly by catalyst acidity [54]. Even under K poisoning conditions, the TSiA-1K-Ce/AC catalyst exhibits markedly stronger acid strength than 1K-Ce/AC. TSiA incorporation significantly mitigates Brønsted acid site depletion in the K-poisoned Ce/AC catalyst, thus enhancing low-temperature catalytic activity, which is evidenced by corresponding improvements in the NO conversion rates.
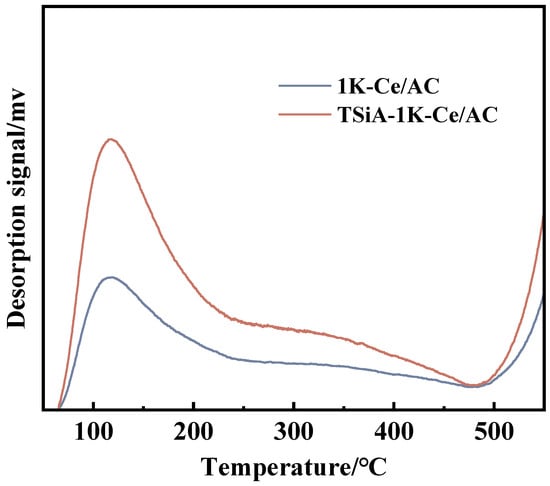
Figure 13.
NH3-TPD profiles of 1K-Ce/AC and TSiA-1K-Ce/AC catalysts.
3.3. TSiA-Ce/AC Catalyst Anti-K Poisoning Kinetics Results
In order to further explore the effect of TSiA modification on the denitrification activity and anti-K toxicity performance of the catalyst, Figure 14 and Table 3 show the results of the Arrhenius curve of Ce/AC, TSiA-Ce/AC, 1K-Ce/AC, and TSiA-1K-Ce/AC. The Ea of TSiA-Ce/AC was marginally higher than that of Ce/AC, suggesting that the TSiA modification preserved the original SCR reaction mechanism. However, the increased A value indicated an expansion of active sites, which consequently enhanced the NO conversion efficiency. Although 1K-Ce/AC exhibits the lowest activity, primarily due to a reduced A value from attenuated active site density, its decreased activation energy indicates that the catalytic centers, rate-limiting steps, or both are somewhat different [55]. This shift arises from severe Brønsted acid site depletion post-K poisoning, underscoring that reaction kinetics are greatly influenced by Brønsted acidity. These kinetic observations, combined with the EDS analysis results, collectively demonstrate that potassium deposition leads to coverage of active sites on the Ce/AC catalyst. K appeared to preferentially deactivate high-energy active sites, causing the remaining low-energy sites to dominate the catalytic process, consequently lowering the Ea. Furthermore, K-induced pore blockage reduced the quantity of accessible active sites, leading to a direct decrease in A value. However, TSiA-1K-Ce/AC exhibited only a marginal decrease in the reaction rate while maintaining stable Ea. This suggests that the TSiA modifier preferentially adsorbed K species, thereby protecting the Ce/AC active sites and preserving the original reaction pathway. The TSiA-modified catalyst maintained its acidic sites, as evidenced by only a modest reduction in the A value. This demonstrates that TSiA modification effectively enhanced the K resistance of the catalyst.
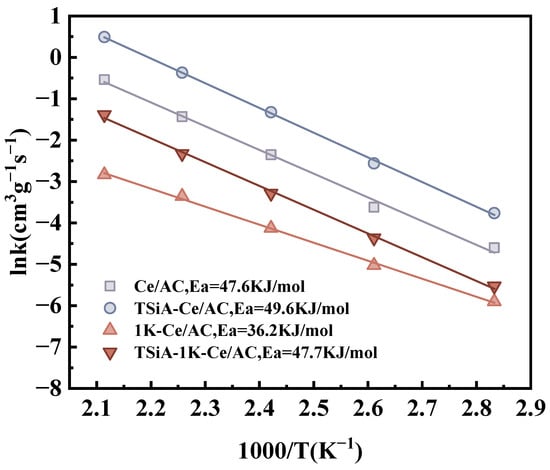
Figure 14.
Arrhenius plots of NO conversion over Ce/AC, TSiA-Ce/AC, 1K-Ce/AC, TSiA-1K-Ce/AC catalysts. Reaction conditions: [NO] = [NH3] = 500 ppm, 4 vol% [O2], N2 balance, and GHSV = 20,000 h−1.
Table 3.
Summary of activation energy Ea and intercept lnA of NH3-SCR reaction on Ce/AC, TSiA-Ce/AC, 1K-Ce/AC, and TSiA-1K-Ce/AC catalysts.
3.4. Possible TSiA-Ce/AC Catalyst Anti-K Poisoning Mechanism
Figure 15 presents the proposed mechanism by which the TSiA-Ce/AC catalyst resists K poisoning. Previous studies have demonstrated that K species degrade SCR catalyst performance by diminishing both redox properties and surface acidity [10,39,41]. Specifically for Ce-based catalysts, K+ preferentially interacts with B acid sites [56,57], which are critical for NH3 adsorption. Consequently, the depletion of acidic sites results in ineffective NH3 adsorption at the active centers, thereby suppressing catalytic activity. The NH3-TPD analysis revealed that the TSiA-Ce/AC catalyst exhibited abundant B acid sites, demonstrating enhanced acid site concentration and strength compared to the unmodified catalyst, even under K poisoning conditions. The FT-IR analysis demonstrated that TSiA retained its Keggin structure, and its modification introduced TSiA-derived B acid sites and abundant surface oxygen functional groups. The XRD analysis revealed that crystalline WO3 phases persist within the TSiA structure of the catalyst. When stabilized in an HPA matrix, these WO3 species exhibited higher activity than in their decomposed state. These features likely facilitated K+ dispersion and chemical stabilization through interaction with the [SiW12O40]4− framework. This mechanism prevented K+ from neutralizing oxygen-containing groups on the Ce/AC surface, thereby preserving acidic sites and maintaining catalytic activity. The SEM and EDS analyses confirmed that K species preferentially occupy active sites on Ce/AC, while TSiA modification appeared to mitigate this deactivation by preventing K-induced site coverage and structural degradation. This protective effect reduced the detrimental impact of K poisoning on catalytic performance, consequently enhancing the NH3 and NO adsorption capacity. The kinetic analysis suggests that strong interactions likely occur between K species and the abundant polyoxoanions (e.g., [SiW12O40]4−) introduced by TSiA modification. This interaction appeared to shield the active sites from K poisoning, thereby preserving the active sites of Ce/AC and maintaining the original standard SCR reaction pathway. Although K species blocked the acidic sites, the TSiA-Ce/AC catalyst preserved intact Brønsted acid sites. Simultaneously, the inherent Brønsted acidity of TSiA provided supplemental catalytic acid sites for NH3 adsorption and activation, thereby mitigating K poisoning effects. This preservation of abundant active sites enhanced both the low-temperature NH3-SCR activity and K resistance of the Ce/AC catalyst system.
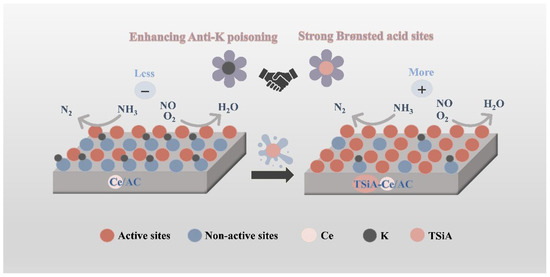
Figure 15.
Schematic illustration of Ce/AC and TSiA-Ce/AC catalysts’ K-resistant mechanism for NH3-SCR reaction.
4. Conclusions
In summary, this study demonstrated that HPA (TPA, MPA, TsiA) modification of Ce/AC catalysts via stepwise incipient wetness impregnation significantly enhanced low-temperature NH3-SCR performance. In addition, HPA modification effectively enhanced the K resistance of Ce/AC systems. Under the K-level of 100 μmol/g, the TSiA-Ce/AC catalyst exhibited a denitrification activity improvement exceeding 20% relative to the Ce/AC catalyst performance. This study demonstrated that TSiA modification significantly mitigated the physical and chemical poisoning effects of K on the catalyst. Acting as a K+ scavenger, TSiA binds K+ ions and effectively preserves Brønsted acid sites associated with oxygen-containing functional groups on the Ce/AC surface. Concurrently, intrinsic Brønsted acidity of TSiA provides supplemental catalytic acid sites, enhancing NH3 adsorption and activation, and thereby improving the low-temperature NH3-SCR activity of the catalyst system. These results demonstrate enhanced K-poisoning resistance in the TSiA-Ce/AC catalyst, providing valuable design principles for synthesizing K-tolerant catalysts for low-temperature NH3-SCR applications in biomass-fired power plants with high alkali-metal flue gas concentrations.
Author Contributions
Conceptualization, J.L.; methodology, T.X.; validation, Q.C.; formal analysis, T.Z.; investigation, T.X. and Y.D.; data curation, T.X.; writing—original draft, T.Z.; writing—review & editing, M.F.; visualization, T.Z. and Q.C.; supervision, M.F. and J.L. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the National Key Research and Development Program of China (2024YFC3712205); Sichuan Science and Technology Program (2023YFS0354); Natural Science Foundation of Sichuan Province, China (2025ZNSFSC1256); CNPC Innovation Fund (2024DQ02-0208); and Integrated Treatment of Multiple Pollutants in Electrolytic Aluminum Flue Gas and Demonstration of Carbon Resource Recycling Technology (24H0881).
Data Availability Statement
The original contributions presented in this study are included in the article. Further inquiries can be directed to the corresponding author.
Conflicts of Interest
Author Yongchun Deng was employed by the Sichuan Aostar Aluminium Co., Ltd. The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
- Liu, S.; Lang, S.; Zhang, S.; Cao, Z.; Yang, J.; Zhou, Y.; Hu, N.; Wu, Y. Effect of three component evolution of cotton stalk on physical properties of its forming fuel during hydrothermal treatment. J. China Coal Soc. 2023, 48, 2410–2418. [Google Scholar]
- Elkaee, S.; Kim, S.Y.; Phule, A.D.; Uz Zaman, M.W.; Lee, S.G.; Park, G.; Hwan Yang, J. Catalysts for fast and NO2 SCR reactions for the removal of nitrogen oxides emitted from various sources: Recent advances, mechanisms, and future directions. J. Environ. Chem. Eng. 2023, 11, 111–131. [Google Scholar] [CrossRef]
- Liu, J.; Lv, D.; Wang, Y.; Zhao, Y.; Li, G.; Zhang, G. Influence of Catalyst Structural Remodelling on The Performance of NH3-SCR Reactions: A Mini Review. ChemCatChem 2024, 16, 21. [Google Scholar] [CrossRef]
- Yao, X.; Zheng, Y.; Zhou, H.; Xu, K.; Xu, Q.; Li, L. Effects of biomass blending, ashing temperature and potassium addition on ash sintering behaviour during co-firing of pine sawdust with a Chinese anthracite. Renew. Energy 2020, 147, 2309–2320. [Google Scholar] [CrossRef]
- Mlonka-Medrala, A.; Magdziarz, A.; Gajek, M.; Nowinska, K.; Nowak, W. Alkali metals association in biomass and their impact on ash melting behaviour. Fuel 2020, 261, 17. [Google Scholar] [CrossRef]
- Lan, X.; Jing, Y.; Xu, R.; Zhao, L.; Hao, H. Insights into the Ce-doping CuCoAlO with superior resistance to alkali metal poisons for CO-SCR removal of NOx. J. Environ. Chem. Eng. 2023, 11, 110251. [Google Scholar] [CrossRef]
- Han, L.; Cai, S.; Gao, M.; Hasegawa, J.; Wang, P.; Zhang, J.; Shi, L.; Zhang, D. Selective Catalytic Reduction of NOx with NH3 by Using Novel Catalysts: State of the Art and Future Prospects. Chem. Rev. 2019, 119, 10916–10976. [Google Scholar] [CrossRef]
- Zhang, K.; Luo, N.; Huang, Z.; Zhao, G.; Chu, F.; Yang, R.; Tang, X.; Wang, G.; Gao, F.; Huang, X. Recent advances in low-temperature NH3-SCR of NOx over Ce-based catalysts: Performance optimizations, reaction mechanisms and anti-poisoning countermeasures. Chem. Eng. J. 2023, 476, 24. [Google Scholar] [CrossRef]
- Chen, Y.L.; Shu, S.; Wang, S.X.; Li, J.J. Mn-HAP SCR Catalyst: Preparation and Sulfur Resistance. J. Inorg. Mater. 2022, 37, 1065–1072. [Google Scholar] [CrossRef]
- Chen, L.; Li, J.; Ge, M. The poisoning effect of alkali metals doping over nano V2O5-WO3/TiO2 catalysts on selective catalytic reduction of NOx by NH3. Chem. Eng. J. 2011, 170, 531–537. [Google Scholar] [CrossRef]
- Klimczak, M.; Kern, P.; Heinzelmann, T.; Lucas, M.; Claus, P. High-throughput study of the effects of inorganic additives and poisons on NH3-SCR catalysts—Part I: V2O5–WO3/TiO2 catalysts. Appl. Catal. B Environ. 2010, 95, 39–47. [Google Scholar] [CrossRef]
- Kang, K.; Yao, X.; Huang, Y.; Cao, J.; Rong, J.; Zhao, W.; Luo, W.; Chen, Y. Insights into the co-doping effect of Fe3+ and Zr4+ on the anti-K performance of CeTiOx catalyst for NH3-SCR reaction. J. Hazard. Mater. 2021, 416, 125821. [Google Scholar] [CrossRef] [PubMed]
- Ilbeygi, H.; Jaafar, J. Recent Progress on Functionalized Nanoporous Heteropoly Acids: From Synthesis to Applications. Chem. Rec. 2024, 24, 32. [Google Scholar] [CrossRef]
- Sazama, P.; Wichterlova, B.; Tabor, E.; Stastny, P.; Sathu, N.K.; Sobalik, Z.; Dedecek, J.; Sklenak, S.; Klein, P.; Vondrova, A. Tailoring of the structure of Fe-cationic species in Fe-ZSM-5 by distribution of Al atoms in the framework for N2O decomposition and NH3-SCR-NOx. J. Catal. 2014, 312, 123–138. [Google Scholar] [CrossRef]
- Ashrafi, S.; Taghizadeh, M.; Rezaei, N.; Firouzjaee, M.H. Enhancement of Stability and Activity of Pt-Loaded HSiW/UiO-66 Acid Catalyst and Kinetic Modeling for n-Butane Hydroisomerization. Ind. Eng. Chem. Res. 2025, 64, 4809–4822. [Google Scholar] [CrossRef]
- Li, H.; Yu, Z.; Zhang, Z.; Li, S.; Shi, Z.; Ni, C.; Cao, L.; Du, H.; Luo, Q.; Wang, F. In-Depth Understanding of Highly Active Silicotungstic Acid Catalysts for Ethanol Dehydration to Ethylene under Industrially Favorable Conditions. Ind. Eng. Chem. Res. 2024, 63, 7624–7635. [Google Scholar] [CrossRef]
- Putluru, S.S.R.; Schill, L.; Godiksen, A.; Poreddy, R.; Mossin, S.; Jensen, A.D.; Fehrmann, R. Promoted V2O5/TiO2 catalysts for selective catalytic reduction of NO with NH3 at low temperatures. Appl. Catal. B Environ. 2016, 183, 282–290. [Google Scholar] [CrossRef]
- Gupta, S.; Jain, R.; Malpani, S.K.; Yadav, A.; Goyal, D.; Bharti, S.; Shukla, S.K.; Ulucan-Karnak, F. Immobilization of Heteropolyacids on Zeolite Surface for Catalytic Conversion of Glucose to 5-Hydroxymethyl Furfural (5-HMF). Top. Catal. 2024, 17. [Google Scholar] [CrossRef]
- Zhou, X.; Jiao, J.; Jiao, W.; Wang, R. Oxidative desulfurization of model oil over the bowl-shaped N-doped carbon material loaded by the defective silicotungstic acid. Sep. Purif. Technol. 2023, 310, 11. [Google Scholar] [CrossRef]
- Dong, Y.; Ran, M.; Zhang, X.; Lin, S.; Li, W.; Yang, Y.; Song, H.; Wu, W.; Liu, S.; Zheng, C.; et al. Promoting effect of polyoxometallic acid modification on the NH3-SCR performance of Mn-based catalysts. J. Environ. Chem. Eng. 2024, 12, 10. [Google Scholar] [CrossRef]
- Geng, Y.; Lian, Z.; Zhang, Y.; Liu, J.; Jin, D.; Shan, W. Heteropoly acid-grafted iron oxide catalysts for efficient selective catalytic reduction of NOx with NH3. Catal. Sci. Technol. 2024, 14, 3064–3075. [Google Scholar] [CrossRef]
- Xue, H.; Guo, X.; Guo, Q.; Xue, Z.; Yu, J.; Meng, T.; Mao, D. Promotional effects of phosphotungstic acid on the alkali metals poisoning resistance of MnOx catalyst for NH3-SCR. Fuel 2025, 390, 12. [Google Scholar] [CrossRef]
- Huang, Y.; Li, P.; Zhang, R.; Wei, Y. Efficiency of Phosphotungstic Acid Modified Mn-Based Catalysts to Promote Activity and N2 Formation for Selective Catalytic Reduction of NO with Ammonia. Int. J. Chem. React. Eng. 2019, 17, 16. [Google Scholar] [CrossRef]
- Wu, J.; Jin, S.; Wei, X.; Gu, F.; Han, Q.; Lan, Y.; Qian, C.; Li, J.; Wang, X.; Zhang, R.; et al. Enhanced sulfur resistance of H3PW12O40-modified Fe2O3 catalyst for NH3-SCR: Synergistic effect of surface acidity and oxidation ability. Chem. Eng. J. 2021, 412, 16. [Google Scholar] [CrossRef]
- Gao, F.; Walter, E.D.; Kollar, M.; Wang, Y.; Szanyi, J.; Peden, C.H.F. Understanding ammonia selective catalytic reduction kinetics over Cu/SSZ-13 from motion of the Cu ions. J. Catal. 2014, 319, 1–14. [Google Scholar] [CrossRef]
- Qu, R.; Gao, X.; Cen, K.; Li, J. Relationship between structure and performance of a novel cerium-niobium binary oxide catalyst for selective catalytic reduction of NO with NH3. Appl. Catal. B-Environ. 2013, 142, 290–297. [Google Scholar] [CrossRef]
- Jiang, H.; Zhou, J.; Wang, C.; Li, Y.; Chen, Y.; Zhang, M. Effect of Cosolvent and Temperature on the Structures and Properties of Cu-MOF-74 in Low-temperature NH3-SCR. Ind. Eng. Chem. Res. 2017, 56, 3542–3550. [Google Scholar] [CrossRef]
- Zhang, D.; Zhang, L.; Shi, L.; Fang, C.; Li, H.; Gao, R.; Huang, L.; Zhang, J. In situ supported MnOx-CeOx on carbon nanotubes for the low-temperature selective catalytic reduction of NO with NH3. Nanoscale 2013, 5, 1127–1136. [Google Scholar] [CrossRef]
- Zhu, H.; Wang, R. Phosphotungstic acid-promoted Mn-Fe bimetal oxide with high sulfur resistance for low-temperature selective catalytic reduction of nitrogen oxides with NH3. J. Alloys Compd. 2023, 936, 168272. [Google Scholar] [CrossRef]
- Wei-Liang, F.; Yue-Song, S.; She-Min, Z.; Shu-Bao, S. Promotional Effect of Tungsten Incorporation on Catalytic Performance of Ti0.8Zr0.2Ce0.2O2.4/Al2O3-TiO2-SiOz for Selective Catalytic Reduction of NOx by NH3. J. Inorg. Mater. 2014, 29, 1294–1300. [Google Scholar] [CrossRef]
- Yang, J.; Ren, S.; Zhang, T.; Su, Z.; Long, H.; Kong, M.; Yao, L. Iron doped effects on active sites formation over activated carbon supported Mn-Ce oxide catalysts for low-temperature SCR of NO. Chem. Eng. J. 2020, 379, 11. [Google Scholar] [CrossRef]
- Yang, Z.; Huang, B.; Zhang, G.; Dai, M.; Wen, Z.; Li, W. Low-temperature Denitration Mechanism of NH3-SCR over Fe/AC Catalyst. J. Wuhan Univ. Technol. 2023, 38, 475–484. [Google Scholar] [CrossRef]
- Guo, Q.; Jing, W.; Hou, Y.; Huang, Z.; Ma, G.; Han, X.; Sun, D. On the nature of oxygen groups for NH3-SCR of NO over carbon at low temperatures. Chem. Eng. J. 2015, 270, 41–49. [Google Scholar] [CrossRef]
- Wang, D.; Huang, B.; Shi, Z.; Long, H.; Li, L.; Yang, Z.; Dai, M. Influence of cerium doping on Cu-Ni/activated carbon low-temperature CO-SCR denitration catalysts. RSC Adv. 2021, 11, 18458–18467. [Google Scholar] [CrossRef]
- Saghandali, F.; Kazemeini, M.; Sadjadi, S. Halloysite-supported silicotungstic acid as an efficient catalyst for dehydration of fructose to 5-hydroxymethylfurfural. J. Phys. Chem. Solids 2024, 184, 12. [Google Scholar] [CrossRef]
- Ai, L.; Zhang, D.; Wang, Q.; He, F.; Yang, H.; Wu, Q. Preparation of Ti-heteropolyacid/TiO2 and its rapid photocatalytic degradation of X-3B. J. Mater. Sci. Mater. Electron. 2020, 31, 3166–3171. [Google Scholar] [CrossRef]
- Liu, C.; Bi, Y.; Wang, H.; Zhang, Z.; Wang, J.; Guo, M.; Liu, Q. Promotional Effects on NH3-SCR Performance of CeO2-SnO2 Catalysts Doped by TiO2: A Mechanism Study. Catal. Surv. Asia 2021, 25, 48–57. [Google Scholar] [CrossRef]
- Ke, Y.; Huang, W.; Li, S.; Liao, Y.; Li, J.; Qu, Z.; Yan, N. Surface acidity enhancement of CeO2 catalysts via modification with a heteropoly acid for the selective catalytic reduction of NO with ammonia. Catal. Sci. Technol. 2019, 9, 5774–5785. [Google Scholar] [CrossRef]
- Jiang, Y.; Liu, T.; Lai, C.; Yang, Z.; Lin, R.; Wang, X.; Zhu, X. Deactivation of CeO2-TiO2 catalyst by K2SO4 for NH3-SCR: An experimental and DFT study. Appl. Surf. Sci. 2021, 547, 149196. [Google Scholar] [CrossRef]
- Zhao, W.; Rong, J.; Luo, W.; Long, L.; Yao, X. Enhancing the K-poisoning resistance of CeO2-SnO2 catalyst by hydrothermal method for NH3-SCR reaction. Appl. Surf. Sci. 2022, 579, 152176. [Google Scholar] [CrossRef]
- Jiang, Y.; Lai, C.; Li, Q.; Gao, W.; Yang, L.; Yang, Z.; Lin, R.; Wang, X.; Zhu, X. The poisoning effect of KCl and K2O on CeO2-TiO2 catalyst for selective catalytic reduction of NO with NH3. Fuel 2020, 280, 118638. [Google Scholar] [CrossRef]
- Du, X.; Gao, X.; Qu, R.; Ji, P.; Luo, Z.; Cen, K. The Influence of Alkali Metals on the Ce-Ti Mixed Oxide Catalyst for the Selective Catalytic Reduction of NOx. ChemCatChem 2012, 4, 2075–2081. [Google Scholar] [CrossRef]
- Wang, M.; Su, B.; Ren, S.; Liu, W.; Yang, J.; Chen, Z.; Chen, L. Different lead species deactivation on Mn-Ce activated carbon supported catalyst for low-temperature SCR of NO with NH3: Comparison of PbCl2, Pb(NO3)2 and PbSO4. J. Colloid Interface Sci. 2022, 622, 549–561. [Google Scholar] [CrossRef] [PubMed]
- Xue, H.; Guo, X.; Mao, D.; Meng, T.; Yu, J.; Ma, Z. Phosphotungstic Acid-Modified MnOx for Selective Catalytic Reduction of NOx with NH3. Catalysts 2022, 12, 20. [Google Scholar] [CrossRef]
- Liao, M.; Cai, Y.; Chen, L.; Zou, Y.; Li, Y.; Li, G.; Liu, W.; Zhang, H.; Zhang, S.; Lu, S.; et al. Discovering the remarkable deNOx activity and anti-K poisoning of MnFeOx/H-Beta composite catalyst. Sep. Purif. Technol. 2024, 337, 11. [Google Scholar] [CrossRef]
- Ren, S.; Li, S.; Su, Z.; Yang, J.; Long, H.; Kong, M.; Yang, J.; Cai, Z. Poisoning effects of KCl and As2O3 on selective catalytic reduction of NO with NH3 over Mn-Ce/AC catalysts at low temperature. Chem. Eng. J. 2018, 351, 540–547. [Google Scholar] [CrossRef]
- Burg, P.; Fydrych, P.; Cagniant, D.; Nanse, G.; Bimer, J.; Jankowska, A. The characterization of nitrogen-enriched activated carbons by IR, XPS and LSER methods. Carbon 2002, 40, 1521–1531. [Google Scholar] [CrossRef]
- Zhang, L.; Cui, S.; Guo, H.; Ma, X.; Luo, X. The influence of K+ cation on the MnOx-CeO2/TiO2 catalysts for selective catalytic reduction of NOx with NH3 at low temperature. J. Mol. Catal. A Chem. 2014, 390, 14–21. [Google Scholar] [CrossRef]
- Kong, M.; Liu, Q.; Zhou, J.; Jiang, L.; Tian, Y.; Yang, J.; Ren, S.; Li, J. Effect of different potassium species on the deactivation of V2O5-WO3/TiO2 SCR catalyst: Comparison of K2SO4, KCl and K2O. Chem. Eng. J. 2018, 348, 637–643. [Google Scholar] [CrossRef]
- Karatepe, N.; Orbak, I.; Yavuz, R.; Ozyuguran, A. Sulfur dioxide adsorption by activated carbons having different textural and chemical properties. Fuel 2008, 87, 3207–3215. [Google Scholar] [CrossRef]
- Zhao, Y.; Shi, L.; Shen, Y.; Zhou, J.; Jia, Z.; Yan, T.; Wang, P.; Zhang, D. Self-Defense Effects of Ti-Modified Attapulgite for Alkali-Resistant NOx Catalytic Reduction. Environ. Sci. Technol. 2022, 56, 4386–4395. [Google Scholar] [CrossRef]
- Zheng, L.; Zhou, M.; Huang, Z.; Chen, Y.; Gao, J.; Ma, Z.; Chen, J.; Tang, X. Self-Protection Mechanism of Hexagonal WO3-Based DeNOx Catalysts against Alkali Poisoning. Environ. Sci. Technol. 2016, 50, 11951–11956. [Google Scholar] [CrossRef]
- Yao, M.; Wang, Z.; Li, Y.; Niu, X.; Zhu, Y. Enhanced resistance to K poisoning and SO2&H2O on Ce0.63TiOx catalyst by sulfation treatment. Sep. Purif. Technol. 2024, 348, 127733. [Google Scholar]
- Jiang, Y.; Lai, C.; Li, Q.; Gao, W.; Yang, L.; Yang, Z.; Lin, R.; Wang, X.; Zhu, X. The enhanced Pb resistance of CeO2/TiO2 catalyst for selective catalytic reduction of NO with NH3 by the modification with W. Mol. Catal. 2021, 514, 11. [Google Scholar] [CrossRef]
- Gao, F.; Washton, N.M.; Wang, Y.; Kollar, M.; Szanyi, J.; Peden, C.H.F. Effects of Si/Al ratio on Cu/SSZ-13 NH3-SCR catalysts: Implications for the active Cu species and the roles of Bronsted acidity. J. Catal. 2015, 331, 25–38. [Google Scholar] [CrossRef]
- Peng, Y.; Li, J.; Chen, L.; Chen, J.; Han, J.; Zhang, H.; Han, W. Alkali Metal Poisoning of a CeO2-WO3 Catalyst Used in the Selective Catalytic Reduction of NOx with NH3: An Experimental and Theoretical Study. Environ. Sci. Technol. 2012, 46, 2864–2869. [Google Scholar] [CrossRef]
- Zhou, Z.; Lan, J.; Liu, L.; Liu, Z. Enhanced alkali resistance of sulfated CeO2 catalyst for the reduction of NOx from biomass fired flue gas. Catal. Commun. 2021, 149, 106230. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).