
Recycling Positive Electrode Materials of Li-Ion Batteries by Creating a pH Gradient Within Aqueous Sodium Chloride Electrolyser


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Experimental Section
2.1. Electrochemical Cell
2.2. Quantification of the Cl2 Produced at the Anode Chamber of the Electrolyser
2.3. Characterisation
3. Results and Discussion
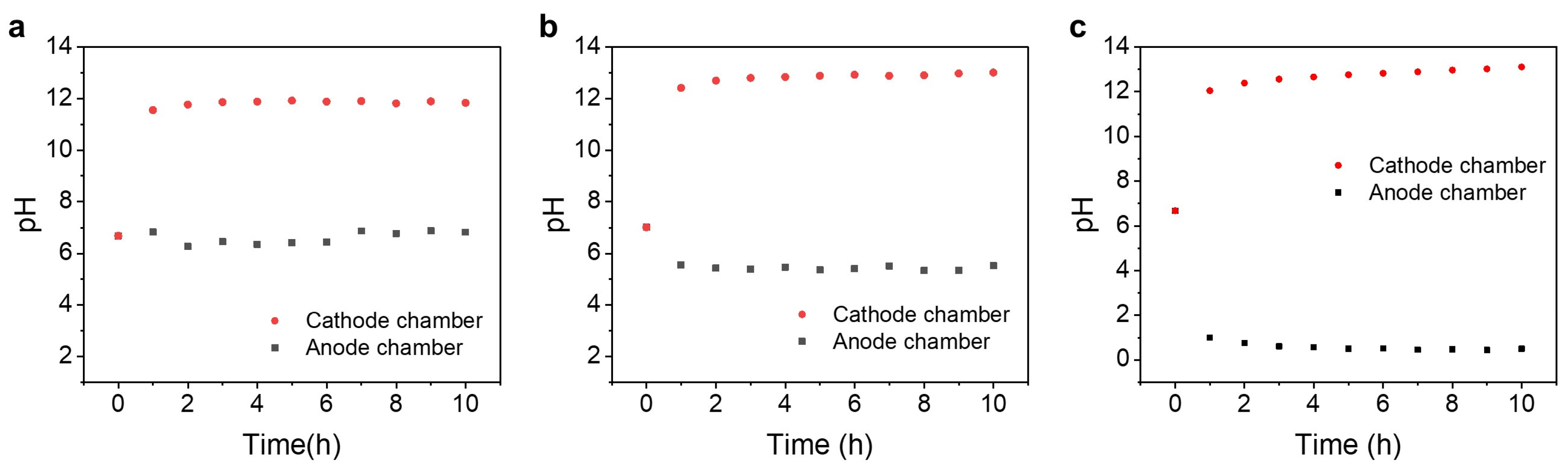
3.1. pH Value Change in the Anode and Cathode Chambers
3.2. Quantification of the Cl2 Product
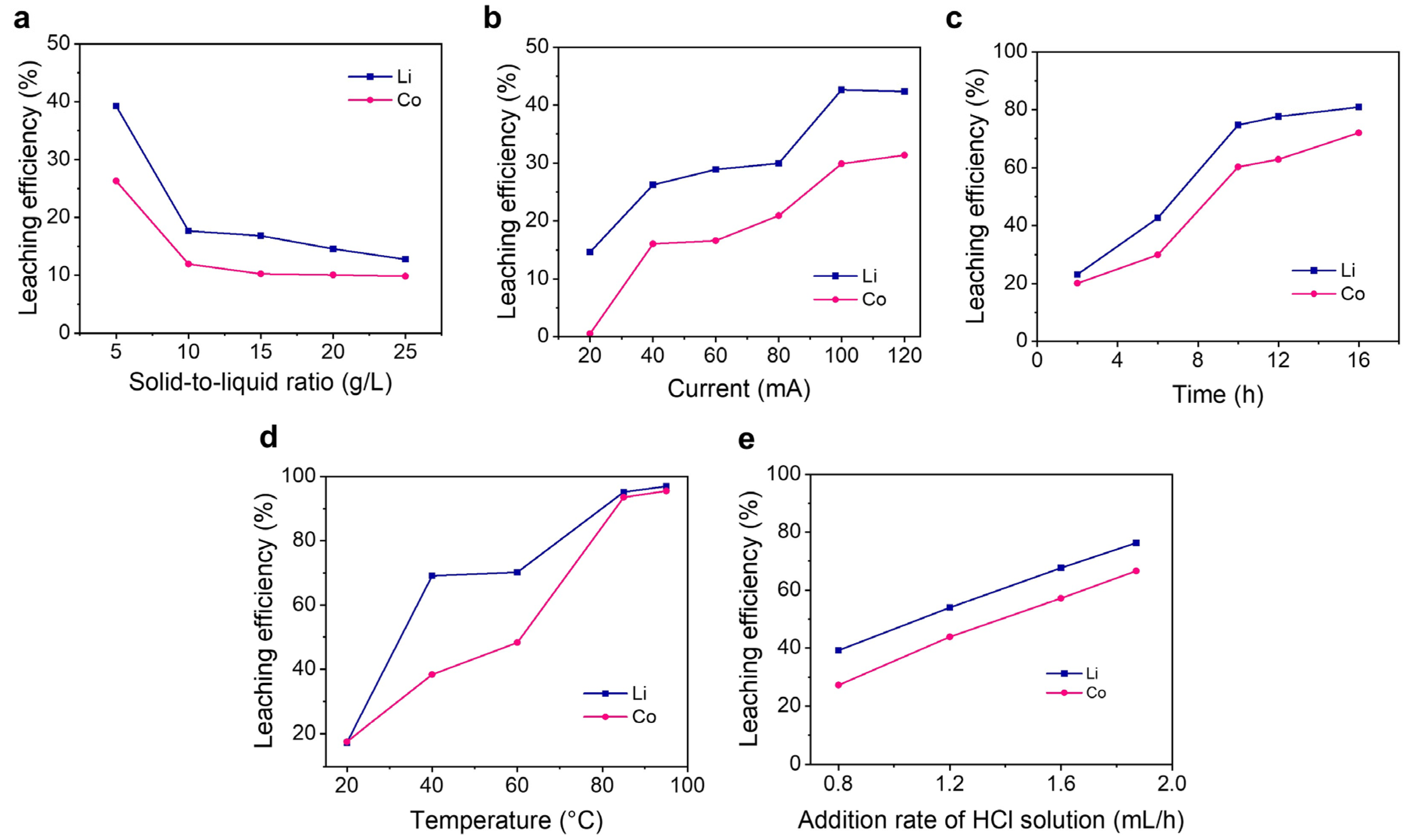
3.3. Effects of Parameters on Leaching Efficiencies of Li and Co
3.3.1. Effect of Solid-to-Liquid Ratio
3.3.2. Effects of Applied Current and Duration
3.3.3. Effect of Temperature
3.3.4. Effect of the HCl Addition Rate
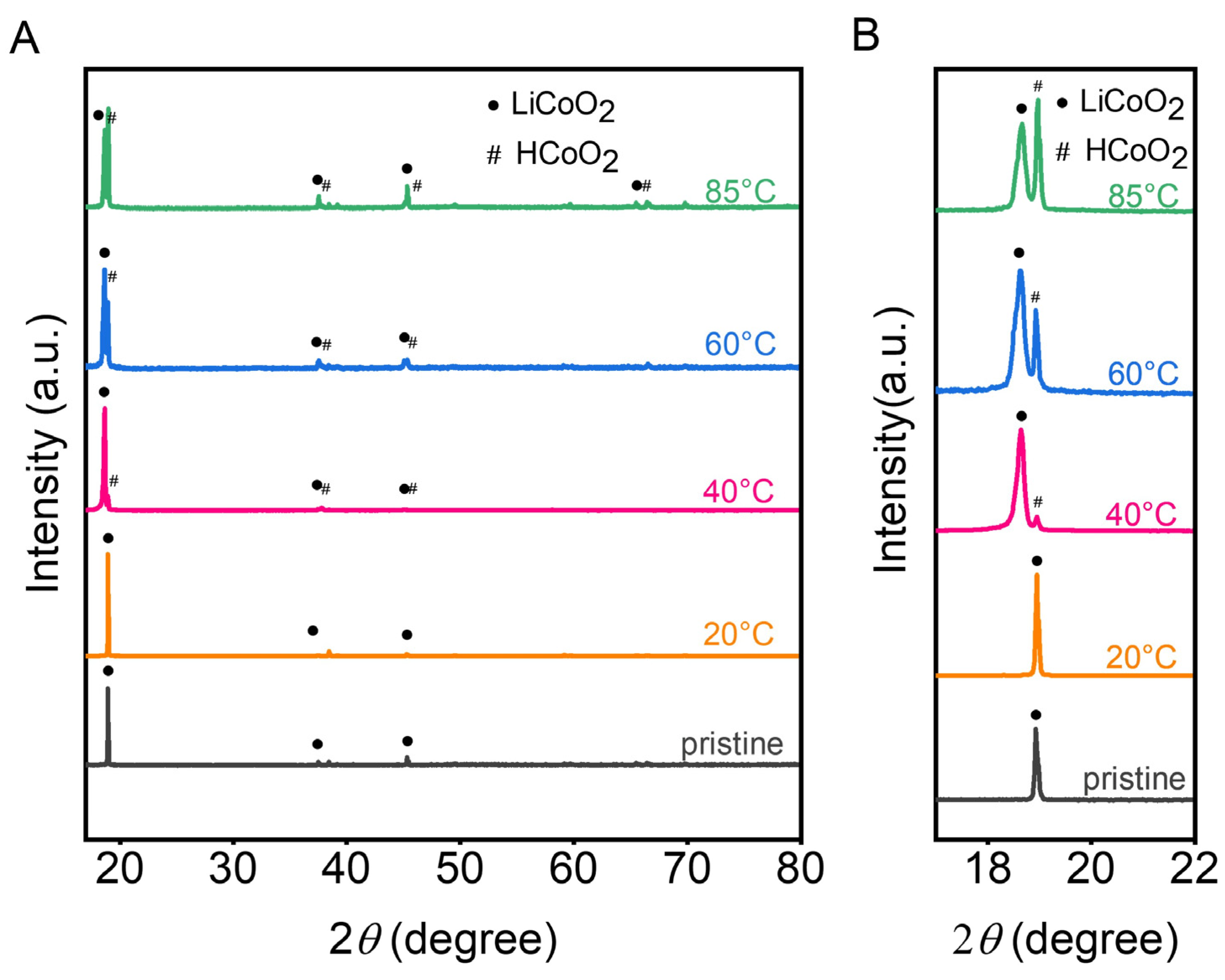
3.4. Materials Characterisation
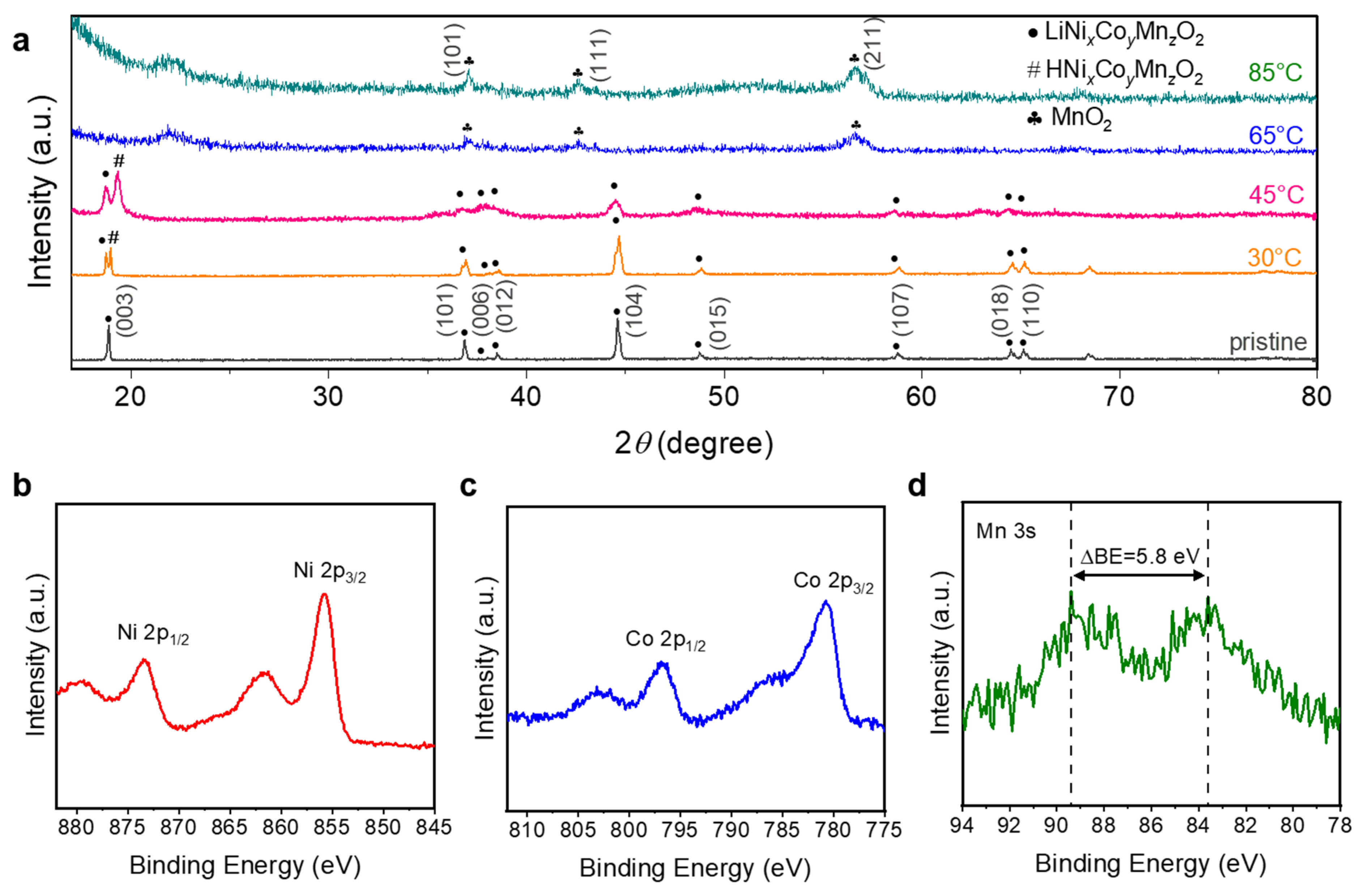
3.4.1. The Residue Powders in the Anode Chambers
3.4.2. Precipitates in the Cathode Chamber
3.4.3. The DSA in the Anode Chamber
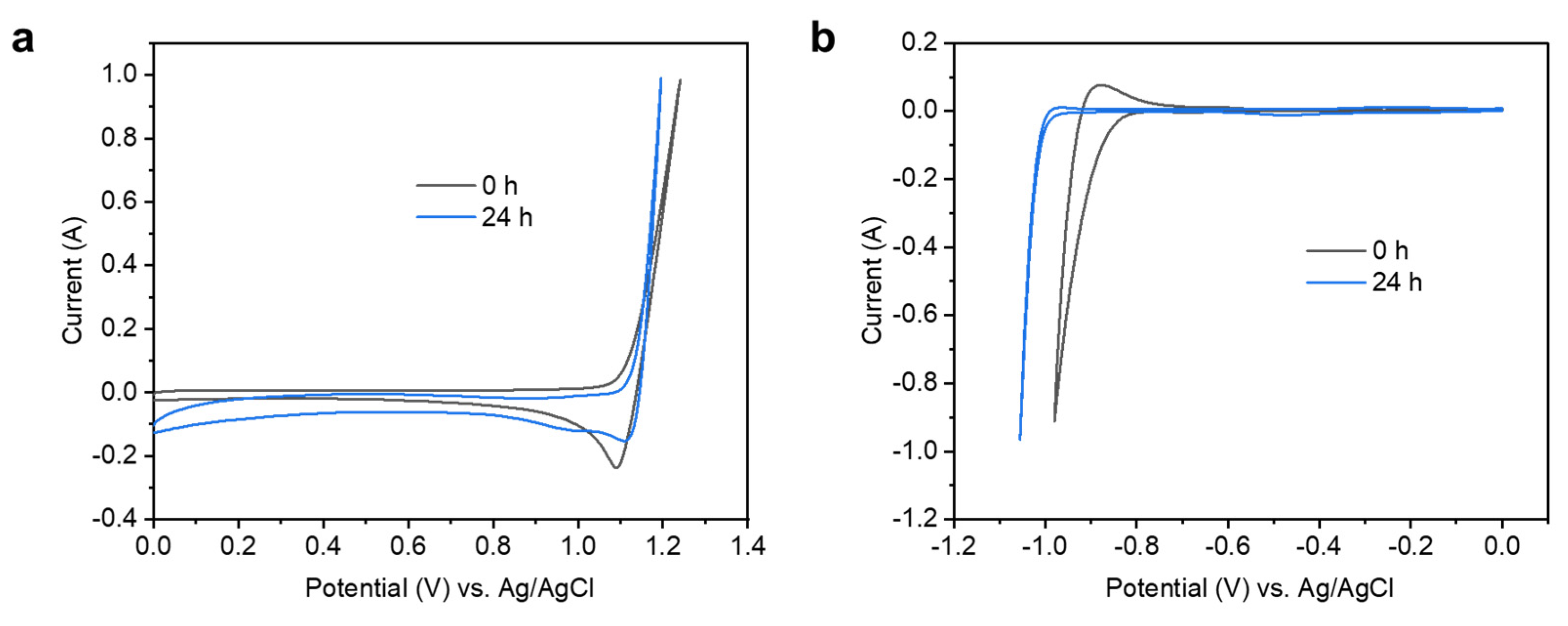
3.5. Electrochemical Characterisation of the Electrolytic Cell
3.6. Recycling of LiNi0.5Co0.2Mn0.3O2 (NCM523)
LiCl + 0.5NiCl2 + 0.2CoCl2 + 0.3MnCl2 + 0.5Cl2 + H2O
3.7. Discussion on Challenges to Address for Large-Scale Applications
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tarascon, J.M.; Armand, M. Issues and challenges facing rechargeable lithium batteries. Nature 2001, 414, 359–367. [Google Scholar] [CrossRef] [PubMed]
- Goodenough, J.B.; Kim, Y. Challenges for Rechargeable Li Batteries. Chem. Mater. 2010, 22, 587–603. [Google Scholar] [CrossRef]
- Kebede, A.A.; Kalogiannis, T.; Van Mierlo, J.; Berecibar, M. A comprehensive review of stationary energy storage devices for large scale renewable energy sources grid integration. Renew. Sustain. Energy Rev. 2022, 159, 112213. [Google Scholar] [CrossRef]
- Velázquez-Martínez, O.; Valio, J.; Santasalo-Aarnio, A.; Reuter, M.; Serna-Guerrero, R. A Critical Review of Lithium-Ion Battery Recycling Processes from a Circular Economy Perspective. Batteries 2019, 5, 68. [Google Scholar] [CrossRef]
- Li, H.; Berbille, A.; Zhao, X.; Wang, Z.; Tang, W.; Wang, Z.L. A contact-electro-catalytic cathode recycling method for spent lithium-ion batteries. Nat. Energy 2023, 8, 1137–1144. [Google Scholar] [CrossRef]
- Sederholm, J.G.; Li, L.; Liu, Z.; Lan, K.-W.; Cho, E.J.; Gurumukhi, Y.; Dipto, M.J.; Ahmari, A.; Yu, J.; Haynes, M.; et al. Emerging Trends and Future Opportunities for Battery Recycling. ACS Energy Lett. 2025, 10, 107–119. [Google Scholar] [CrossRef]
- Fu, Y.; He, Y.; Li, J.; Qu, L.; Yang, Y.; Guo, X.; Xie, W. Improved hydrometallurgical extraction of valuable metals from spent lithium-ion batteries via a closed-loop process. J. Alloys Compd. 2020, 847, 156489. [Google Scholar] [CrossRef]
- Gao, W.; Song, J.; Cao, H.; Lin, X.; Zhang, X.; Zheng, X.; Zhang, Y.; Sun, Z. Selective recovery of valuable metals from spent lithium-ion batteries—Process development and kinetics evaluation. J. Clean. Prod. 2018, 178, 833–845. [Google Scholar] [CrossRef]
- Assefi, M.; Maroufi, S.; Yamauchi, Y.; Sahajwalla, V. Pyrometallurgical recycling of Li-ion, Ni–Cd and Ni–MH batteries: A minireview. Curr. Opin. Green Sustain. Chem. 2020, 24, 26–31. [Google Scholar] [CrossRef]
- Havlík, T. Chapter 16—The Current State and Prospects of Hydrometallurgical Processes. In Hydrometallurgy; Havlík, T., Ed.; Woodhead Publishing: Sawston, UK, 2008; pp. 479–534. [Google Scholar]
- Minimol, M.; Shetty, V.; Saidutta, M.B. Biohydrometallurgical Methods and the Processes Involved in the Bioleaching of WEEE; Elsevier: Boston, MA, USA, 2021; pp. 89–107. [Google Scholar]
- Larouche, F.; Tedjar, F.; Amouzegar, K.; Houlachi, G.; Bouchard, P.; Demopoulos, G.P.; Zaghib, K. Progress and Status of Hydrometallurgical and Direct Recycling of Li-Ion Batteries and Beyond. Materials 2020, 13, 801. [Google Scholar] [CrossRef]
- Vieceli, N.; Benjamasutin, P.; Promphan, R.; Hellström, P.; Paulsson, M.; Petranikova, M. Recycling of Lithium-Ion Batteries: Effect of Hydrogen Peroxide and a Dosing Method on the Leaching of LCO, NMC Oxides, and Industrial Black Mass. ACS Sustain. Chem. Eng. 2023, 11, 9662–9673. [Google Scholar] [CrossRef]
- Guimarães, L.F.; Botelho Junior, A.B.; Espinosa, D.C.R. Sulfuric acid leaching of metals from waste Li-ion batteries without using reducing agent. Miner. Eng. 2022, 183, 107597. [Google Scholar] [CrossRef]
- Zhou, J.; Ni, J.; Guan, X. The critical role of H2 reduction roasting for enhancing the recycling of spent Li-ion battery cathodes in the subsequent neutral water electrolysis. RSC Sustain. 2023, 1, 2241–2253. [Google Scholar] [CrossRef]
- Zhou, C.; Ni, J.; Chen, H.; Guan, X. Harnessing electrochemical pH gradient for direct air capture with hydrogen and oxygen by-products in a calcium-based loop. Sustain. Energy Fuels 2021, 5, 4355–4367. [Google Scholar] [CrossRef]
- Zhou, J.; Guan, X. Closed-Loop and Precipitation-Free CO2 Capture Process Enabled by Electrochemical pH Gradient. ChemSusChem 2025, 18, e202401533. [Google Scholar] [CrossRef]
- Drajlin, D.S.; Suarez, D.S.; Toro, N.; Gálvez, E.D.; Pinna, E.G.; Rodriguez, M.H. Comparative Study of the Dissolution of LCO in HCl Medium with and without H2O2. Metals 2022, 12, 727. [Google Scholar] [CrossRef]
- Freitas, M.B.J.G.; Garcia, E.M. Electrochemical recycling of cobalt from cathodes of spent lithium-ion batteries. J. Power Sources 2007, 171, 953–959. [Google Scholar] [CrossRef]
- Ito, H.; Manabe, A. Chapter 8—Chlor–Alkali Electrolysis. In Electrochemical Power Sources: Fundamentals, Systems, and Applications; Smolinka, T., Garche, J., Eds.; Elsevier: Boston, MA, USA, 2022; pp. 281–304. [Google Scholar]
- Fauvarque, J. The Chlorine Industry. Pure Appl. Chem. 1996, 68, 1713–1720. [Google Scholar] [CrossRef]
- Karlsson, R.K.B.; Cornell, A. Selectivity between Oxygen and Chlorine Evolution in the Chlor-Alkali and Chlorate Processes. Chem. Rev. 2016, 116, 2982–3028. [Google Scholar] [CrossRef]
- Zhou, J.; Bing, J.; Ni, J.; Wang, X.; Guan, X. Recycling the waste LiMn2O4 of spent Li-ion batteries by pH gradient in neutral water electrolyser. Mater. Today Sustain. 2022, 20, 100205. [Google Scholar] [CrossRef]
- Ni, J.; Zhou, J.; Bing, J.; Guan, X. Recycling the cathode materials of spent Li-ion batteries in a H-Shaped neutral water electrolysis cell. Sep. Purif. Technol. 2021, 278, 119485. [Google Scholar] [CrossRef]
- Fu, A.; Zhang, Z.; Lin, J.; Zou, Y.; Qin, C.; Xu, C.; Yan, P.; Zhou, K.; Hao, J.; Yang, X.; et al. Highly stable operation of LiCoO₂ at cut-off ≥ 4.6 V enabled by synergistic structural and interfacial manipulation. Energy Storage Mater. 2022, 46, 406–416. [Google Scholar] [CrossRef]
- Kumar, A.; Phillips, K.R.; Thiel, G.P.; Schröder, U.; Lienhard, J.H. Direct electrosynthesis of sodium hydroxide and hydrochloric acid from brine streams. Nat. Catal. 2019, 2, 106–113. [Google Scholar] [CrossRef]
- Dreyhsig, G.H.; Voßnacker, P.; Kleoff, M.; Baunis, H.; Limberg, N.; Lu, M.; Schomäcker, R.; Riedel, S. Bichloride-based ionic liquids for the merged storage, processing, and electrolysis of hydrogen chloride. Sci. Adv. 2024, 10, eadn5353. [Google Scholar] [CrossRef] [PubMed]
- Carvela, M.; Raschitor, A.; Rodrigo, M.A.; Lobato, J. Recent Progress in Catalysts for Hydrogen-Chlorine Regenerative Fuel Cells. Catalysts 2020, 10, 1263. [Google Scholar] [CrossRef]
- Wang, Y.; Li, M.; Jiang, Y.; Zhang, C.; Chang, W.; Shi, Y. Investigation of swirler effect on combustion and exergy efficiency of hydrogen/chlorine combustor based on entropy generation analysis. Energy 2024, 300, 131593. [Google Scholar] [CrossRef]
- Gupta, A.K.; Parker, R.Z.; Hanrahan, R.J. Gas phase formation of hydrogen chloride by thermal chlorine-steam reaction. Int. J. Hydrogen Energy 1991, 16, 677–682. [Google Scholar] [CrossRef]
- Meng, J.-X.; Wang, X.-B.; Ruan, G.-L.; Li, G.-Q.; Deng, Z.-X. Determination of chlorine in atmosphere by kinetic spectrophotometry. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2005, 61, 823–827. [Google Scholar] [CrossRef]
- Xu, K.; Liu, D.; Feng, L.; Jin, J.; Xiong, Z.; Ni, M.; Liu, Z.; Liu, Q.; Hou, F. Mercury removal by Co3O4@TiO2@Fe2O3 magnetic core-shell oxygen carrier in chemical-looping combustion. Fuel 2021, 306, 121604. [Google Scholar] [CrossRef]
- Bajpai, P. Chapter Six—Chlorine Dioxide Bleaching. In Environmentally Benign Approaches for Pulp Bleaching, 2nd ed.; Bajpai, P., Ed.; Elsevier: Boston, MA, USA, 2012; pp. 135–165. [Google Scholar]
- Noguchi, D. Methyl orange: A brief note on its structural changes. Int. Res. J. Sci. Technol. Educ. Manag. 2024, 4, 50–57. [Google Scholar]
- Le Luu, T.; Kim, J.; Yoon, J. Physicochemical properties of RuO2 and IrO2 electrodes affecting chlorine evolutions. J. Ind. Eng. Chem. 2015, 21, 400–404. [Google Scholar] [CrossRef]
- Exner, K.S. Beyond Dimensionally Stable Anodes: Single-Atom Catalysts with Superior Chlorine Selectivity. ChemElectroChem 2020, 7, 1528–1530. [Google Scholar] [CrossRef]
- Costa Reis, M. Ion activity models: The Debye-Hückel equation and its extensions. ChemTexts 2021, 7, 9. [Google Scholar] [CrossRef]
- Ruocco, G. Mass Transfer by Diffusion and Convection. In Introduction to Transport Phenomena Modeling: A Multiphysics, General Equation-Based Approach; Ruocco, G., Ed.; Springer International Publishing: Cham, Switzerland, 2018; pp. 201–239. [Google Scholar]
- Schalenbach, M.; Hecker, B.; Schmid, B.; Durmus, Y.E.; Tempel, H.; Kungl, H.; Eichel, R.-A. Ionic transport modeling for liquid electrolytes—Experimental evaluation by concentration gradients and limited currents. Electrochem. Sci. Adv. 2023, 3, e2100189. [Google Scholar] [CrossRef]
- Kim, K.; Raymond, D.; Candeago, R.; Su, X. Selective cobalt and nickel electrodeposition for lithium-ion battery recycling through integrated electrolyte and interface control. Nat. Commun. 2021, 12, 6554. [Google Scholar] [CrossRef]
- Guo, Y.; Li, F.; Zhu, H.; Li, G.; Huang, J.; He, W. Leaching lithium from the anode electrode materials of spent lithium-ion batteries by hydrochloric acid (HCl). Waste Manag. 2016, 51, 227–233. [Google Scholar] [CrossRef]
- Sakaguchi, I. Fick’s Law. In Encyclopedia of Geochemistry: A Comprehensive Reference Source on the Chemistry of the Earth; White, W.M., Ed.; Springer International Publishing: Cham, Switzerland, 2018; pp. 483–484. [Google Scholar]
- Lee, K.S.; Myung, S.T.; Amine, K.; Yashiro, H.; Sun, Y.K. Structural and Electrochemical Properties of Layered Li [Ni1 − 2x Cox Mnx]O2 (x = 0.1–0.3) Positive Electrode Materials for Li-Ion Batteries. J. Electrochem. Soc. 2007, 154, A971. [Google Scholar] [CrossRef]
- Ma, M.; Chernova, N.A.; Toby, B.H.; Zavalij, P.Y.; Whittingham, M.S. Structural and electrochemical behavior of LiMn0.4Ni0.4Co0.2O2. J. Power Sources 2007, 165, 517–534. [Google Scholar] [CrossRef]
- Reimers, J.N.; Dahn, J.R. Electrochemical and In Situ X-Ray Diffraction Studies of Lithium Intercalation in Lix CoO2. J. Electrochem. Soc. 1992, 139, 2091. [Google Scholar] [CrossRef]
- Ngala, J.K.; Chernova, N.A.; Ma, M.; Mamak, M.; Zavalij, P.Y.; Whittingham, M.S. The synthesis, characterization and electrochemical behavior of the layered LiNi0.4Mn0.4Co0.2O2 compound. J. Mater. Chem. 2004, 14, 214–220. [Google Scholar] [CrossRef]
- Gupta, R.; Manthiram, A. Chemical Extraction of Lithium from Layered LiCoO2. J. Solid State Chem. 1996, 121, 483–491. [Google Scholar] [CrossRef]
- Billy, E.; Joulié, M.; Laucournet, R.; Boulineau, A.; De Vito, E.; Meyer, D. Dissolution Mechanisms of LiNi1/3Mn1/3Co1/3O2 Positive Electrode Material from Lithium-Ion Batteries in Acid Solution. ACS Appl. Mater. Interfaces 2018, 10, 16424–16435. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Rodríguez, J.M.; Hernán, L.; Morales, J.; Tirado, J.L. Low-temperature hydrothermal transformations of LiCoO2 and HCoO2. Mater. Res. Bull. 1988, 23, 899–904. [Google Scholar] [CrossRef]
- Santana, I.L.; Moreira, T.F.M.; Lelis, M.F.F.; Freitas, M.B.J.G. Photocatalytic properties of Co3O4/LiCoO2 recycled from spent lithium-ion batteries using citric acid as leaching agent. Mater. Chem. Phys. 2017, 190, 38–44. [Google Scholar] [CrossRef]
- Ravina; Dalela, S.; Kumar, S.; Choudhary, B.L.; Alvi, P.A. Structural, optical and Raman studies of Co3O4 nano-particles. Mater. Today Proc. 2023, 79, 165–168. [Google Scholar] [CrossRef]
- Clarke, T.A.; Rizkalla, E.N. X-ray photoelectron spectroscopy of some silicates. Chem. Phys. Lett. 1976, 37, 523–526. [Google Scholar] [CrossRef]
- Sheng, H.; Zhang, X.; Ma, Y.; Wang, P.; Zhou, J.; Su, Q.; Lan, W.; Xie, E.; Zhang, C.J. Ultrathin, Wrinkled, Vertically Aligned Co(OH)2 Nanosheets/Ag Nanowires Hybrid Network for Flexible Transparent Supercapacitor with High Performance. ACS Appl. Mater. Interfaces 2019, 11, 8992–9001. [Google Scholar] [CrossRef]
- Zhang, F.; Zhou, J.; Chen, X.; Zhao, S.; Zhao, Y.; Tang, Y.; Tian, Z.; Yang, Q.; Slavcheva, E.; Lin, Y.; et al. The Recent Progresses of Electrodes and Electrolysers for Seawater Electrolysis. Nanomaterials 2024, 14, 329. [Google Scholar] [CrossRef]
- Zhang, H.-M.; Zuo, L.; Li, J.; Zhang, S.; Guo, J.; Li, X.-P.; Liu, G.; Wang, P.; Sun, J. Research and strategies for efficient electrocatalysts towards anodic oxygen evolution reaction in seawater electrolysis system. J. Mater. Sci. Technol. 2024, 187, 123–140. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, Y.; Wiley, D.; Zhao, S.; Tang, Z. Recent advances in electrocatalytic chloride oxidation for chlorine gas production. J. Mater. Chem. A 2021, 9, 18974–18993. [Google Scholar] [CrossRef]
- Haynes, W.M. (Ed.) CRC Handbook of Chemistry and Physics, 97th ed.; CRC Press: Boca Raton, FL, USA, 2016. [Google Scholar]
- Fu, Z.; Hu, J.; Hu, W.; Yang, S.; Luo, Y. Quantitative analysis of Ni2+/Ni3+ in Li[NixMnyCoz]O2 cathode materials: Non-linear least-squares fitting of XPS spectra. Appl. Surf. Sci. 2018, 441, 1048–1056. [Google Scholar] [CrossRef]
- Zheng, S.; Xu, S.; Wang, Z.; Duan, H.; Chen, D.; Long, M.; Li, Y. Efficient leaching of valuable metals from spent lithium-ion batteries using green deep eutectic solvents: Process optimization, mechanistic analysis, and environmental impact assessment. J. Clean. Prod. 2024, 480, 144128. [Google Scholar] [CrossRef]
- Yang, J.; Ren, S.; Su, B.; Zhou, Y.; Hu, G.; Jiang, L.; Cao, J.; Liu, W.; Yao, L.; Kong, M.; et al. Insight into N2O Formation Over Different Crystal Phases of MnO2 During Low-Temperature NH3–SCR of NO. Catal. Lett. 2021, 151, 2964–2971. [Google Scholar] [CrossRef]
- Toupin, M.; Brousse, T.; Bélanger, D. Influence of Microstucture on the Charge Storage Properties of Chemically Synthesized Manganese Dioxide. Chem. Mater. 2002, 14, 3946–3952. [Google Scholar] [CrossRef]
- Cifuentes, L.A.M.; Crisóstomo, G. Corrosion behaviour and catalytic effectiveness of Pb–Ca–Sn, RuO2–IrO2/Ti and IrO2–Ta2O5/Ti anodes for copper electrowinning. Corros. Eng. Sci. Technol. 2011, 46, 737–744. [Google Scholar] [CrossRef]
- Xie, Y.; Zhong, L.; Sun, G. Optimization of Conditions for Chlorine Determination by Methyl Orange Spectrophotometry. Light Ind. Sci. Technol. 2010, 26, 8–9+16. (In Chinese) [Google Scholar]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, Y.; Guan, X. Recycling Positive Electrode Materials of Li-Ion Batteries by Creating a pH Gradient Within Aqueous Sodium Chloride Electrolyser. Processes 2025, 13, 1525. https://doi.org/10.3390/pr13051525
Chen Y, Guan X. Recycling Positive Electrode Materials of Li-Ion Batteries by Creating a pH Gradient Within Aqueous Sodium Chloride Electrolyser. Processes. 2025; 13(5):1525. https://doi.org/10.3390/pr13051525
Chicago/Turabian StyleChen, Yue, and Xiaofei Guan. 2025. "Recycling Positive Electrode Materials of Li-Ion Batteries by Creating a pH Gradient Within Aqueous Sodium Chloride Electrolyser" Processes 13, no. 5: 1525. https://doi.org/10.3390/pr13051525
APA StyleChen, Y., & Guan, X. (2025). Recycling Positive Electrode Materials of Li-Ion Batteries by Creating a pH Gradient Within Aqueous Sodium Chloride Electrolyser. Processes, 13(5), 1525. https://doi.org/10.3390/pr13051525