3.3. Thermodynamic Model of Vapor–Liquid Equilibrium
According to the different calculation methods for the fugacity of liquid-phase mixtures, the thermodynamic methods for calculating vapor–liquid equilibrium can be divided into the equation of state method and the activity coefficient method.
The equation of state method relies on the equation of state (EOS) and mixing rules when calculating the vapor–liquid equilibrium. The calculation of two-phase fugacity needs to apply the same equation of state and the same mixing rule at the same time. Theoretically, this method is applicable to the calculation of vapor–liquid equilibrium from normal pressure to high pressure. However, due to the influence of fluid polarity on the mixing rules, the advantages of the equation of state method will become more prominent under selected pressure conditions, especially under high-pressure conditions and near-critical conditions. The widely used equation of state includes the cubic equation of state and multiparametric equation.
The cubic equation includes the van der Waals (vdW), Redlich–Kwong (RK), Soave–Redlich–Kwong (SRK), and Peng–Robinson (PR) equations [
26]. The vdW equation is the first equation of state applicable to a real gas. It is based on the ideal gas law and modified by introducing the intermolecular gravitational parameter a and the molecular volume parameter b. On the basis of the vdW equation, the RK equation introduces temperature parameters to correct it, which greatly improves the calculation accuracy compared to the vdW equation. However, there is still a significant deviation in the calculation of highly polar substances and hydrogen bonds. Soave corrected the temperature parameters of the RK equation and introduced an eccentricity factor ω, improving the calculation accuracy of the RK equation for substances with high polarity and hydrogen bonding. In 1976, Peng and Robinson improved the SRK equation, proposing a PR equation that better reflects the characteristics of real fluids. In addition, they improved the accuracy of the SRK equation in calculating saturated vapor pressure and liquid density.
In addition to the cubic equation of state, researchers also proposed multiparametric equations using specific systems, such as the Bendict–Webb–Rubin (BWR) equation [
27] for hydrocarbon systems and the Martin–Hou (MH) equation [
28] that is widely used in the field of synthetic ammonia. Compared with the simple cubic equation of state, the multiparametric equation has more characteristic constants to describe the system and can accurately describe the vapor–liquid equilibrium relationship of the system for a wider range of temperatures and pressures. However, the complexity of this equation has increased the difficulty and number of calculations required.
In the activity coefficient method, the properties of the vapor–liquid phase are described by the equation of state method and the activity coefficient method. Different from the equation of state, the activity coefficient method can be applied to many types of systems and is not affected by mixing rules. It has higher applicability and accuracy under medium- and low-pressure conditions. The activity coefficient method can be divided into the regular solution model, the local composition equation based on the local composition theory, and the group contribution model according to the different classifications of solutions.
In a regular solution, the collision of molecules is completely random, and the composition distribution of each part in the solution is uniform. However, due to different intermolecular interactions, the collision of molecules is not completely random. The equations based on this theory mainly include the Scatchard–Hildebrand equation, Whol equation, Scatchard–Hammer equation, etc., and their applicability is very limited.
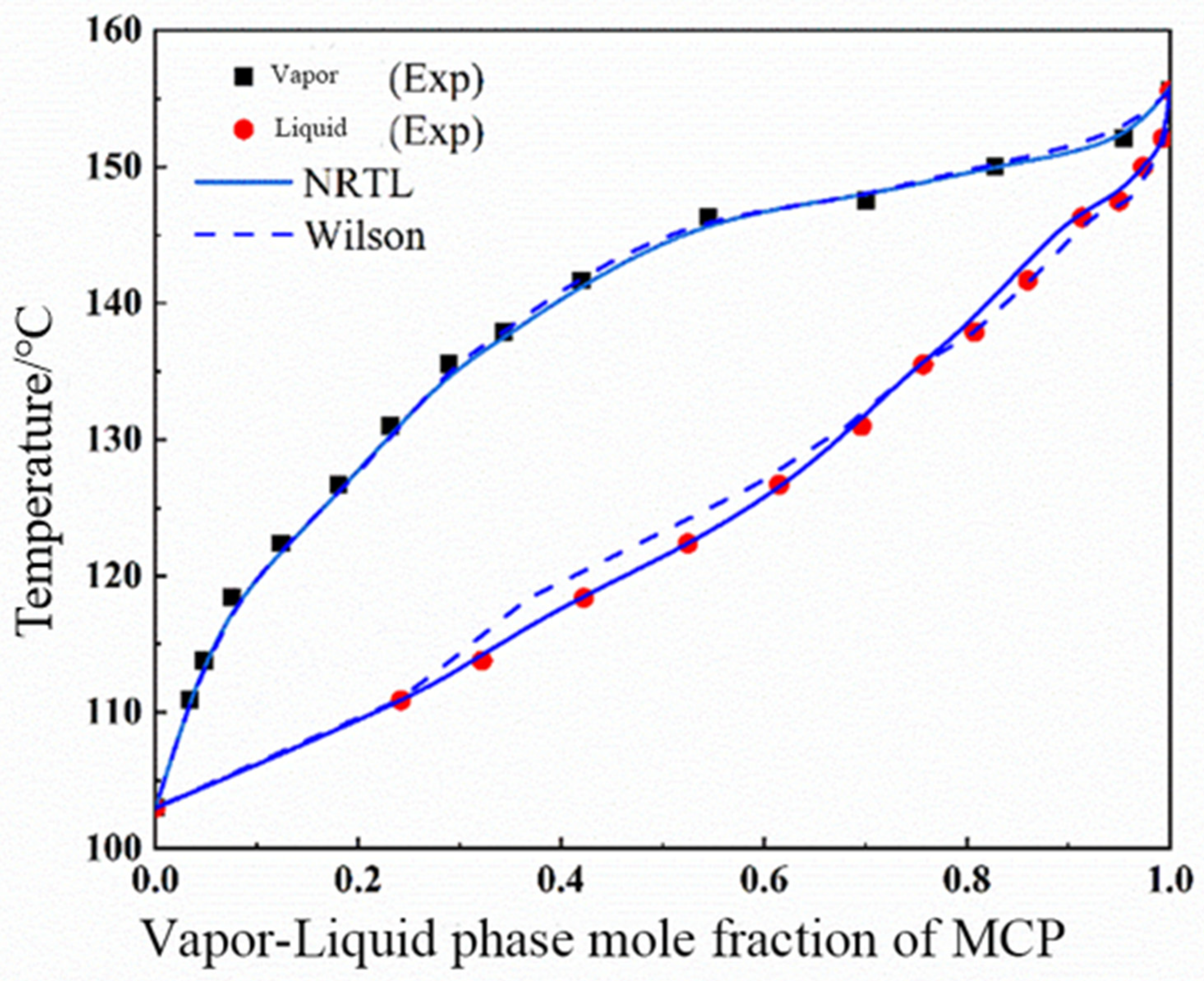
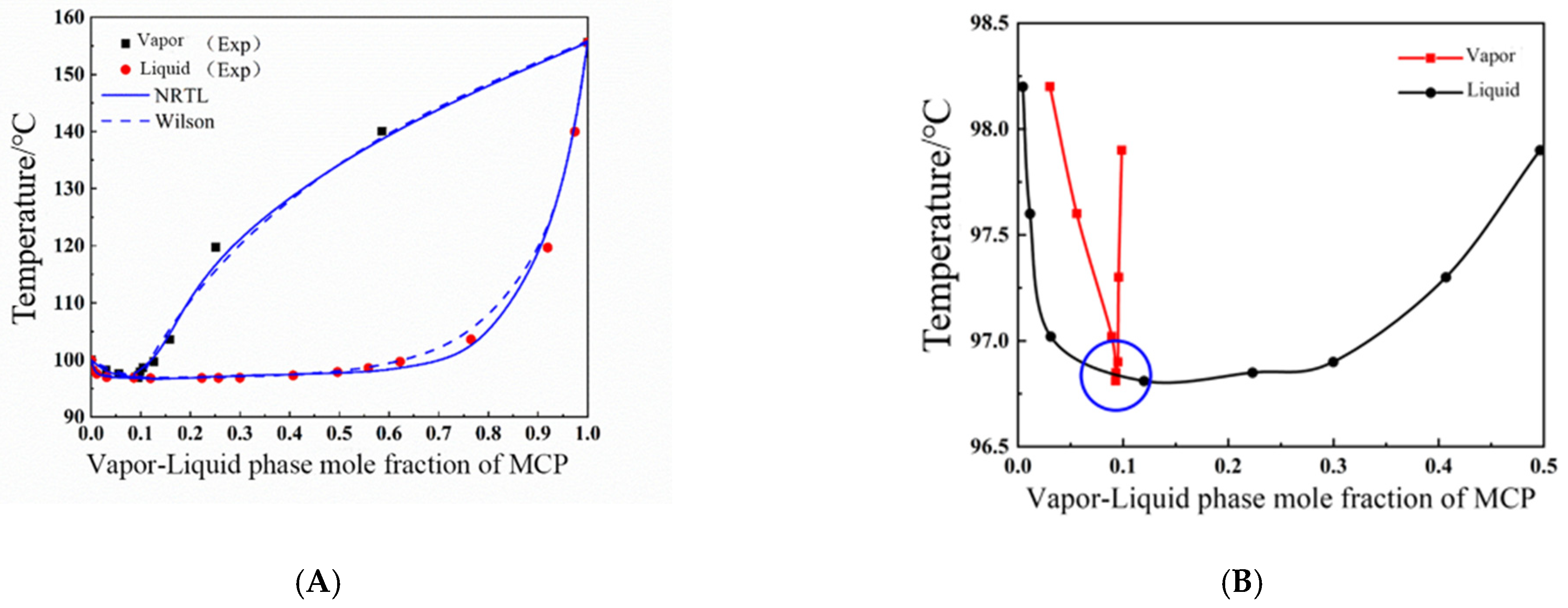
The theory of local composition suggests that the local composition of a solution is not equal to its overall composition. In a solution, the probability of other component atoms appearing around a certain component atom is influenced by the combination of component concentration and intermolecular interactions. The Wilson equation is the first equation established based on the local theory, in addition to the Non-Random Two Liquid (NRTL) equation applicable to partially miscible systems, as well as the Universal Mass Chemical Model (UNIQUAC) equation proposed based on the concept of local composition and lattice-like models.
In actual chemical production, there are often combinations between numerous components. It is unrealistic to measure the vapor–liquid equilibrium data of each multi-component system using experimental methods. The group contribution method is a phase-equilibrium estimation method. In the group contribution method, the physical properties of a mixture are obtained by the joint contribution of the groups that make up this system. In different systems, the contribution of the same group to the system is the same. The widely used group contribution-based equation is UNIQUAC (UNIQUAC functional group activity coefficients).
Due to the widespread application of local composition models, the brief introduction to their main equations is as follows:
The Wilson equation can accurately describe the activity coefficient in non-polar systems and polar miscible systems, especially in hydrocarbon alcohol systems, but the Wilson equation is not suitable for calculating the activity coefficient of partially miscible systems. The Wilson equation of binary systems is shown in Equations (3) and (4).
Based on the above equation, we obtain the following equation:
In the above equation, Λij is the Wilson parameter, which is a dimensionless quantity, Λij > 0, and Λij ≠ Λji; however, Λi = Λj = 1. (gij-gji) is the energy parameter for the binary interaction, and gij = gji.
- (2)
In 1968, Renon and Prausnitz further developed the concept of local composition, introducing non-random parameters that can reflect the characteristics of different components
α12, and the NRTL equation was obtained. This equation can be used to calculate partially miscible systems, especially liquid–liquid layered systems. The NRTL equation is shown in Equation (7):
In the above equation, τij is the parameter of the NRTL equation; Gji is the interaction energy of the solution; αij is the characteristic function of the solution. Here, α is 0.3, and for most solutions, it is between 0.2 and 0.47.
- (3)
In 1975, Abrams and Prausnitz further introduced the size and shape parameters of molecules based on the quasilattice model and local composition model and derived the UNIQUAC equation using the double-liquid theory. Due to the introduction of molecular microscopic characteristic parameters, the equation theoretically has a wider range of applications, but currently, the microscopic parameters of some substances cannot be provided.
The UNIQUAC equation divides the activity coefficient into two parts, i.e., combination and remainder, which are used to reflect the molecular shape and the effect of shape, size, and intermolecular interaction on the activity coefficient. This equation is expressed as follows:
In the above equation, θi and φi represent the average area fraction and volume fraction of pure substance i; ri and qi are pure material parameters calculated based on the van der Waals volume and surface area of the molecule; z is the lattice coordination number; uij is the interaction energy between molecules and i-j, determined using the experimental data.
3.4. Thermodynamic Consistency Verification
When measuring the complete vapor–liquid equilibrium data of a material system through experimental methods, errors cannot be completely avoided due to the influence of reagent purity, measuring instruments, system environment, and analytical detection. Therefore, it is necessary to use certain methods to determine whether the vapor–liquid equilibrium data of each group measured in the experiment are reliable. The Gibbs–Duhem equation shows that the activity coefficients of all components in the mixture are related to each other. If the activity coefficient of all components in the system can be measured, these data should comply with the Gibbs–Duhem equation. If a group of experimental data does not conform to the Gibbs–Duhem equation, it indicates that this group of data is incorrect and there is no correlation between the components. Instead, the method of using the activity coefficient form of the Gibbs–Duhem equation to check the quality of experimental data and whether the vapor–liquid two-phase data are related is called the thermodynamic consistency check of vapor–liquid equilibrium data.
For binary systems, the Gibbs–Duhem equation used for testing is in the following form:
The vapor–liquid equilibrium in this study is measured under constant-pressure conditions; therefore, d
p = 0, and Equation (14) can be simplified as follows:
By integrating the above equation from x
1 = 0 to x
1 = 1, we conclude the following:
In the above equation, HE represents the excess mixing enthalpy, which usually varies with composition and temperature and is difficult to obtain experimentally. In 1947, Herington proposed a semi-empirical method based on the Gibbs–Duhem equation to perform consistency checks on the constant-pressure vapor–liquid equilibrium data of binary systems. The principle is to calculate the activity coefficients of each substance in the system from the experimental data and plot ln(γ
1/γ
2) versus x1. Then, we can calculate the value of D according to Equation (17). Using the maximum temperature T
max and the minimum temperature T
min of the system, J can be calculated according to Equation (18). If (D-J) < 10, it is considered that the constant-pressure vapor–liquid equilibrium data set is thermodynamically consistent.
According to the phase-equilibrium criterion, when the two vapor and liquid phases reach equilibrium, where each component is
i(
i = 1, 2, ...,
N), the fugacity in all directions is equal:
According to the definitions of fugacity and the fugacity coefficient, as well as activity and the activity coefficient, the fugacity of component i in the mixture can be expressed by the fugacity coefficient or by the activity coefficient. In the activity coefficient method, if the liquid f
iL is expressed through the activity coefficient, then the following is true:
In the above equation,
fiθ represents the fugacity of component i in the standard state. According to the Lewis–Randall rule, the fugacity of the components in a mixture is equal to the fugacity of the pure liquid i at the equilibrium temperature T and pressure P; therefore, we obtain the following equation:
In Equation (21), P is the pressure under phase-equilibrium conditions; yi is the mole fraction of component i in the vapor phase; xi is the mole fraction of component i in the liquid phase; φiV is the fugacity coefficient in the vapor-phase mixture of component i; Pis is the saturated vapor pressure of pure substance i at phase-equilibrium temperature; φis is the fugacity coefficient of component i which is a pure gas at the equilibrium temperature T and saturated vapor pressure Pis; γi is the activity coefficient of component i; ViL is the liquid-phase molar volume of pure substance i.
When the system is at low pressure, the vapor phase can be regarded as an ideal gas. Equation (21) can be simplified as follows:
The saturated vapor pressure in Equation (22) is usually calculated using the extended Antonie Equation (23), and
Table 7 lists the parameters of the extended Antonie equation for each component. The Wagner equation is considered to be the most accurate and explicit functional equation for fitting experimental data. As it uses critical properties as parameters, the Wagner equation can more accurately describe the saturated vapor pressure data of most working fluids over a relatively large temperature range, even the data in the critical region [
32]. In this section, the saturated vapor pressure of MCP is fitted using the Wagner equation, as shown in Equations (24) and (25). The parameters of the Wagner equation are in
Table 8.
The integration test method is simple and easy to perform, but it is a comprehensive test of experimental data across the entire composition range. Therefore, there may be situations where the errors generated by different experimental points cancel out each other, causing the surface test to pass. If you want to identify “bad points” in the experimental data, you need to conduct point-by-point testing on all experimental data.
In 1973, Van Ness et al. proposed to use the △
y value for point-by-point verification of the vapor–liquid equilibrium measurement results of a mixture system. The representative equation of △y is shown in Equation (26). If △
y is less than 1, it indicates that the data have passed the test.



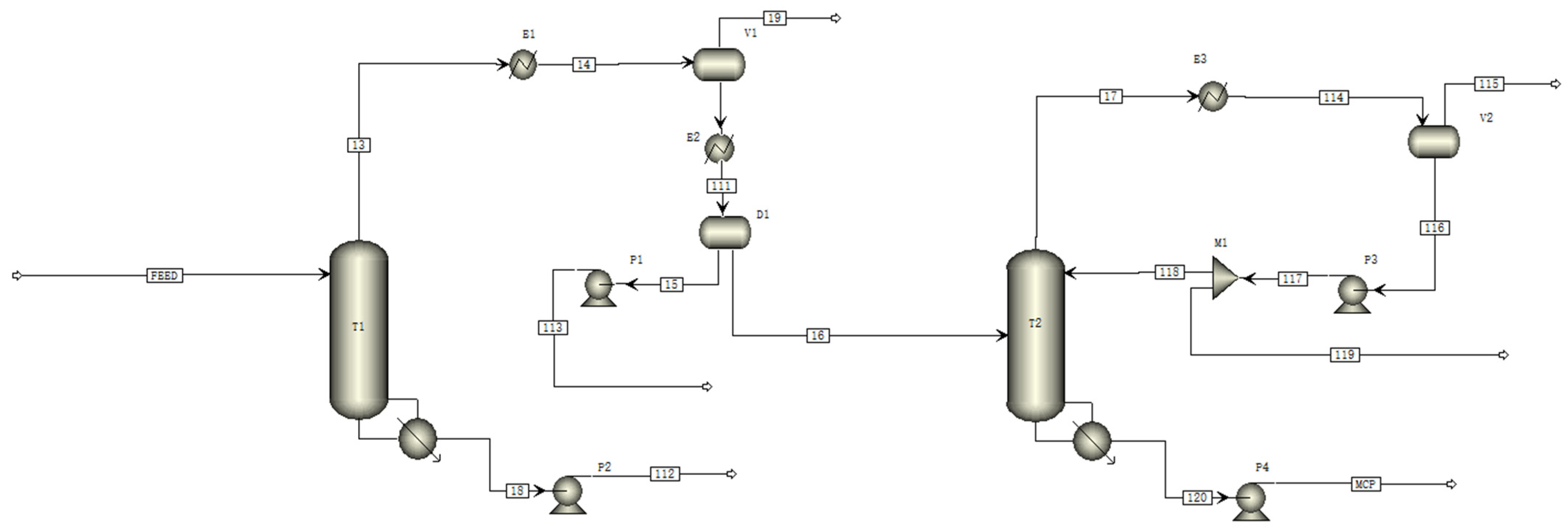
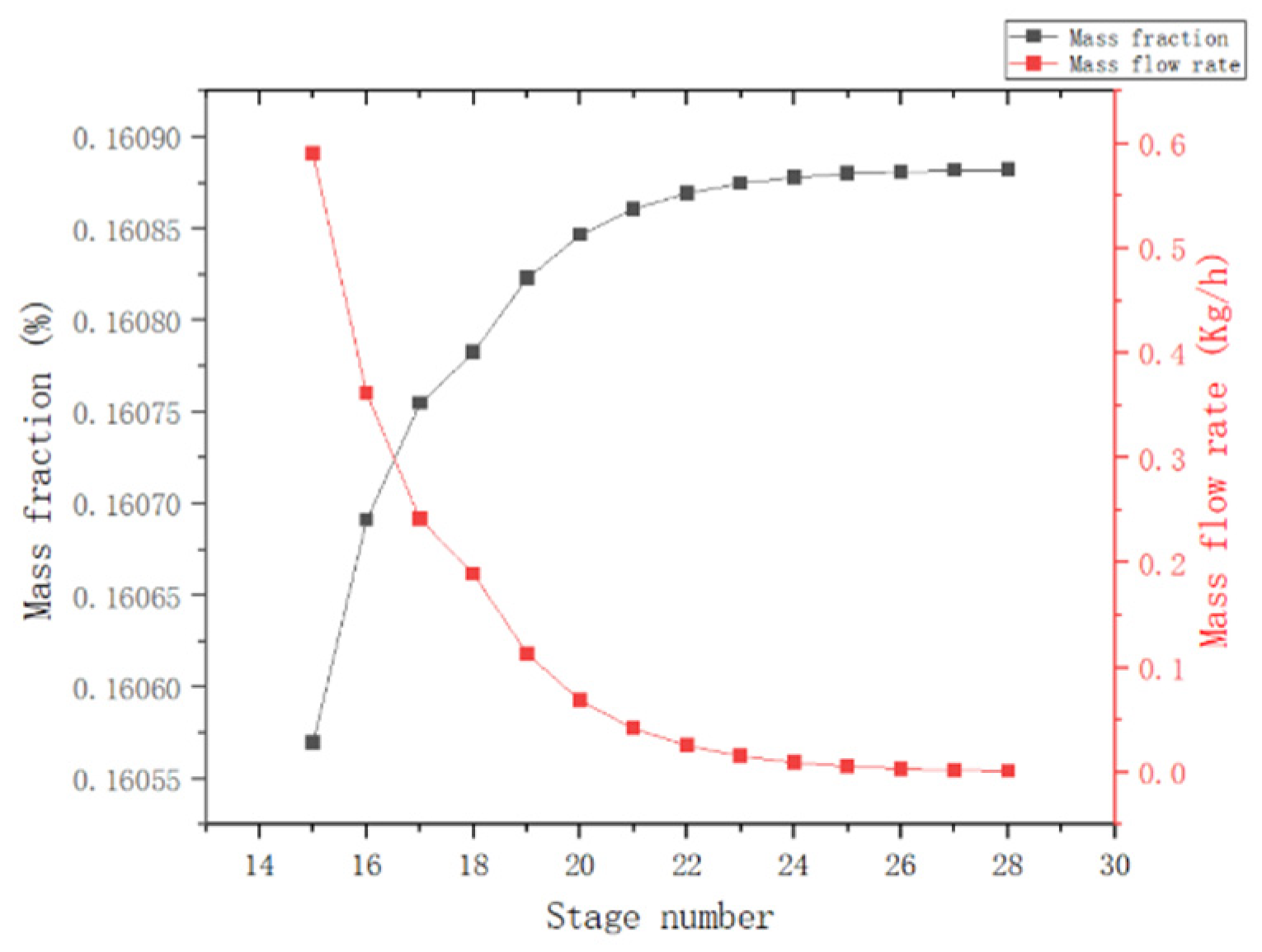
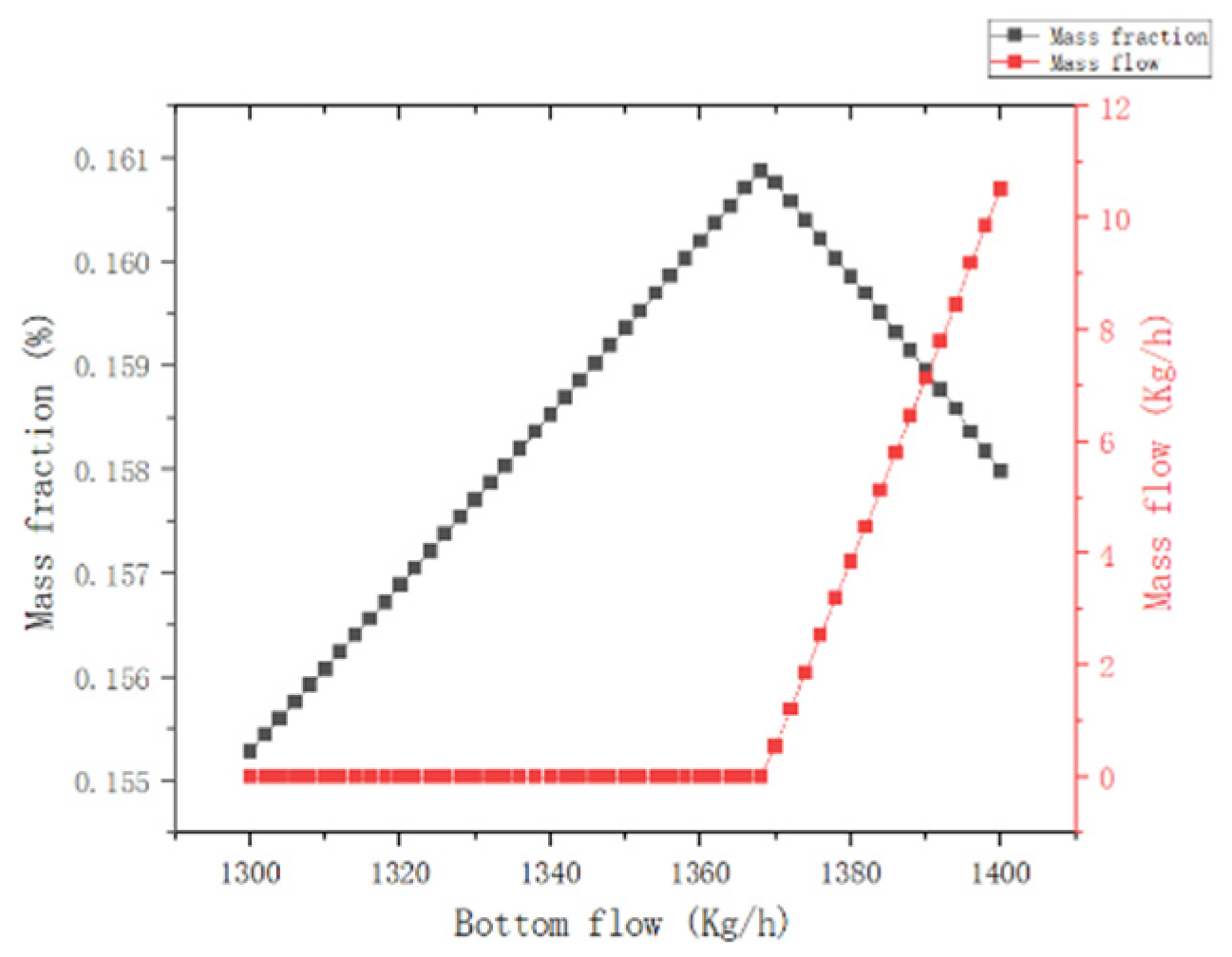
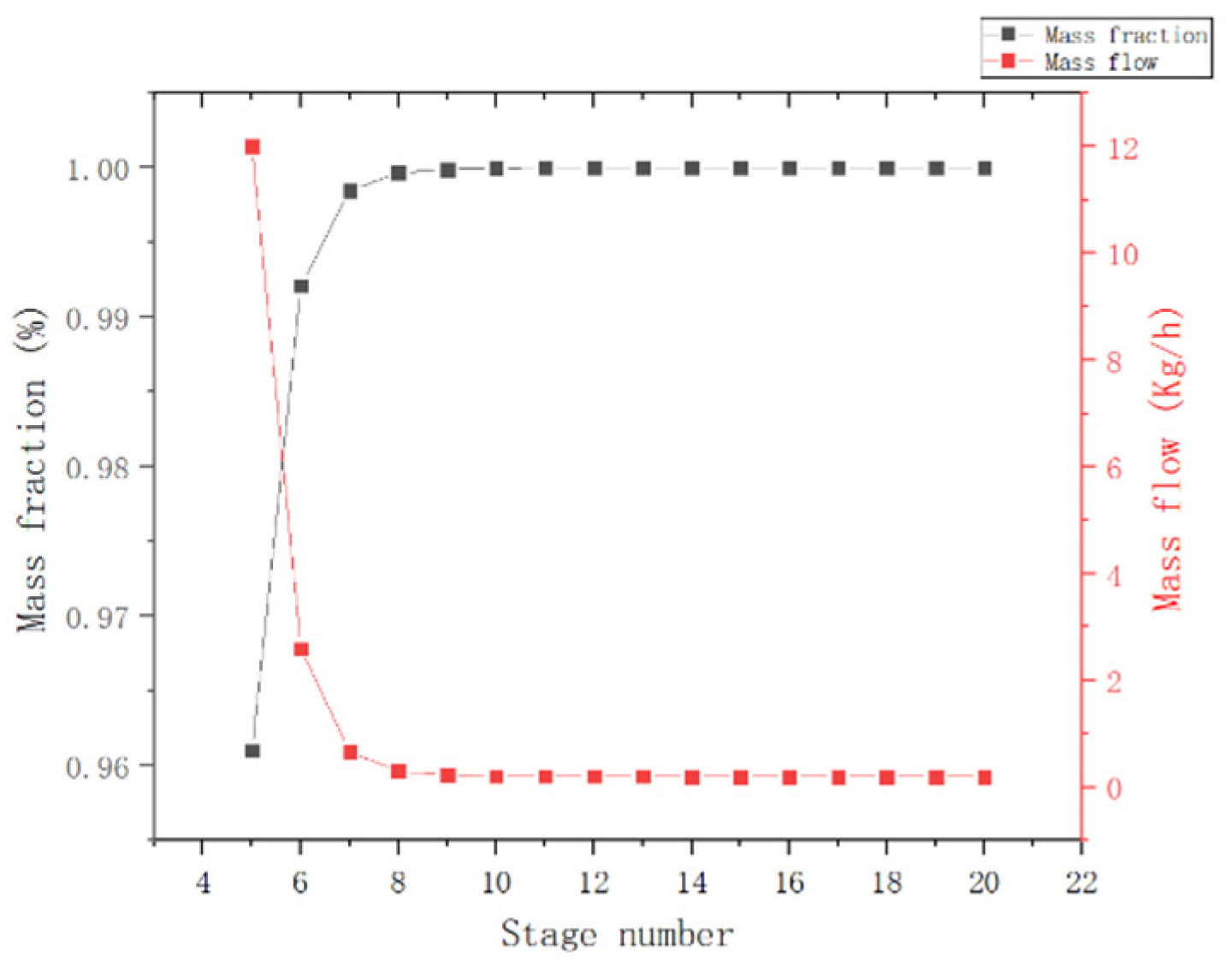

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}