Abstract
The Qinghai–Tibetan Plateau (QTP) is under serious desertification stress, which has been receiving increasing attention. Although the restoration of surface vegetation is crucial, the growth of plants is often hindered by unfavorable nutrient-deficient conditions. The plant-associated endophytic microbiome is considered the secondary genome of the host and plays a significant role in host survival under environmental stresses. However, the community compositions and functions of plant-endophytic microorganisms in the QTP desertification environments remain unclear. Therefore, this study investigated the endophytic microbiome of the pioneer plant Gueldenstaedtia verna on the QTP and its contribution to host growth under stressful conditions. The results showed that nutrient-deficient stresses strongly influenced the microbial community structures in the rhizosphere. The impacts of these stresses, however, decreased from the rhizosphere community to the plant endophytes, resulting in consistent plant endophytic microbial communities across different sites. Members of Halomonas were recognized as keystone taxa in the endophytic microbiome of G. verna. Correlation analysis, metagenome-assembled genomes (MAGs), and comparative genome analyses have shown that the keystone taxa of the plant endophytic microbiome may promote plant growth through pathways such as nitrogen fixation, IAA, and antioxidant production, which are important for improving plant nutrient acquisition and tolerance. This finding may provide a crucial theoretical foundation for future phytoremediation efforts in desertification environments on the Qinghai-Tibet Plateau.
1. Introduction
The Qinghai–Tibetan Plateau (QTP), known as the “Roof of the World” and the “Third Pole”, is the largest high-altitude grassland worldwide [1,2,3]. It is one of the largest carbon reservoirs in the world and serves as a major water source for one-fifth of the global population [2,4,5]. Therefore, it is important to regulate the regional and global climates [6,7]. Despite its importance, the QTP is suffering from desertification stresses. Many studies have shown that approximately 90% of the grasslands in the QTP are degraded [8,9,10,11], 33–50% of which are highly degraded [12]. In addition, the QTP suffers from semi-drought and nutrient depletion stresses, which results in a decline in biodiversity and a loss of soil nutrients (including about 42% reduction in soil organic carbon storage and about 33% reduction in nitrogen), leading to the widespread nutrient-deficient stresses [8,13,14,15,16]. The restoration of QTP desertified areas is urgently needed.
Pioneer plants are the colonizers of new or disturbed sites or pristine soils [17,18]. Owing to their rapid growth rates and high tolerance to nutrient-deficient conditions, pioneer plants play critical roles during the recovery of bare environments [19,20,21]. Gueldenstaedtia verna is among the most common pioneer plants found colonizing bare lands in the QTP [22,23,24]. G. verna has strong tolerance, which facilitates its fast colonization and rapid growth on degraded soils [25,26]. Therefore, G. verna may serve as a key pioneer plant for restoring the desertification environments in the QTP.
The nutrient-deficient nature of desertification environments is one of the major barriers that hinders their revegetation. In turn, the pioneer plants could recruit appropriate endophytic microbiomes to overcome such stresses. Endophytic microbiomes could colonize plant roots and stems and promote the fitness of host plants under environmental stresses by performing diverse plant growth promotion services [27,28]. For example, the plant-associated endophytic microbiome can supply nitrogen to plants through biological nitrogen fixation, thereby promoting host plant growth [29]. In addition, the plant-associated endophytic microbiome can enhance host survival and stimulate plant growth by producing 3-indole acetic acid (IAA) [30] and antioxidants [31,32]. Briefly, plant-associated endophytic microorganisms can increase the biomass, chlorophyll content, and protein content of host plants by providing nutrients uptake and improving plant resilience [33,34]. Therefore, the host plants are better able to cope with environmental stresses, including drought, disease, and high salinity [29,33,35]. Further consideration of pioneer plants in the QTP (such as G. verna) and their endophytic microbiomes may provide valuable information for future phytoremediation practices in QTP desertification environments.
To investigate the endophytic microbiome and its response to environmental stresses of desertification, G. verna was collected from two desertification sites on the QTP. A non-desertification site from the QTP was sampled as the control site. A combination of physicochemical analyses, 16S rRNA amplicon sequencing, and metagenomic sequencing was performed to decipher the microbial community compositions and functions across different plant compartments (i.e., rhizosphere soil, roots, and stems). The current study aimed to (1) characterize the diversity and composition of the G. verna-associated endophytic microbiome, (2) identify the keystone taxa of the endophytic microbiome and their interactions with physicochemical conditions, and (3) analyze the metabolic potential of the endophytic keystone taxa and investigate their roles in revegetation in desertification environments.
2. Materials and Methods
2.1. Sample Collection and Pretreatment
G. verna plants were collected from two desertification sites (abbreviated as “DS”) in Lhasa City, Tibet, China (29°44′24″ N, 91°40′47″ E, DS1 and 29°44′21″ N, 91°41′21″ E, DS2, respectively). Control samples were collected from a non-desertification site in Lhasa City, Tibet, China (29°43′36″ N, 91°40′26″ E, CK). At each site, 10–15 plant samples and their corresponding rhizosphere soils were collected using an ethanol-sterilized shovel. Samples were immediately stored on ice and subsequently returned to the laboratory, where they were stored at −80 °C.
Each G. verna plant was divided into different parts, including the rhizosphere soil, roots, and stems. Briefly, rhizosphere soil strongly attached to the roots was washed using a TE buffer (pH = 7.5) and Tween 20 (2%), followed by recovery through centrifugation. Subsequently, different parts of each G. verna plant were partitioned into two parts for physicochemical and microbial analyses [36]. In addition, the roots and stems of each G. verna plant were first surface-disinfected with NaClO (2%) and then washed five times with 70% ethanol and sterile water, respectively, to completely remove microorganisms attached to the surfaces of the roots and stems. All samples were stored at −80 °C until further analysis.
2.2. Physicochemical Analysis
The rhizosphere soil of each G. verna was freeze-dried, mashed, and sieved through a 2 mm sieve. To measure the pH, a 2 g sieved sample was mixed with 5 mL of ultrapure water and placed in a shaking incubator for 2 h. The mixed slurry was allowed to stand for 0.25 h, and the pH of the supernatant was measured using an HQ30d pH meter (HACH, Loveland, CO, USA). The total carbon (TC), total organic carbon (TOC), and inorganic carbon (IC) were measured using a total organic carbon meter (TOC-L CPH, Shimadzu, Japan). Total nitrogen (TN) and total sulfur (TS) were measured using Ion chromatography (ICS-600, Thermo Fisher, Waltham, MA, USA) after digestion of 0.1 g of a sieved sample with 10 mL of 30% H2O2 and 0.05 mL of formic acid for 2 h [37]. For the ammonium nitrogen (N_NH4+) and nitrate nitrogen (N_NO3−) measurements, 3 g of the sieved sample was mixed with 15 mL of 1 mol/L KCl solution and shaken for 2 h. The mixed solutions were then centrifuged using a centrifuge (5000 rpm, 0.25 h). The N_NH4+ in the supernatant was quantified using a chromogenic reaction with sodium nitroprusside-phenol, while N_NO3− was measured using sulfonamide and N-(1-naphthyl)-ethylenediamine hydrochloride [38]. The chromogenic samples were analyzed using a full-wavelength microplate reader (Muitiskan Sky, Thermo Fisher Scientific, Waltham, MA, USA). For the root and stem of G. verna, the dry weight (DW) of each plant’s root (R_DW) and stem (S_DW) was measured after freeze-drying. The plant material was then crushed and passed through a 2 mm sieve. The TN content in the roots (R_TN) and stems (S_TN) was determined using the same method as described for rhizosphere soil TN analysis.
2.3. DNA Sequencing and Analyses
A total of 0.25 g of lyophilized rhizosphere soil of each G. verna was extracted using a DNeasy PowerSoil kit (Qiagen, Hilden, Germany) following the manufacturer’s protocol. In addition, the disinfected and cleaned roots and stems of each G. verna plant were frozen in liquid nitrogen and ground using a pestle. Tissues and endophytes from the mashed roots and stems of each G. verna plant were extracted using a DNeasy PowerSoil kit (Qiagen, Hilden, Germany) following the manufacturer’s protocol. To analyze the microbial communities in each compartment of each G. verna, the 515F/806R primer set (515F: 5′-GTGCCAGCMGCCGCGGTAA-3′ and 806R: 5′-GGACTACVSGGGTATCTAAT-3′) was used to target the V4 hypervariable region of the 16S rRNA gene [39,40]. The resulting amplicons were barcoded, pooled, and sequenced using the Illumina MiSeq System (Personal Biotechnology Company, Shanghai, China). The sequencing libraries were analyzed in QIIME2 [41]. Briefly, the primers were trimmed, the sequences (trimmed) were filtered to obtain low-quality reads, and chimeras were removed. The high-quality sequences were grouped into amplicon sequence variants (ASVs) using DADA2 [42,43]. The resulting representative sequences were searched against the SILVA (132) database for taxonomic assignment [44]. Downstream analyses of the amplicon sequencing libraries were conducted in R using the package “phyloseq” (version 1.30.0) [45].
The quantification of 16S rRNA and nifH functional genes was performed using the quantitative PCR (qPCR) StepOnePlus system (Applied Biosystems, Foster City, CA, USA). The primers of 16S rRNA and nifH used for RT-qPCR are listed in Table S1 [46,47]. The relative abundance of nifH was calculated as nifH gene copies per 16S rRNA gene copy [46]. In all experiments, negative controls using PCR-grade water were included in each run to exclude possible DNA contamination. The minimum R2 value of the standard curve was 99%, and the efficiency was >80%.
2.4. Metagenomic Sequencing and Analysis
In order to better obtain MAGs related to keystone taxa of Halomonas through metagenomic binning and to analyze the metabolic potential related to Halomonas, a plant endophyte DNA sample (XP_Y7) with the highest relative abundance of Halomonas from the QTP desertification site was selected for metagenomic sequencing using the Illumina MiSeq System at the Personal Biotechnology Company (Shanghai, China). A total of 280,880,148 raw reads were trimmed and quality controlled using Trimmomatic, implementing the recommended parameters (leading: 3, trailing: 3, sliding window: 4:15, and minlen: 100) [48]. The clean reads were purified using Kraken2 (version 2.0.9beta) and MetaWRAP (version 1.3.2) to remove plant host interference [49,50]. After filtering, the remaining 195,700,247 reads were assembled using Megahit (k = 21−121, step = 12) [51]. The metagenome-assembled genomes (MAGs) were retrieved in MetaWRAP using the default parameters [50]. The quality of MAGs was estimated using CheckM (version 1.0.12), and only MAGs with completeness > 80% and contamination < 5% were retained for downstream analysis [52]. The Genome Taxonomy Database (GTDB) was used to assign the taxonomic affiliations of the retrieved MAGs [53]. The functional genes of the retrieved MAGs were annotated using KofamKOALA (version 1.3.0) against the KEGG database [54]. For comparative genomics analysis, fifteen Halomonas-associated reference genomes were obtained from the NCBI database (Table S2), and functional genes were annotated using KofamKOALA. Subsequently, the functional genes (such as nitrogen-fixation genes) of these 15 reference genomes related to Halomonas were compared with the two Halomonas-related MAGs obtained by metagenomic sequencing analysis.
2.5. Statistical Analysis and Data Visualization
The statistical analysis results of 10–15 biological replicates of each were presented in the format of “mean ± standard deviation”. The alpha diversity measurements, including Observed species and the Shannon index, of the rarified sample libraries were performed. The chloroplast and mitochondria of the samples were removed by the “subset_taxa” command in the “phyloseq” package in R. The beta diversity was visualized as a Bray–Curtis distance matrix in an NMDS plot. This distance matrix was further analyzed using multivariate analysis of variance (PERMANOVA) in the R package vegan to analyze the differences between sample groups. The statistical differences in microbial communities were calculated using an ANOVA. ASVs with a relative abundance greater than 1/1000 in roots or stems were screened for keystone taxa analysis. The keystone taxa were calculated using Spearman’s correlation (ρ) > |0.7| and p < 0.05 in the R package “igraph” (version 1.2.6) and then imported to “Cytoscape” (version 3.8.0) for visualization [55,56,57]. Meanwhile, the key indicators of keystone taxa (i.e., betweenness and degree) were calculated using the R package “igraph” [55,58,59]. The interactions between environmental parameters and the microbial communities (Env−Bio) were calculated using Spearman’s correlation (ρ) > |0.6| and p < 0.05 and visualized in “Cytoscape” [57]. An ANOVA analysis was used to statistically compare the physicochemical parameters and microbial communities in different sites or locations. Correlation was calculated based on the Pearson coefficient between the measurements of the Halomonas and nifH genes, TN, and DW. R > |0.7| and p < 0.05 were considered strong correlations [46]. This study’s 16S rRNA and shogun metagenome sequencing data were submitted to GenBank with the accession number PRJNA1192419.
3. Results
3.1. Physicochemical Conditions at Three Sites
The physicochemical conditions of 10–15 biological replicates at each of the three sampling sites were characterized (Figure 1). Among the three sampling sites, DS1 and DS2 were desertification sites (DSs), and the CK was a non-desertification control site in this study. For rhizosphere soil (Figure 1A), the pH at the three sites were all weakly alkaline, with average pH values of 7.4 ± 0.4, 8.3 ± 0.1, and 8.0 ± 0.1 in DS1, DS2, and the CK, respectively. The overall pH, however, was not statistically notable between the DS and CK. Similarly, there was no appreciable difference in the total sulfur content (TS) between the CK (341.7 ± 40.5 mg/kg) and the DS (249.5 ± 122.9 mg/kg). The total carbon content (TC), total organic carbon (TOC), and inorganic carbon (IC) were markedly higher (p < 0.05) in the CK (3.4 ± 1.2%, 1.4 ± 0.5%, and 1.9 ± 0.8%, respectively) than those of DS1 (0.8 ± 0.4%, 0.7 ± 0.4%, and 0.1 ± 0.1%, respectively) and DS2 (0.6 ± 0.3%, 0.4 ± 0.3%, and 0.2 ± 0.1%, respectively) (p < 0.05). Compared with the CK (93.4 ± 42.5 mg/kg), the total nitrogen (TN) in DS1 (57.5 ± 31.1 mg/kg) and DS2 (64.1 ± 48.2 mg/kg) were relatively depleted. In addition, the ammonium nitrogen (N_NH4+) of the CK (4.5 ± 0.8 mg/kg) was notably lower than that of DS1 (7.3 ± 2.6 mg/kg) and DS2 (11.8 ± 3.7 mg/kg) (p < 0.05). On the contrary, the nitrate nitrogen (N_NO3−) of the CK (0.2 ± 0.1 mg/kg) was considerably higher than the ammonium nitrogen of DS1 (0.1 ± 0.1 mg/kg) and DS2 (0.1 ± 0.1 mg/kg) (p < 0.05). In general, the TC, TOC, IC, TN, and N_NO3− of the CK were substantially higher than those of the DS, but the N_NH4+ of the CK was notably lower than that of the DS (p < 0.05).
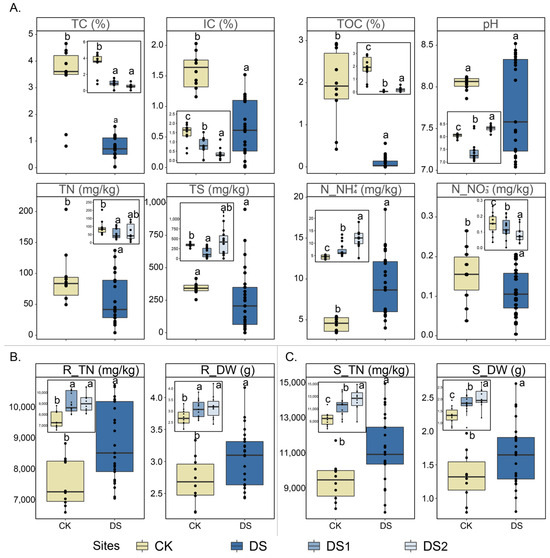
Figure 1.
Physicochemical parameters of the soil (A), root (B), and stem (C) samples (10–15 biological replicates from each site) collected from two desertification sites (DS1 and DS2) and one control site (CK). Different letters (i.e., a, b, and c) suggest statistically distinct differences (p < 0.05) by ANOVA. Abbreviations: TN, total nitrogen; IC, inorganic carbon; TOC, total organic carbon; TS, total sulfur content; and N_NH4+, ammonium nitrogen. R_TN, total nitrogen of root; R_DW, dry weight of root; S_TN, total nitrogen of stem; and S_DW, dry weight of stem.
The total nitrogen (R_TN) and dry weight (R_DW) in the roots were considerably higher in DS1 (8629.2 ± 1308.8 mg/kg and 2.9 ± 0.4 g, respectively) and DS2 (9151.1 ± 819.7 mg/kg and 3.2 ± 0.5 g, respectively) compared to the CK (7539.1 ± 741.1 mg/kg and 2.3 ± 0.3 g, respectively) (Figure 1B). A similar trend was observed in the stems, where DS1 exhibited a pronounced increase in R_TN and R_DW (10,256.2 ± 1568.2 mg/kg and 1.8 ± 0.3 g, respectively), as did DS2 (12,466.1 ± 1602.2 mg/kg and 2.1 ± 0.3 g, respectively) relative to the CK (9451.6 ± 1231.1 mg/kg and 1.5 ± 0.3 g, respectively) (Figure 1C).
3.2. Microbial Community Compositions
A total of 105 libraries were generated from Illumina Miseq sequencing of the 16S rRNA genes, with a total of 8,384,999 raw reads. After removing chloroplasts and mitochondria, a total of 5,454,075 interference-free reads remained. Although the community alpha diversity in the roots and stems of DS1 was higher than that of DS2, the alpha diversity of the CK was markedly greater than that of DS1 and DS2 (p < 0.05 for Observed species and the Shannon index), indicating elevated community richness and evenness of roots and stems in the CK compared to the DS (Figure 2). Similarly, the community alpha diversity measurements of the rhizosphere were also substantially higher at the CK site than at the DS (p < 0.05 for Observed species and the Shannon index). A beta diversity analysis showed that the compositions of the microbial community in the rhizosphere soil of different sites were different (p < 0.05), but the compositions of the endophytic microbial community in plants of different sites were clustered (Figure 3). A PERMANOVA indicated that different plant compartments (rhizosphere soil and the plant endosphere) were the major factors affecting the microbial community, accounting for 19.41% of the variation. The geographical origin of the samples markedly affected the community structure (11.49%, p < 0.001).
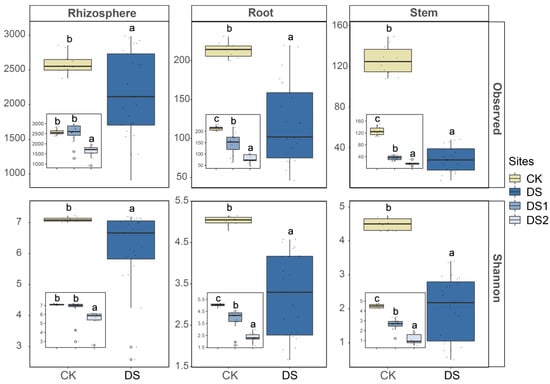
Figure 2.
The alpha diversity was measured in the rhizosphere soil, root, and stem samples (10–15 biological replicates from each site) collected from two desertification sites (DS1 and DS2) and one control site (CK). Different letters (i.e., a, b, and c) suggest statistically distinct differences (p < 0.05) by ANOVA.
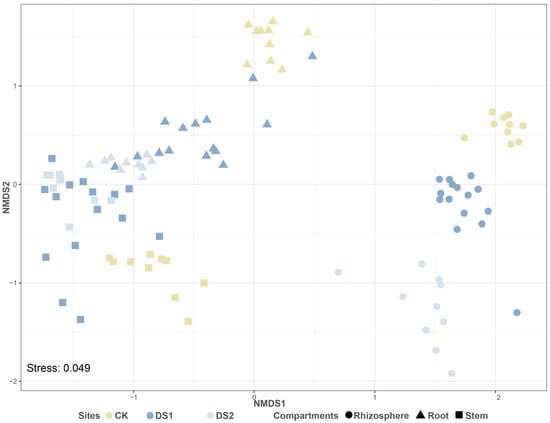
Figure 3.
The NMDS plot of beta diversity measured as Bray–Curtis distances for different sites and different compartments.
The relative abundances of the top 12 ASVs were compared to elucidate the distribution of major microbial populations at different sites (Figure 4). Compared with the CK, Halomonas_ASV1 (36.2% ± 22.2%, p < 0.05), Hyphomonadaceae_uncultured_ASV9 (2.4% ± 1.5%, p < 0.05), Mycobacterium_ASV61 (2.6% ± 2.4%, p < 0.05), and Pseudonocardia_ASV18 (5.4% ± 4.2%, p < 0.05) were enriched in the roots of the DS. For the stems, Halomonas_ASV1 (45.8% ± 28.3%, p < 0.05), Hyphomonadaceae_uncultured_ASV9 (3.5% ± 2.1%, p < 0.05), Mycobacterium_ASV61 (0.6% ± 0.1%, p < 0.05), and Arthrobacter_ASV8 (2.2% ± 0.3%, p < 0.05) were enriched in the stems of the DS compared to the CK. Furthermore, compared with the CK, Bacillus_ASV3 (6.3% ± 1.6%, p < 0.05) and Arthrobacter_ASV8 (1.7% ± 0.3%, p < 0.05) were enriched in the rhizosphere soil of the DS. In contrast, RB41_ASV29 (4.5% ± 0.6%, p < 0.05) was enriched in the rhizosphere soil of the CK.

Figure 4.
Bubble plots demonstrated the distribution of the relative abundances of the dominant microbial genera among two different types of sites (DS and CK). Different letters (i.e., a and b) suggest statistically distinct differences (p < 0.05) by ANOVA.
3.3. Identification of the Keystone Taxa in the Endophyte of G. verna
The identification of keystone taxa was based on the criteria of nodes with high degrees and low betweenness centralities [58,59]. Under this criterion, three nodes were identified as the keystone taxa of the roots’ (Figure 5A) and stems’ (Figure 5B) endophytes. To further explore the taxonomic classification and biotic interactions of the keystone taxa in the root (Figure 5C and Tables S3 and S4) and stem (Figure 5D and Tables S5 and S6) endophytes, two co-occurrence networks were constructed. In the root network, three ASVs from the class Gammaproteobacteria were identified as keystone taxa, namely members of the genera Halomonas, Achromobacter, and Serratia. The keystone taxa identified in the network for stems and roots were similar. In particular, one of the three keystone taxa from the class Gammaproteobacteria also belonged to the genus Halomonas, while the other two were Pantoea and Luteimonas, respectively. In addition, the keystone taxa in both roots and stems exhibited strong positive correlations with other microorganisms.
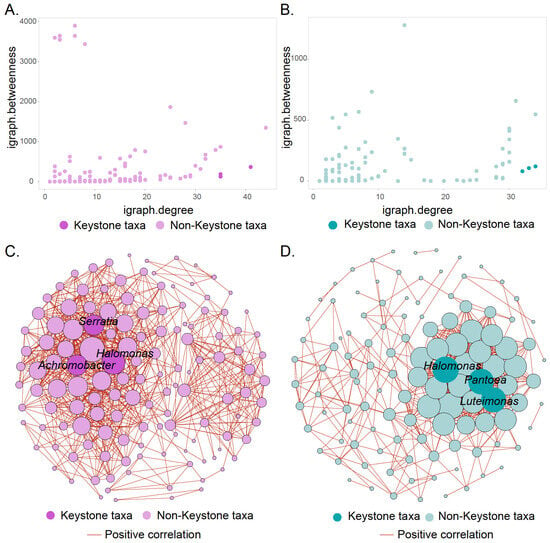
Figure 5.
Identification of endophytic keystone taxa in the roots and stems. The scatter plot shows the criteria for selecting the keystone taxa of roots (A) and stems (B), where high node degree and low betweenness centrality are the established standards for identifying keystone taxa. Co-occurrence network analysis showing the taxonomic classification of keystone taxa and biological interactions in the roots’ (C) and stems’ (D) endophyte. Among them, edges show only strong (Spearman correlation > |0.7| and p < 0.05) connections. The size of the nodes represents its degree. The color of edges shows positive correlation.
3.4. Interactions Between Environmental Factors and Microbial Communities in Desertification Environments
To demonstrate the effect of physicochemical conditions on the colonization of microbial populations in various compartments of G. verna in desertification environments, co-occurrence networks were used to visualize the interactions between the rhizosphere soil physicochemical parameters and the microbial populations inhabiting each compartment (Figure 6 and Tables S7 and S8). The physicochemical parameters exerted 246 pairs of (Spearman correlation > |0.6| and p < 0.05) connections on 222 microbial populations, including 155 microbial populations from rhizosphere soil, 37 endophytic microbiomes from roots, and 30 endophytic microbiomes from stems. Among all the physicochemical parameters, N_NH4+ was the most influential factor, affecting 99 individual microbial ASVs, followed by the TOC, which showed 72 strong and notable correlations in the co-occurrence network. The TS and pH had pronounced effects on 16 and 55 ASVs, respectively. The keystone taxa responded particularly to changes in the N_NH4+ and TOC environmental factors. Halomonas (two ASVs from the root and one ASV from the stem) in the plant endophytic microbial community exhibited a robust and highly correlated association with N_NH4+, while two Halomonas-related genera (one ASV from the root and one ASV from the stem) showed strong and substantial correlations with the TOC.
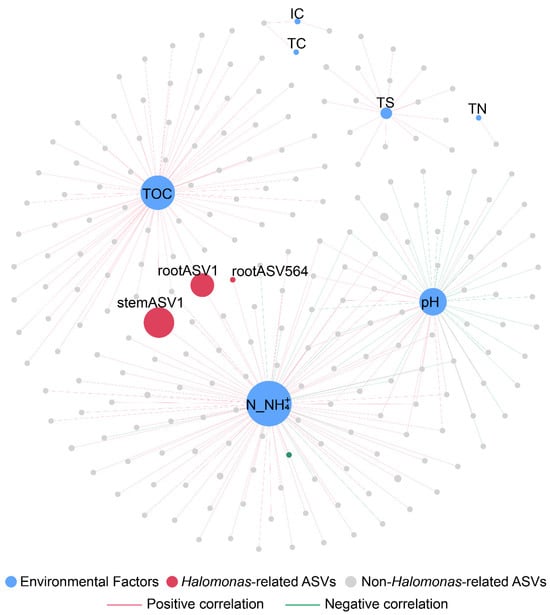
Figure 6.
Co-occurrence network analysis showing the correlations between the physicochemical parameters and the individual microbial ASVs. Edges show only strong (Spearman correlation > |0.6| and p < 0.05) connections. The node size of ASVs represents the abundance of ASVs. The node size of the environment factor represents the size of the degree. The color of edges shows positive and negative correlation. Size of the node is proportional to the number of connections. Abbreviations: TN, total nitrogen; IC, inorganic carbon; TOC, total organic carbon; TS, total sulfur content; and N_NH4+, ammonium nitrogen.
3.5. Quantification of nifH Genes and Their Association with Halomonas
The relative abundance of nifH in plant roots (8.98% ± 5.87%) and stems (11.58% ± 6.75%) was markedly higher than that in rhizosphere soil (1.96% ± 1.27%) (p < 0.05). Moreover, at the DS, the relative abundance of nifH was notably higher across all plant compartments compared to the CK (Figure 7). To further investigate the relationship between the endophyte Halomonas and nitrogen fixation, a correlation analysis was conducted between nifH and Halomonas in the plant roots and stems, respectively (Figure 8). The results revealed a strong positive correlation between Halomonas and nifH in both the roots and stems of plants. Additionally, further correlation analysis indicated that Halomonas in the roots and stems exhibited a strong positive correlation with both the TN and DW of the plants (Figure 8), suggesting a potential role in promoting plant growth.
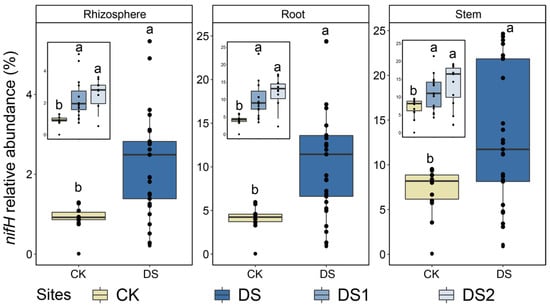
Figure 7.
The relative abundance of nifH was measured in the rhizosphere soil (10–15 replicates from each site) and root and stem endosphere (10–15 replicates from each site). Different letters (i.e., a and b) suggest significant differences (p < 0.05) by ANOVA.
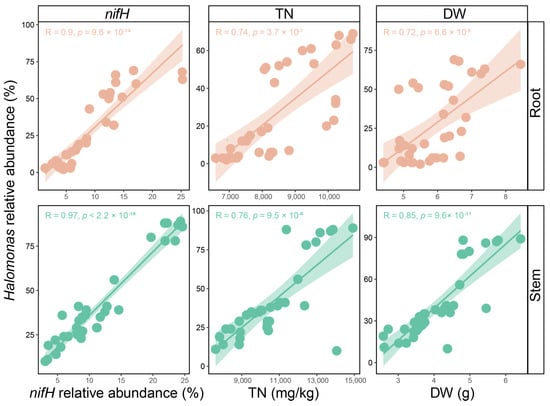
Figure 8.
Correlations between the Halomonas and nifH functional gene, TN, and DW in roots and stems, respectively. Each point represents an individual sample. The solid line indicates the linear regression of the Halomonas and nifH functional gene, TN, and DW, respectively. The light color areas indicate the 95% confidence intervals. Pearson correlation (R) and the statistical difference (p) are shown. Abbreviations: TN, total nitrogen and DW, dry weight.
3.6. Metagenomic Analysis and Binning
Metagenome binning was performed to reveal the metabolic capabilities of the endophytic microbial community members associated with desertification sites. Two high-quality MAGs (completeness > 80% and contamination < 5%) were retrieved, and single-copy gene classification based on GTDB-TK indicated that both MAG01_Halomonas_endophyte_desert_2 and MAG02_Halomonas_endophyte_desert_2 were related to the genus Halomonas (Table S9). Genes for which Halomonas-related MAGs may be involved in plant growth promotion, including nitrogen fixation, auxin IAA biosynthesis, and antioxidant production, were annotated and identified using kofamscan in the KEGG database (Figure 9 and Table S10).
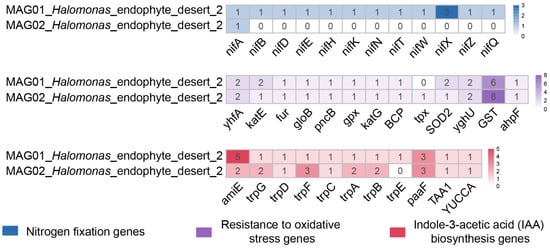
Figure 9.
Halomonas-associated MAGs encoded genes for nitrogen fixation, IAA biosynthesis, and antioxidant production. The heatmap exhibits the number of ORFs assigned to the genes involved in nitrogen fixation, IAA biosynthesis, and antioxidant production.
Nitrogen fixation potential. Endophytic keystone taxa can provide nutrients for plant growth through their nitrogen fixation potential. A metagenomic analysis revealed that MAG01_Halomonas_endophyte_desert_2 encoded complete nitrogen-fixation-related genes, including nitrogen fixation protein (nifB, nifT, nifX, nifZ, and nifQ), Nif-specific regulatory protein genes (nifA), nitrogenase iron protein (nifH), a nitrogenase molybdenum-iron protein alpha chain (nifD), nitrogenase molybdenum-cofactor synthesis protein (nifE), nitrogenase molybdenum-iron protein (nifN), a nitrogenase molybdenum-iron protein beta chain (nifK), and nitrogenase-stabilizing/protective protein (nifW). MAG02_Halomonas_endophyte_desert_2 only encoded Nif-specific regulatory protein genes (nifA), which may be due to the incompleteness of its MAG (83.23%) (Table S9).
Other plant growth promoting potential. Both MAG01_Halomonas_endophyte_desert_2 and MAG02_Halomonas_endophyte_desert_2 retrieved nearly identical auxin IAA biosynthesis-related genes, such as tryptophan biosynthesis genes (trpABCDEFG), the amidase gene (amiE), enoyl-coa hydratase gene (echA), L-tryptophan---pyruvate aminotransferase (TAA1), and indole-3-pyruvate monooxygenase (YUCCA), which play an important role in plant growth promotion [60,61,62]. Similarly, the genes related to antioxidant production retrieved from MAG01_Halomonas_endophyte_desert_2 and MAG02_Halomonas_endophyte_desert_2 were almost identical. Glutathione S-transferase was the most abundant enzyme retrieved in MAG01_Halomonas_endophyte_desert_2 and MAG02_Halomonas_endophyte_desert_2, followed by GSH-dependent disulfide bond oxidoreductase, putative redox protein, the superoxide dismutase Fe-Mn family, thioredoxin-dependent thiol peroxidase, catalase-peroxidase, glutathione peroxidase, nicotinate phosphoribosyltransferase, hydroxyacylglutathione hydrolase, ferric uptake regulation protein, and catalase.
3.7. Comparative Genome Analysis of Halomonas MAGs/Genomes
In addition to the two MAGs assembled in this study, 15 reference genomes of Halomonas spp., isolated and purified from various environments, such as saline wetland soil, salt marsh sediment, plant stem, coastal rhizosphere soil, deep sea metal sulfide rock, saline–alkaline soils, etc., were downloaded from the NCBI database and functionally annotated (Figure 10 and Table S11). A comparative genome analysis revealed that these Halomonas populations have numerous core metabolic functions. For instance, genes essential for plant growth promotion, including IAA biosynthesis, and antioxidant production were retrieved from all Halomonas-associated MAGs/genomes. Although genes related to nitrogen fixation (nif) were not proposed as core gene sets, the presence of these genes was found in several Halomonas genomes/MAGs, one of which was assembled in this study and one from the NCBI database (i.e., Halomonas endophytica MC28).
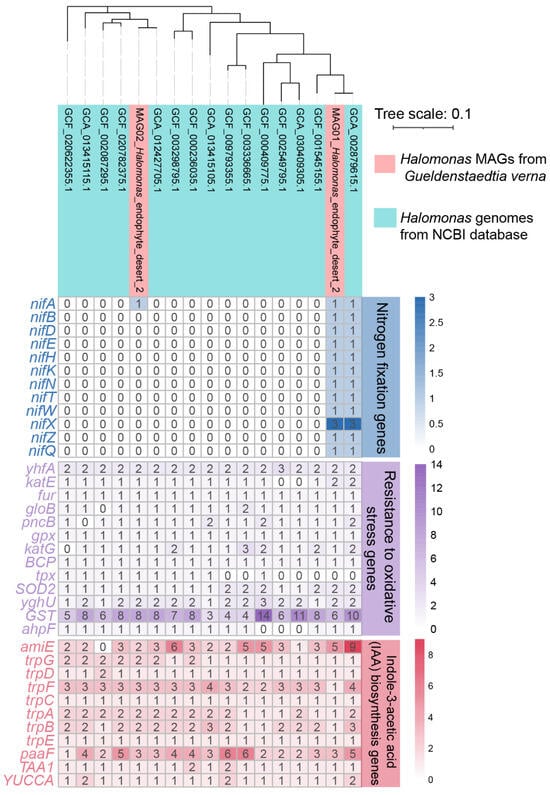
Figure 10.
Phylogenomic tree of two MAGs associated with Halomonas spp. and the Halomonas spp. reference genomes obtained from NCBI. The heatmap exhibits the number of ORFs assigned to the genes involved in nitrogen fixation, IAA biosynthesis, and antioxidant production.
4. Discussion
As the world’s largest carbon pool and largest freshwater storage area outside the Arctic and Antarctic, the QTP is suffering from nutrient-deficient stress owing to desertification [8,13,14,15,16]. The desertification of the QTP has led to a decrease in its water-holding capacity and has caused the loss of major nutrients [63,64], further intensifying the expansion of desertification. Therefore, it is critical to re-establish surface vegetation and stop the desertification process in the QTP [65,66,67]. The plant-associated endophytic microbiome is considered a genome secondary to its host and plays a critical role in host survival under environmental stress [29,68]. In the current study, the endophytic microbiome associated with the pioneer plant G. verna, one of the few plants that survive in the desertification environment of the QTP, has been investigated to elucidate its contribution to the host under hostile environmental conditions.
4.1. Soil Nutrient Deficiencies in the Desertification Environment of QTP
Physicochemical analyses indicated that the soils of G. verna on the QTP desertification sites (DS1 and DS2) were nutrient-deficient (Figure 1). Among the nutrients, decreased TC, TOC, IC, and TN concentrations were observed. In contrast, the rhizosphere of G. verna growing at the control site (CK) showed notably higher concentrations of nutrients. These results indicated that QTP desertification sites were severely deficient in carbon and nitrogen. Indeed, the degradation and desertification of the QTP often lead to a serious loss of nutrients, such as carbon and nitrogen [9,69,70], consistent with current desertification sites. Therefore, the current desertification sites are representative of investigations on plant-associated endophytic microbiome responses to plateau desertification environments.
4.2. Microbial Community and Diversity
A higher alpha diversity was observed in all compartments of the control site than in the QTP desertification sites, suggesting that nutrient-deficient stress had a strong impact on the microbial community (Figure 2). The community diversity detected in the plant endosphere of G. verna at all sites was markedly lower than that in the rhizosphere soil, indicating that the endophytic microbial communities in G. verna were less complex [71]. A beta diversity analysis showed that the key factors affecting microbial communities differed between plant compartments (i.e., rhizosphere soil and the plant endosphere), implying that the endophytic microbial communities of G. verna may shape microbial populations that differ from their rhizosphere soils (Figure 3) [19]. Furthermore, unlike the rhizosphere soil, the plant endophytic microbial community formed a unique cluster, suggesting that the plant endophytic microbial community may be highly conserved regardless of the site.
4.3. Halomonas as the Keystone Taxa of Plant Endophytes
Community structure analyses revealed that Halomonas was a highly abundant endophytic microorganism in roots and stems but also a differential microorganism (Figure 4), indicating that it may be a crucial plant endophyte [72,73,74]. In addition to a basic community analysis, the investigation of biological networks would provide an opportunity to identify keystone taxa [75,76]. The identification of keystone taxa is crucial because they play an important role in regulating the structure of the microbial community and maintaining ecosystem stability [77,78]. High-degree nodes and low betweenness are recognized criteria for identifying keystone taxa [79,80]. According to these criteria and taxonomic classification, similar keystone taxa were found in the root and stem endophytes (Figure 5 and Tables S3–S6). All keystone taxa found in roots and stems belonged to the class Gammaproteobacteria, indicating consistency in root and stem endophytes. Notably, members of Halomonas were identified as keystone taxa in both roots and stems and are therefore considered to play a critical role in plant endophytes. Indeed, members of Halomonas have been reported to dominate the endophytic microbiome of various plants, such as Salicornioideae [81], Cyperus laevigatus [82], and Arthrocnemum macrostachyum [83], and are well known for their role as plant growth promoters, providing essential nutrients to host plants [73,82].
4.4. Microbial Community Interacts with the Environmental Conditions
An analysis of the interactions between microbial communities and physicochemical parameters in the desertification environment of the QTP revealed that different physicochemical parameters have varying effects on microbial communities. Among the physicochemical parameters, N_NH4+ was the most influential (Figure 6 and Tables S7 and S8), indicating that N_NH4+ may be an important indicator of desertification reclamation. Conditions severely lacking N may drive microorganisms, especially diazotrophs, to mediate biological nitrogen fixation processes [84,85]. These processes play important roles in the initial accumulation of bioavailable N_NH4+ in nutrient-deficient desertification sites on the QTP [86,87]. Meanwhile, the relative abundances of a minor portion of the endophytes were impacted by the N_NH4+. Among these microbial communities, endophytic keystone taxa (i.e., Halomonas) were strongly positively correlated with N_NH4+, implying that Halomonas may be closely related to nitrogen cycles and may mediate the nitrogen fixation process [88,89], which is of great significance for the promotion of plant growth in QTP desertification environments.
4.5. Plant Growth Promotion Potential of Keystone Taxa
Endophytic microorganisms exhibited a higher relative abundance of nitrogen fixation functional genes (nifH) compared to those in rhizosphere soil in this study (Figure 7), suggesting the presence of a substantial number of nitrogen-fixing microorganisms within the plant [90]. Moreover, the keystone taxa Halomonas showed a strong positive correlation with nifH, R_TN, and S_TN (Figure 8), suggesting that the endophytic keystone taxa Halomonas may possess nitrogen fixation potential, thereby promoting plant growth [46,91]. The Halomonas MAGs obtained from the metagenomic analysis provide an important opportunity to explore the nitrogen fixation potential of keystone taxa. Fortunately, a complete nitrogen fixation pathway was found in MAG01_Halomonas_endophyte_desert_2, including the key nitrogenase gene nifHDK (Figure 9) [92,93], further indicating that Halomonas may have the potential to fix nitrogen. Additionally, although nitrogen fixation was not identified as a core gene in the selected Halomonas genomes (Figure 10 and Table S11), plant endophytic Halomonas are well known for their nitrogen-fixing capabilities. For instance, Halomonas sp. JG12 from the roots of Salicornia brachiata, Halomonas sp. MC1 from Mesembryanthemum crystallinum, and Halomonas endophytica MC28 from the stems of Populus euphratica have been shown to possess nitrogen fixation capabilities, along with other plant-growth-promoting traits such as IAA biosynthesis [73,94,95,96]. These findings indicate that nitrogen fixation may serve as a crucial ecological service for Halomonas within plants.
In addition to nitrogen fixation, keystone taxa have been shown to promote plant growth and enhance plant survival [29,97], which may facilitate the phytoremediation process in desertification environments [98,99]. In this study, Halomonas showed a strong positive correlation with plant dry weight, suggesting that Halomonas may play a role in plant growth (Figure 8) [90]. This is further supported by the fact that all Halomonas-related MAGs/genomes encoded genes associated with plant growth promotion, such as IAA (auxin) biosynthesis and oxidative stress resistance (Figure 9 and Figure 10). Among these plant growth promotion traits, IAA mainly mediates the synthesis of tryptophan by trpABCDEFG, followed by the conversion of the synthesized tryptophan into IAA by TAA1 and YUCCA [60,61,62]. IAA regulates various processes, including cell elongation, root development, and vascular tissue differentiation, thereby comprehensively enhancing plant growth, development, and environmental adaptability [100,101,102]. The production of antioxidants can reduce oxidative damage to plants, indicating that keystone endophytes may promote plant growth under various environmental stresses, such as heat, drought, etc. [97,103]. The identification of plant growth promotion genes in the keystone taxa is consistent with previous findings. Halomonas has been shown to notably facilitate plant growth by IAA [104] and antioxidants [105]. These observations further highlight that keystone endophytes may play a critical role in enhancing host fitness through various plant growth promotion services [19,106].
5. Conclusions
During plant growth, the endophytic microbiomes associated with plants play a leading role in providing significant ecological services, including nutrient acquisition and resistance to adverse physicochemical conditions, and are believed to be the secondary genome of the host plant [29,107]. Knowledge of plant endophytes is still limited compared with that of the rhizosphere microbiome. Investigations of the pioneer plant G. verna at desertification and control sites have revealed that nutrient-deficient conditions strongly influence the soil microbiome and that their effects on endophytic communities are less pronounced. In addition, members of Halomonas have been identified as keystone taxa in the plant endophytic microbiomes. Although the Halomonas-related strains were not successfully isolated in this study, a correlation analysis revealed that Halomonas may have the potential to fix nitrogen and promote plant growth. The metagenomic and comparative genome analyses further emphasized the potential of Halomonas to promote plant growth through pathways such as nitrogen fixation, IAA biosynthesis, and antioxidant production. Notably, these findings reveal the ecological function of the keystone taxa inhabiting G. verna, and the manipulation of these keystone taxa may improve the reclamation strategy of the QTP desertification environment. This study provides a novel perspective by investigating the metabolism of important plant endophytic keystone taxa to understand the ecological processes involved in the reclamation of QTP desertification environments.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/pr13041199/s1.
Author Contributions
Conceptualization, T.K., B.L. and P.G.; methodology, T.K. and P.G.; software, T.K.; validation, T.K. and Y.H.; formal analysis, T.K. and Y.W.; investigation, T.K., B.L. and Y.H.; resources, B.L. and H.L.; data curation, T.K. and H.L.; visualization, T.K.; supervision, T.K., W.S. and P.G.; project administration, W.S. and P.G.; funding acquisition, W.S.; writing—original draft preparation, T.K.; writing—review and editing, B.L., X.S., W.S. and P.G. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the GDAS’ Project of Science and Technology Development (grant nos. 2020GDASYL-20200102014, 2023GDASZH-2023010103, and 2022GDASZH-2022010106), the National Natural Science Foundation of China (grant nos. 32161143018, U21A2035, 42277247, 42321005, and 42107230), the Guangdong Basic and Applied Basic Research Foundation (grant nos. 2022B1515120033, 2023B1515040007, and 2022A1515011429), and the Guangdong Foundation for the Program of Science and Technology Research (grant no. 2023B1212060044).
Data Availability Statement
This study’s 16S rRNA and shogun metagenome sequencing data were submitted to GenBank with the accession number PRJNA1192419.
Conflicts of Interest
The authors declare no competing interests.
References
- Qiu, J. China: The third pole. Nature 2008, 454, 393–397. [Google Scholar] [CrossRef] [PubMed]
- Qiu, J. Trouble in Tibet: Rapid changes in Tibetan grasslands are threatening Asia’s main water supply and the livelihood of nomads. Nature 2016, 529, 142–146. [Google Scholar] [CrossRef] [PubMed]
- Dong, S.; Shang, Z.; Gao, J.; Boone, R.B. Enhancing sustainability of grassland ecosystems through ecological restoration and grazing management in an era of climate change on Qinghai-Tibetan Plateau. Agric. Ecosyst. Environ. 2020, 287, 106684. [Google Scholar] [CrossRef]
- Ni, J. Carbon storage in grasslands of China. J. Arid. Environ. 2002, 50, 205–218. [Google Scholar] [CrossRef]
- Wang, G.; Qian, J.; Cheng, G.; Lai, Y. Soil organic carbon pool of grassland soils on the Qinghai-Tibetan Plateau and its global implication. Sci. Total Environ. 2002, 291, 207–217. [Google Scholar] [CrossRef]
- Yanai, M.; Li, C.; Song, Z. Seasonal heating of the Tibetan Plateau and its effects on the evolution of the Asian summer monsoon. J. Meteorol. Soc. Jpn. 1992, 70, 319–351. [Google Scholar] [CrossRef]
- Duan, A.; Wu, G. Role of the Tibetan Plateau thermal forcing in the summer climate patterns over subtropical Asia. Clim. Dyn. 2005, 24, 793–807. [Google Scholar] [CrossRef]
- Li, X.L.; Gao, J.; Brierley, G.; Qiao, Y.M.; Zhang, J.; Yang, Y.W. Rangeland degradation on the Qinghai-Tibet plateau: Implications for rehabilitation. Land Degrad. Dev. 2013, 24, 72–80. [Google Scholar] [CrossRef]
- Liu, S.; Zamanian, K.; Schleuss, P.-M.; Zarebanadkouki, M.; Kuzyakov, Y. Degradation of Tibetan grasslands: Consequences for carbon and nutrient cycles. Agric. Ecosyst. Environ. 2018, 252, 93–104. [Google Scholar] [CrossRef]
- Miehe, G.; Schleuss, P.-M.; Seeber, E.; Babel, W.; Biermann, T.; Braendle, M.; Chen, F.; Coners, H.; Foken, T.; Gerken, T. The Kobresia pygmaea ecosystem of the Tibetan highlands–Origin, functioning and degradation of the world’s largest pastoral alpine ecosystem: Kobresia pastures of Tibet. Sci. Total Environ. 2019, 648, 754–771. [Google Scholar] [CrossRef]
- Harris, R.B. Rangeland degradation on the Qinghai-Tibetan plateau: A review of the evidence of its magnitude and causes. J. Arid. Environ. 2010, 74, 1–12. [Google Scholar] [CrossRef]
- Cao, J.; Adamowski, J.F.; Deo, R.C.; Xu, X.; Gong, Y.; Feng, Q. Grassland degradation on the Qinghai-Tibetan Plateau: Reevaluation of causative factors. Rangel. Ecol. Manag. 2019, 72, 988–995. [Google Scholar] [CrossRef]
- Wen, L.; Dong, S.; Li, Y.; Wang, X.; Li, X.; Shi, J.; Dong, Q. The impact of land degradation on the C pools in alpine grasslands of the Qinghai-Tibet Plateau. Plant Soil 2013, 368, 329–340. [Google Scholar] [CrossRef]
- Wang, Y.; Ren, Z.; Ma, P.; Wang, Z.; Niu, D.; Fu, H.; Elser, J.J. Effects of grassland degradation on ecological stoichiometry of soil ecosystems on the Qinghai-Tibet Plateau. Sci. Total Environ. 2020, 722, 137910. [Google Scholar] [CrossRef]
- Vogt, J.; Safriel, U.; Von Maltitz, G.; Sokona, Y.; Zougmore, R.; Bastin, G.; Hill, J. Monitoring and assessment of land degradation and desertification: Towards new conceptual and integrated approaches. Land Degrad. Dev. 2011, 22, 150–165. [Google Scholar] [CrossRef]
- Bardgett, R.D.; Bullock, J.M.; Lavorel, S.; Manning, P.; Schaffner, U.; Ostle, N.; Chomel, M.; Durigan, G.; Fry, E.L.; Johnson, D. Combatting global grassland degradation. Nat. Rev. Earth Environ. 2021, 2, 720–735. [Google Scholar] [CrossRef]
- Connell, J.H.; Slatyer, R.O. Mechanisms of succession in natural communities and their role in community stability and organization. Am. Nat. 1977, 111, 1119–1144. [Google Scholar] [CrossRef]
- Zhang, Q.; Araya, M.M.; Astorga-Eló, M.; Velasquez, G.; Rilling, J.I.; Campos, M.; Sadowsky, M.J.; Jorquera, M.A.; Acuña, J.J. Composition and potential functions of rhizobacterial communities in a pioneer plant from Andean altiplano. Diversity 2022, 14, 14. [Google Scholar] [CrossRef]
- Sun, X.; Song, B.; Xu, R.; Zhang, M.; Gao, P.; Lin, H.; Sun, W. Root-associated (rhizosphere and endosphere) microbiomes of the Miscanthus sinensis and their response to the heavy metal contamination. J. Environ. Sci. 2021, 104, 387–398. [Google Scholar] [CrossRef]
- Sun, X.; Zhou, Y.; Tan, Y.; Wu, Z.; Lu, P.; Zhang, G.; Yu, F. Restoration with pioneer plants changes soil properties and remodels the diversity and structure of bacterial communities in rhizosphere and bulk soil of copper mine tailings in Jiangxi Province, China. Environ. Sci. Pollut. Res. 2018, 25, 22106–22119. [Google Scholar] [CrossRef]
- Liao, C.; Liu, B.; Xu, Y.; Li, Y.; Li, H. Effect of topography and protecting barriers on revegetation of sandy land, Southern Tibetan Plateau. Sci. Rep. 2019, 9, 6501. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Ma, Y.; Wang, X.; Xu, L. Plant community assembly mechanisms of a subalpine meadow community along different successional time. Rangel. Ecol. Manag. 2021, 77, 118–125. [Google Scholar] [CrossRef]
- Cai, W.; Guan, T.; Li, H.; Lai, L.; Zhang, X.; Zhou, J.; Jiang, L.; Zheng, Y. Vegetation succession of abandoned croplands in Ruanliang and Yingliang in the Ordos Plateau. Acta Ecol. Sin. 2018, 38, 21–28. [Google Scholar] [CrossRef]
- Yang, Y.; Niu, K.; Hu, Z.; Niklas, K.J.; Sun, S. Linking species performance to community structure as affected by UV-B radiation: An attenuation experiment. J. Plant Ecol. 2018, 11, 286–296. [Google Scholar] [CrossRef]
- Wu, G.L.; Liu, Y.; Tian, F.P.; Shi, Z.H. Legumes functional group promotes soil organic carbon and nitrogen storage by increasing plant diversity. Land Degrad. Dev. 2017, 28, 1336–1344. [Google Scholar] [CrossRef]
- Li, Y.-G.; Li, L.-H.; Jiang, G.-M.; Niu, S.-L.; Liu, M.-Z.; Gao, L.-M.; Peng, Y.; Jiang, C.-D. Traits of chlorophyll fluorescence in 99 plant species from the sparse-elm grassland in Hunshandak Sandland. Photosynthetica 2004, 42, 243–249. [Google Scholar] [CrossRef]
- Schlaeppi, K.; Bulgarelli, D. The plant microbiome at work. Mol. Plant-Microbe Interact. 2015, 28, 212–217. [Google Scholar] [CrossRef]
- Gao, M.; Xiong, C.; Gao, C.; Tsui, C.K.; Wang, M.-M.; Zhou, X.; Zhang, A.-M.; Cai, L. Disease-induced changes in plant microbiome assembly and functional adaptation. Microbiome 2021, 9, 187. [Google Scholar] [CrossRef]
- Trivedi, P.; Leach, J.E.; Tringe, S.G.; Sa, T.; Singh, B.K. Plant–microbiome interactions: From community assembly to plant health. Nat. Rev. Microbiol. 2020, 18, 607–621. [Google Scholar] [CrossRef]
- Kuklinsky-Sobral, J.; Araújo, W.L.; Mendes, R.; Geraldi, I.O.; Pizzirani-Kleiner, A.A.; Azevedo, J.L. Isolation and characterization of soybean-associated bacteria and their potential for plant growth promotion. Environ. Microbiol. 2004, 6, 1244–1251. [Google Scholar] [CrossRef]
- Zhang, H.; Sun, X.; Dai, M. Improving crop drought resistance with plant growth regulators and rhizobacteria: Mechanisms, applications, and perspectives. Plant Commun. 2022, 3, 100228. [Google Scholar] [CrossRef] [PubMed]
- Abdelkrim, S.; Jebara, S.H.; Jebara, M. Antioxidant systems responses and the compatible solutes as contributing factors to lead accumulation and tolerance in Lathyrus sativus inoculated by plant growth promoting rhizobacteria. Ecotoxicol. Environ. Saf. 2018, 166, 427–436. [Google Scholar] [CrossRef] [PubMed]
- Bodenhausen, N.; Bortfeld-Miller, M.; Ackermann, M.; Vorholt, J.A. A synthetic community approach reveals plant genotypes affecting the phyllosphere microbiota. PLoS Genet. 2014, 10, e1004283. [Google Scholar] [CrossRef] [PubMed]
- Eckert, B.; Weber, O.B.; Kirchhof, G.; Halbritter, A.; Stoffels, M.; Hartmann, A. Azospirillum doebereinerae sp. nov., a nitrogen-fixing bacterium associated with the C4-grass Miscanthus. Int. J. Syst. Evol. Microbiol. 2001, 51, 17–26. [Google Scholar] [CrossRef]
- Sun, Q.; Yamada, T.; Han, Y.; Takano, T. Influence of salt stress on C4 photosynthesis in Miscanthus sinensis Anderss. Plant Biol. 2021, 23, 44–56. [Google Scholar] [CrossRef]
- Sun, X.; Kong, T.; Huang, D.; Chen, Z.; Zhang, Y.; Häggblom, M.M.; Soleimani, M.; Liu, H.; Ren, Y.; Wang, Y.; et al. Microbial sulfur and arsenic oxidation facilitate the establishment of biocrusts during reclamation of degraded mine tailings. Environ. Sci. Technol. 2024, 58, 12441–12453. [Google Scholar] [CrossRef]
- Colina, M.; Gardiner, P. Simultaneous determination of total nitrogen, phosphorus and sulphur by means of microwave digestion and ion chromatography. J. Chromatogr. A 1999, 847, 285–290. [Google Scholar] [CrossRef]
- Yang, W.; Ma, J.; Zhen, Y.; Li, W.; Yao, Z.; Feng, W. Community characteristics and functional gene response analysis of phosphorus-metabolizing bacteria in plateau saline lake sediments. Front. Environ. Sci. 2022, 10, 994104. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Lauber, C.L.; Walters, W.A.; Berg-Lyons, D.; Lozupone, C.A.; Turnbaugh, P.J.; Fierer, N.; Knight, R. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc. Natl. Acad. Sci. USA 2011, 108, 4516–4522. [Google Scholar] [CrossRef]
- Sun, X.; Chen, Q.; Häggblom, M.M.; Liu, G.; Kong, T.; Huang, D.; Chen, Z.; Li, F.; Li, B.; Sun, W. Microbially mediated sulfur oxidation coupled with arsenate reduction within oligotrophic mining–impacted habitats. ISME J. 2024, 18, wrae110. [Google Scholar] [CrossRef]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef] [PubMed]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [PubMed]
- Kong, T.; Sun, X.; Gu, Z.; Yang, N.; Huang, Y.; Lan, L.; Gao, P.; Liu, H.; Wang, Y.; Jiang, F.; et al. Differential mechanisms of microbial As(III) and Sb(III) oxidation and their contribution to tailings reclamation. Environ. Sci. Technol. 2024, 58, 11447–11458. [Google Scholar] [CrossRef] [PubMed]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2012, 41, D590–D596. [Google Scholar] [CrossRef]
- McMurdie, P.J.; Holmes, S. phyloseq: An R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef]
- Sun, X.; Kong, T.; Häggblom, M.M.; Kolton, M.; Li, F.; Dong, Y.; Huang, Y.; Li, B.; Sun, W. Chemolithoautotropic diazotrophy dominates the nitrogen fixation process in mine tailings. Environ. Sci. Technol. 2020, 54, 6082–6093. [Google Scholar] [CrossRef]
- Muyzer, G.; de Waal, E.C.; Uitterlinden, A.G. Profiling of complex microbial populations by denaturing gradient gel electrophoresis analysis of polymerase chain reaction-amplified genes coding for 16S rRNA. Appl. Environ. Microbiol. 1993, 59, 695–700. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Wood, D.E.; Lu, J.; Langmead, B. Improved metagenomic analysis with Kraken 2. Genome Biol. 2019, 20, 257. [Google Scholar] [CrossRef]
- Uritskiy, G.V.; DiRuggiero, J.; Taylor, J. MetaWRAP—A flexible pipeline for genome-resolved metagenomic data analysis. Microbiome 2018, 6, 158. [Google Scholar] [CrossRef]
- Li, D.; Liu, C.-M.; Luo, R.; Sadakane, K.; Lam, T.-W. MEGAHIT: An ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 2015, 31, 1674–1676. [Google Scholar] [CrossRef] [PubMed]
- Parks, D.H.; Imelfort, M.; Skennerton, C.T.; Hugenholtz, P.; Tyson, G.W. CheckM: Assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 2015, 25, 1043–1055. [Google Scholar] [CrossRef] [PubMed]
- Parks, D.H.; Chuvochina, M.; Waite, D.W.; Rinke, C.; Skarshewski, A.; Chaumeil, P.-A.; Hugenholtz, P. A standardized bacterial taxonomy based on genome phylogeny substantially revises the tree of life. Nat. Biotechnol. 2018, 36, 996–1004. [Google Scholar] [CrossRef] [PubMed]
- Aramaki, T.; Blanc-Mathieu, R.; Endo, H.; Ohkubo, K.; Kanehisa, M.; Goto, S.; Ogata, H. KofamKOALA: KEGG Ortholog assignment based on profile HMM and adaptive score threshold. Bioinformatics 2020, 36, 2251–2252. [Google Scholar] [CrossRef]
- Csardi, G.; Nepusz, T. The igraph software package for complex network research. InterJournal Complex. Syst. 2006, 1695, 1–9. [Google Scholar]
- Newman, M.E.J. Modularity and community structure in networks. Proc. Natl. Acad. Sci. USA 2006, 103, 8577–8582. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Hu, A.; Ju, F.; Hou, L.; Li, J.; Yang, X.; Wang, H.; Mulla, S.I.; Sun, Q.; Bürgmann, H.; Yu, C.P. Strong impact of anthropogenic contamination on the co-occurrence patterns of a riverine microbial community. Environ. Microbiol. 2017, 19, 4993–5009. [Google Scholar] [CrossRef]
- Sun, X.; Xu, R.; Dong, Y.; Li, F.; Tao, W.; Kong, T.; Zhang, M.; Qiu, L.; Wang, X.; Sun, W. Investigation of the ecological roles of putative keystone taxa during tailing revegetation. Environ. Sci. Technol. 2020, 54, 11258–11270. [Google Scholar] [CrossRef]
- Mano, Y.; Nemoto, K. The pathway of auxin biosynthesis in plants. J. Exp. Bot. 2012, 63, 2853–2872. [Google Scholar] [CrossRef]
- Spaepen, S.; Vanderleyden, J.; Remans, R. Indole-3-acetic acid in microbial and microorganism-plant signaling. FEMS Microbiol. Rev. 2007, 31, 425–448. [Google Scholar] [CrossRef] [PubMed]
- Krome, K.; Rosenberg, K.; Dickler, C.; Kreuzer, K.; Ludwig-Müller, J.; Ullrich-Eberius, C.; Scheu, S.; Bonkowski, M. Soil bacteria and protozoa affect root branching via effects on the auxin and cytokinin balance in plants. Plant Soil 2010, 328, 191–201. [Google Scholar] [CrossRef]
- Li, X.; Jia, X.; Dong, G. Influence of desertification on vegetation pattern variations in the cold semi-arid grasslands of Qinghai-Tibet Plateau, North-west China. J. Arid. Environ. 2006, 64, 505–522. [Google Scholar] [CrossRef]
- Lu, J.; Dong, Z.; Li, W.; Hu, G. The effect of desertification on carbon and nitrogen status in the northeastern margin of the Qinghai-Tibetan Plateau. Environ. Earth Sci. 2014, 71, 807–815. [Google Scholar] [CrossRef]
- Gang, C.; Zhao, W.; Zhao, T.; Zhang, Y.; Gao, X.; Wen, Z. The impacts of land conversion and management measures on the grassland net primary productivity over the Loess Plateau, Northern China. Sci. Total Environ. 2018, 645, 827–836. [Google Scholar] [CrossRef]
- Miehe, G.; Miehe, S.; Böhner, J.; Kaiser, K.; Hensen, I.; Madsen, D.; Liu, J.; Opgenoorth, L. How old is the human footprint in the world’s largest alpine ecosystem? A review of multiproxy records from the Tibetan Plateau from the ecologists’ viewpoint. Quat. Sci. Rev. 2014, 86, 190–209. [Google Scholar] [CrossRef]
- Zhang, H.; Fan, J.; Wang, J.; Cao, W.; Harris, W. Spatial and temporal variability of grassland yield and its response to climate change and anthropogenic activities on the Tibetan Plateau from 1988 to 2013. Ecol. Indic. 2018, 95, 141–151. [Google Scholar] [CrossRef]
- Vandenkoornhuyse, P.; Quaiser, A.; Duhamel, M.; Le Van, A.; Dufresne, A. The importance of the microbiome of the plant holobiont. New Phytol. 2015, 206, 1196–1206. [Google Scholar] [CrossRef]
- Yuan, Z.-Q.; Jiang, X.-J.; Liu, G.-J.; Jin, H.-J.; Chen, J.; Wu, Q.-B. Responses of soil organic carbon and nutrient stocks to human-induced grassland degradation in a Tibetan alpine meadow. Catena 2019, 178, 40–48. [Google Scholar] [CrossRef]
- Peng, F.; Xue, X.; You, Q.; Huang, C.; Dong, S.; Liao, J.; Duan, H.; Tsunekawa, A.; Wang, T. Changes of soil properties regulate the soil organic carbon loss with grassland degradation on the Qinghai-Tibet Plateau. Ecol. Indic. 2018, 93, 572–580. [Google Scholar] [CrossRef]
- Beckers, B.; Op De Beeck, M.; Thijs, S.; Truyens, S.; Weyens, N.; Boerjan, W.; Vangronsveld, J. Performance of 16s rDNA primer pairs in the study of rhizosphere and endosphere bacterial microbiomes in metabarcoding studies. Front. Microbiol. 2016, 7, 650. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Zhou, N.; Zhao, Z.-Y.; Zhang, K.; Tian, C.-Y. High-throughput sequencing analysis of the endophytic bacterial diversity and dynamics in roots of the halophyte Salicornia europaea. Curr. Microbiol. 2016, 72, 557–562. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Wang, P.; Tian, H.; Tao, Z.; Guo, T. Transcriptome analysis of ice plant growth-promoting endophytic bacterium Halomonas sp. strain MC1 to identify the genes involved in salt tolerance. Microorganisms 2020, 8, 88. [Google Scholar] [CrossRef] [PubMed]
- Navarro-Torre, S.; Carro, L.; Rodríguez-Llorente, I.D.; Pajuelo, E.; Caviedes, M.Á.; Igual, J.M.; Klenk, H.-P.; Montero-Calasanz, M.d.C. Halomonas radicis sp. nov., isolated from Arthrocnemum macrostachyum growing in the Odiel marshes (Spain) and emended descriptions of Halomonas xinjiangensis and Halomonas zincidurans. Int. J. Syst. Evol. Microbiol. 2020, 70, 220–227. [Google Scholar] [CrossRef]
- Agler, M.T.; Ruhe, J.; Kroll, S.; Morhenn, C.; Kim, S.-T.; Weigel, D.; Kemen, E.M. Microbial hub taxa link host and abiotic factors to plant microbiome variation. PLoS Biol. 2016, 14, 1002352. [Google Scholar] [CrossRef]
- Dunne, J.A.; Williams, R.J.; Martinez, N.D. Network structure and biodiversity loss in food webs: Robustness increases with connectance. Ecol. Lett. 2002, 5, 558–567. [Google Scholar] [CrossRef]
- Van Der Heijden, M.G.; Hartmann, M. Networking in the plant microbiome. PLoS Biol. 2016, 14, 1002378. [Google Scholar] [CrossRef]
- Banerjee, S.; Schlaeppi, K.; van der Heijden, M.G. Keystone taxa as drivers of microbiome structure and functioning. Nat. Rev. Microbiol. 2018, 16, 567–576. [Google Scholar] [CrossRef]
- Berry, D.; Widder, S. Deciphering microbial interactions and detecting keystone species with co-occurrence networks. Front. Microbiol. 2014, 5, 219. [Google Scholar] [CrossRef]
- Liang, Y.; Zhao, H.; Deng, Y.; Zhou, J.; Sun, B. Long-term oil contamination alters the molecular ecological networks of soil microbial functional genes. Front. Microbiol. 2016, 7, 169724. [Google Scholar] [CrossRef]
- Mora-Ruiz, M.d.R.; Font-Verdera, F.; Díaz-Gil, C.; Urdiain, M.; Rodríguez-Valdecantos, G.; González, B.; Orfila, A.; Rosselló-Móra, R. Moderate halophilic bacteria colonizing the phylloplane of halophytes of the subfamily Salicornioideae (Amaranthaceae). Syst. Appl. Microbiol. 2015, 38, 406–416. [Google Scholar] [CrossRef] [PubMed]
- Enquahone, S.; van Marle, G.; Simachew, A. Plant growth-promoting characteristics of halotolerant endophytic bacteria isolated from Sporobolus specatus (Vahr) Kunth and Cyperus laevigatus L. of Ethiopian rift valley lakes. Arch. Microbiol. 2022, 204, 403. [Google Scholar] [CrossRef] [PubMed]
- Mora-Ruiz, M.d.R.; Font-Verdera, F.; Orfila, A.; Rita, J.; Rosselló-Móra, R. Endophytic microbial diversity of the halophyte Arthrocnemum macrostachyum across plant compartments. FEMS Microbiol. Ecol. 2016, 92, fiw145. [Google Scholar] [CrossRef] [PubMed]
- Shafreen, M.; Vishwakarma, K.; Shrivastava, N.; Kumar, N. Physiology and distribution of nitrogen in soils. In Soil Nitrogen Ecology; Springer: Cham, Switzerland, 2021; pp. 3–31. [Google Scholar] [CrossRef]
- Sun, W.; Xiao, E.; Häggblom, M.; Krumins, V.; Dong, Y.; Sun, X.; Li, F.; Wang, Q.; Li, B.; Yan, B. Bacterial survival strategies in an alkaline tailing site and the physiological mechanisms of dominant phylotypes as revealed by metagenomic analyses. Environ. Sci. Technol. 2018, 52, 13370–13380. [Google Scholar] [CrossRef]
- Zhang, Y.; Cao, C.; Peng, M.; Xu, X.; Zhang, P.; Yu, Q.; Sun, T. Diversity of nitrogen-fixing, ammonia-oxidizing, and denitrifying bacteria in biological soil crusts of a revegetation area in Horqin Sandy Land, Northeast China. Ecol. Eng. 2014, 71, 71–79. [Google Scholar] [CrossRef]
- Deng, S.; Zhang, D.; Wang, G.; Zhou, X.; Ye, C.; Fu, T.; Ke, T.; Zhang, Y.; Liu, Y.; Chen, L. Biological soil crust succession in deserts through a 59-year-long case study in China: How induced biological soil crust strategy accelerates desertification reversal from decades to years. Soil Biol. Biochem. 2020, 141, 107665. [Google Scholar] [CrossRef]
- Llamas, I.; Moral, A.d.; Martínez-Checa, F.; Arco, Y.; Arias, S.; Quesada, E. Halomonas maura is a physiologically versatile bacterium of both ecological and biotechnological interest. Antonie Van Leeuwenhoek 2006, 89, 395–403. [Google Scholar] [CrossRef]
- Faulkner, M.; Hoeven, R.; Kelly, P.P.; Sun, Y.; Park, H.; Liu, L.-N.; Toogood, H.S.; Scrutton, N.S. Chemoautotrophic production of gaseous hydrocarbons, bioplastics and osmolytes by a novel Halomonas species. Biotechnol. Biofuels Bioprod. 2023, 16, 152. [Google Scholar] [CrossRef]
- Li, Y.; Yang, R.; Häggblom, M.M.; Li, M.; Guo, L.; Li, B.; Kolton, M.; Cao, Z.; Soleimani, M.; Chen, Z. Characterization of diazotrophic root endophytes in Chinese silvergrass (Miscanthus sinensis). Microbiome 2022, 10, 186. [Google Scholar] [CrossRef]
- Giller, K.E.; James, E.K.; Ardley, J.; Unkovich, M.J. Science losing its way: Examples from the realm of microbial N2-fixation in cereals and other non-legumes. Plant Soil 2024, 1–24. [Google Scholar] [CrossRef]
- Egener, T.; Sarkar, A.; Martin, D.E.; Reinhold-Hurek, B. Identification of a NifL-like protein in a diazotroph of the β-subgroup of the Proteobacteria, Azoarcus sp. strain BH72. Microbiology 2002, 148, 3203–3212. [Google Scholar] [CrossRef] [PubMed]
- Dixon, R. The oxygen-responsive NIFL-NIFA complex: A novel two-component regulatory system controlling nitrogenase synthesis in γ-Proteobacteria. Arch. Microbiol. 1998, 169, 371–380. [Google Scholar] [CrossRef] [PubMed]
- Jha, B.; Gontia, I.; Hartmann, A. The roots of the halophyte Salicornia brachiata are a source of new halotolerant diazotrophic bacteria with plant growth-promoting potential. Plant Soil 2012, 356, 265–277. [Google Scholar] [CrossRef]
- Chen, C.; Anwar, N.; Wu, C.; Fu, G.; Wang, R.; Zhang, C.; Wu, Y.; Sun, C.; Wu, M. Halomonas endophytica sp. nov., isolated from liquid in the stems of Populus euphratica. Int. J. Syst. Evol. Microbiol. 2018, 68, 1633–1638. [Google Scholar] [CrossRef]
- Wang, L.; Shao, Z. Aerobic denitrification and heterotrophic sulfur oxidation in the genus Halomonas revealed by six novel species characterizations and genome-based analysis. Front. Microbiol. 2021, 12, 652766. [Google Scholar] [CrossRef]
- de Vries, F.T.; Griffiths, R.I.; Knight, C.G.; Nicolitch, O.; Williams, A. Harnessing rhizosphere microbiomes for drought-resilient crop production. Science 2020, 368, 270–274. [Google Scholar] [CrossRef]
- Schmidt, S.; Reed, S.C.; Nemergut, D.R.; Stuart Grandy, A.; Cleveland, C.C.; Weintraub, M.N.; Hill, A.W.; Costello, E.K.; Meyer, A.; Neff, J. The earliest stages of ecosystem succession in high-elevation (5000 metres above sea level), recently deglaciated soils. Proc. R. Soc. B 2008, 275, 2793–2802. [Google Scholar] [CrossRef]
- Bar-Even, A.; Noor, E.; Milo, R. A survey of carbon fixation pathways through a quantitative lens. J. Exp. Bot. 2012, 63, 2325–2342. [Google Scholar] [CrossRef]
- Ahemad, M.; Kibret, M. Mechanisms and applications of plant growth promoting rhizobacteria: Current perspective. J. King Saud. Univ. Sci. 2014, 26, 1–20. [Google Scholar] [CrossRef]
- Goswami, D.; Pithwa, S.; Dhandhukia, P.; Thakker, J.N. Delineating Kocuria turfanensis 2M4 as a credible PGPR: A novel IAA-producing bacteria isolated from saline desert. J. Plant Interact. 2014, 9, 566–576. [Google Scholar] [CrossRef]
- Sagar, L.; Singh, S.; Sharma, A.; Maitra, S.; Attri, M.; Sahoo, R.K.; Ghasil, B.P.; Shankar, T.; Gaikwad, D.J.; Sairam, M.; et al. Role of Soil Microbes against Abiotic Stresses Induced Oxidative Stresses in Plants. In Microbial Symbionts and Plant Health: Trends and Applications for Changing Climate; Springer: Singapore, 2023; pp. 149–177. [Google Scholar] [CrossRef]
- Hamilton, C.E.; Gundel, P.E.; Helander, M.; Saikkonen, K. Endophytic mediation of reactive oxygen species and antioxidant activity in plants: A review. Fungal Divers. 2012, 54, 1–10. [Google Scholar] [CrossRef]
- Oliva, G.; Di Stasio, L.; Vigliotta, G.; Guarino, F.; Cicatelli, A.; Castiglione, S. Exploring the Potential of Four Novel Halotolerant Bacterial Strains as Plant-Growth-Promoting Rhizobacteria (PGPR) under Saline Conditions. Appl. Sci. 2023, 13, 4320. [Google Scholar] [CrossRef]
- Xiao, S.; Wan, Y.; Zheng, Y.; Wang, Y.; Fan, J.; Xu, Q.; Gao, Z.; Wu, C. Halomonas ventosae JPT10 promotes salt tolerance in foxtail millet (Setaria italica) by affecting the levels of multiple antioxidants and phytohormones. Plant-Environ. Interact. 2023, 4, 275–290. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Kong, T.; Huang, D.; Chen, Z.; Kolton, M.; Yang, J.; Huang, Y.; Cao, Y.; Gao, P.; Yang, N.; et al. Arsenic (As) oxidation by core endosphere microbiome mediates As speciation in Pteris vittata roots. J. Hazard. Mater. 2023, 454, 131458. [Google Scholar] [CrossRef]
- Chen, Q.-L.; Hu, H.-W.; Zhu, D.; Ding, J.; Yan, Z.-Z.; He, J.-Z.; Zhu, Y.-G. Host identity determines plant associated resistomes. Environ. Pollut. 2020, 258, 113709. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).