Abstract
The Ara h 6 protein is an important allergen found in peanuts (Arachis hypogaea). Ara h 6 represents a significant risk to human health, given its potential to trigger IgE-mediated anaphylaxis. Seeing as peanuts are often heat-processed prior to consumption, understanding the effect heat application has on the Ara h 6 protein’s structure and function is vital. Therefore, the purpose of this study was to explore, through the application of long-timescale 200 ns GROMACS molecular dynamics simulations, the structural changes that occur in the Ara h 6 allergen during thermal processing at 300 K, 350 K, 400 K and 450 K. Larger fluctuations in the Ara h 6 allergen’s secondary structure, RMSD and RMSF were identified at higher processing temperatures. However, observed decreases in Rg and SASA as processing temperature rose from 300 K to 400 K suggested that these observed fluctuations in the structure may be due to a compaction of the protein’s structure. Overall, the Ara h 6 allergen exhibited high thermostability.
1. Introduction
Peanuts (Arachis hypogaea) are an important source of protein worldwide. They can be consumed in their raw form, although they are often processed prior to consumption [1]. Processing peanuts, in many cases, involves a form of heat treatment, including roasting, boiling, or frying [2]. Although an energy- and nutrient-dense food, peanuts do contain several known allergenic proteins [3]. As a result, they represent a potential danger to human health, as they can trigger severe IgE-mediated allergic reactions. The major allergens identified in peanuts include the Ara h 1, 2, 3 and 6 allergens [4]. The Ara h 2 and 6 allergens appear to be the most proficient at eliciting anaphylaxis and therefore are often considered the dominant allergens in peanuts [5,6]. Ara h 2 and 6 are both 2S albumin seed storage proteins and are moderately homologous with 59% of the same amino acid sequence [7]. Even though Ara h 2 and 6 possess a similar heat and immunologically stable core structure and many homologous IgE-binding epitopes, each of these allergens has its own distinct properties [7,8]. Of the seven linear epitopes of the Ara h 6 peanut allergen identified and located between residues Met3–Ser15, Lys26–Ile36, Tyr43–Arg49, Cys61–Gly72, Cys75–Asp87, Lys99–Gln108 and Cys109–Val121, five are homologous to epitopes on Ara h 2, but two are entirely unique [8].
Given the structural and functional diversity of peanut allergens and the potential risk they pose to human health, understanding how the thermal processing of peanuts may affect protein conformation and functionality positively or negatively is critical. Molecular dynamics (MD) simulations can be employed in the study of this, allowing a preliminary review of the changes that occur at a molecular level within the allergen during stages of thermal treatment. GROningen MAchine for Chemical Simulations (GROMACS 2023.2), a high-performance molecular dynamics software package, is well adapted to the study of food proteins. An obstacle to the broader application of GROMACS MD simulations in the study of food allergens undergoing treatment is the large computational power requirement necessary to run long-timescale simulations, comparable to real-life processes. Although computationally costly, the execution and analysis of GROMACS MD simulations in the study of food allergens can be crucial in advancing discovery by allowing more directed and pertinent future experimental work.
GROMACS MD simulation studies of the Ara h 2 and 6 allergens have been performed in the past. This study aims to build upon those preceding works. Hollingsworth et al. [9] performed long-timescale 1000 ns GROMACS MD simulations of the Ara h 2 allergen to evaluate the structural mechanism of its allergenicity and the efficiency of different GPU/CPU configurations in running the simulations. Earlier, Vanga et al. [10] performed a 1 ns GROMACS MD simulation study of the Ara h 6 allergen examining the impact thermal and electric field treatment had on the allergen’s conformation. Vanga et al. [10] highlight in their work the need for long-timescale MD studies to allow further insight into the Ara h 6 allergen. As the Ara h 2 allergen has already been studied over a long-timescale GROMACS MD simulation and to fill the gap identified by Vanga et al. [10], the Ara h 6 allergen will be the focus of this study. The Ara h 6 allergen is made up of 127 amino acid residues and weighs 14.98 kDa [7]. The amino acid sequence of the Ara h 6 allergen is depicted in Figure 1A below, and the allergen’s surface properties are depicted in Figure 1B. The Ara h 6 allergen possesses a highly stable structural conformation with multiple α-helices joined by turns and coils, as seen in Figure 1C. The structure is further stabilized by the presence of five disulfide bonds.

Figure 1.
(A) VMD Sequence Viewer analysis of the Ara h 6 allergen in peanuts, (B) VMD 3-D structure of the Ara h 6 allergen in peanuts colored based on residue type, and (C) VMD 3-D structure of the Ara h 6 allergen in peanuts colored based on secondary structure.
Taking into consideration that heat treatment is the process most commonly used in the processing of peanuts and the gap in knowledge identified by Vanga et al. [10], the primary aim of this study will be to ascertain the effect heat treatment has on the conformation of the Ara h 6 allergen through the application of a long-timescale 200 ns GROMACS MD simulation.
2. Materials and Methods
The methodology applied in this study was adapted from that employed by Vanga et al. [10], Vagadia et al. [11], Saxena et al. [12], Wang et al. [13] and Zhu et al. [14] in their molecular dynamics studies of various food proteins. GROMACS tutorial “Lysozyme in Water” by Lemkul [15] was also an important resource aiding in the development of this protocol.
2.1. Molecular Dynamics (MD) Simulations
Classical molecular dynamics (MD) simulations were performed using the latest version of the GROningen MAchine for Chemical Simulations software package, version 2023.2 [16,17,18,19]. The Ara h 6 protein, an allergen found in peanuts (Arachis hypogaea), was used in all simulations. The Ara h 6 allergen’s structure was procured from the Research Collaboratory for Structural Bioinformatics Protein Data Bank (RCSB PDB) under PDB entry ID 1W2Q [7,20]. To ensure there were no non-protein residues present in the starting configuration of the Ara h 6 protein obtained from the RCSB PDB, the structure was loaded into the ChimeraX visualization program (version 1.6) for preliminary visualization [21,22]. The validated PDB structure was then uploaded to the CHARMM-GUI PDB Reader and Manipulator and converted to a CHARMM-readable format to ensure compatibility with the chosen force field [23,24,25,26,27]. The CHARMM36m force field (updated July 2022) and the CHARMM-modified TIP3P water model were used in all simulations [28]. The Ara h 6 allergen was encapsulated in a cubic simulation box of 10.615 nm × 10.615 nm × 10.615 nm with periodic boundary conditions. The distance between the protein and the simulation box edge was set at 1.2 nm to ensure that the minimum image convention was observed, per the chosen CHARMM36m force field. As illustrated in Figure 2 below, 38,270 water molecules were added into the simulation box to solvate the Ara h 6 protein. The system was then neutralized, as 3 of the 38,270 solute molecules were removed from the simulation box and 3 Na+ ions were added in their place.
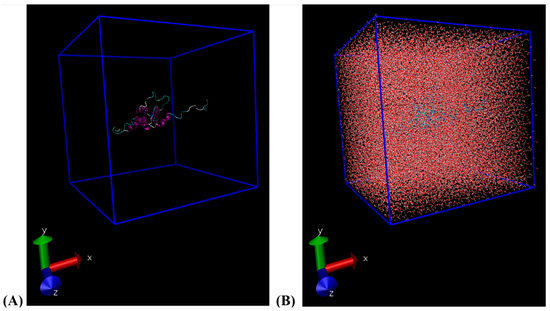
Figure 2.
(A) VMD visualization of the cubic simulation box built (10.615 nm × 10.615 nm × 10.615 nm); (B) VMD visualization of the water-solvated cubic simulation box built.
The assembled system was then energy-minimized using the steepest descent algorithm. With a maximum step-size (emstep) set at 0.01 nm, the minimization converged to a maximum force of less than 100 kJ mol−1 nm−1 (emtol) in 8,867 steps. The figure below, Figure 3, illustrates how the potential energy of the system successfully converged over the 8,867 minimization steps performed.
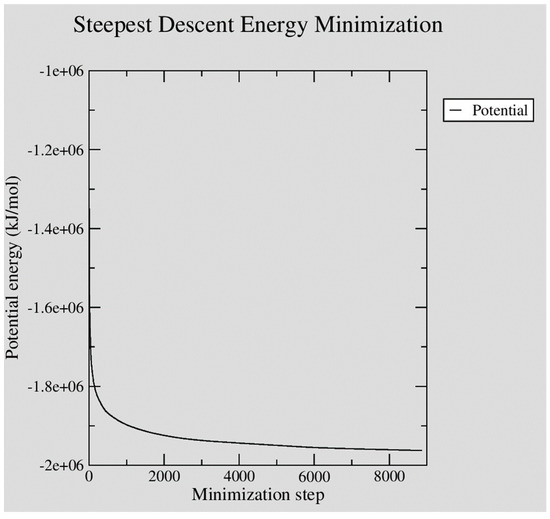
Figure 3.
XMGRACE plot illustrating the steady convergence of potential energy during GROMACS steepest descent energy minimization of the neutralized Ara h 6 protein in water system built.
The energy-minimized system, constructed as indicated above, was used as the starting assembly in all simulations. Simulations were all performed in triplicate. Each replicate began with a constant-volume constant-temperature equilibration (NVT) of 200 ps, during which the energy-minimized Ara h 6 protein was position-restrained. In NVT, the velocity-rescaling temperature coupling was used to maintain the system at the treatment temperature under investigation, either 300 K, 350 K, 400 K, or 450 K [29]. These four specific thermal treatment temperatures were selected following a thorough review of previously performed MD experiments of food proteins [30]. Similar temperature ranges to those investigated in these previous MD studies were maintained in this experiment to facilitate an analysis and discussion of findings relative to the broader literature available. Another factor considered as the thermal treatment temperatures to be investigated in this study were selected was the conditions industrially processed dry-roasted peanuts may be exposed to. The 400 K and 450 K treatment temperatures chosen reflect the thermal conditions dry-roasted peanuts may be subjected to during industrial processing [31,32]. Initial velocities of the Ara h 6 protein particles were randomly generated during the NVT equilibration step, according to a Maxwell–Boltzmann velocity distribution. Following NVT, the system then underwent a constant-pressure constant-temperature equilibration (NPT) of 200 ps, during which the NVT-equilibrated Ara h 6 protein was position-restrained. In NPT, the Parinello–Rahman barostat was chosen to maintain the pressure at 1 bar [33]. The integration step used for NVT, NPT and the production run was 2 fs. The coupling time constants, tau_t and tau_p, used during NVT, NPT and the production run were set at 0.1 ps and 2.0 ps, respectively. The isothermal compressibility of water was set to 4.5 × 10−5 bar−1. The parameters specified in all .mdp files used during the simulation setup, NVT and NPT equilibration and production runs were selected based on the CHARMM36m force field requirements for GROMACS. Long-range electrostatic interactions were computed using the smooth Particle Mesh Ewald (PME) method [34]. Cubic interpolation and a Fourier grid spacing of 0.12 nm were used for PME. Short-range electrostatic and Van der Waals interactions were cut off at a distance of 1.2 nm. Periodic boundary conditions were applied in all three directions, the x-, y- and z-axes. Finally, the bonds of the protein were constrained using the LINear Constraint Solver (LINCS) algorithm [35]. Twelve MD simulation production runs of a duration of 200 ns were executed, as each of the four processing temperatures (300 K, 350 K, 400 K or 450 K) were run in triplicate with a new NVT (and thus, initial Maxwell distributed velocity profile), NPT, and production run undertaken for each replicate. Compressed coordinates were written to the output .xtc file every 1 ps. Figure 4 below provides further detail on the particular GROMACS commands used in the methodology described above.

Figure 4.
Summary of computational steps taken to set up and run the GROMACS simulations.
2.2. Analysis of Molecular Dynamics (MD) Simulations
The generated trajectories were processed using the GROMACS trajectory converter (gmx trjconv) to remove periodic boundary conditions and isolate the Ara h 6 protein from the solvent. VMD version 1.9.3 molecular visualization software was used to verify and visualize the converted trajectories and to generate snapshots of the Ara h 6 protein’s structure [36]. GROMACS analysis tools were used to evaluate root-mean-square deviation (gmx rms), root-mean-square fluctuation and B-factors (gmx rmsf), radius of gyration (gmx gyrate), hydrogen bonds (gmx hbond) and solvent-accessible surface area (gmx sasa). The XMGRACE version 5.1.25 graphing software was used to generate graphs for all simulations [37]. A secondary structure analysis of all MD trajectories was performed using the STRIDE algorithm, an element of the VMD (version 1.9.3) plugin TIMELINE [38].
To establish the statistical significance of the observed trends, the data obtained were analyzed by one-way analysis of variance (ANOVA) using IBM SPSS Statistics software platform, version 29.0.1.1. A significance level of 0.05 (p < 0.05) was used throughout. The effect size, Eta-squared (η2), was interpreted as follows: η2 < 0.01 is a negligible effect, 0.01 ≤ η2 < 0.06 is a small effect, 0.06 ≤ η2 < 0.14 is a medium effect and η2 ≥ 0.14 is a large effect. Assumptions in one-way ANOVA were validated using the Shapiro–Wilk test of normality and the Levene test for homogeneity of variances [39,40]. To validate for the absence of spurious outliers in the data, stem-and-leaf plots were analyzed for isolated values at the ends. If the Levene test indicated unequal variances, Welch and Brown–Forsythe robust tests of equality of means were used, along with Tamhane’s T2 post hoc test. If the Levene test proved equal variances, post hoc analysis to determine which groups differ significantly was performed using Tukey’s HSD (honestly significant difference) multiple comparison test. Post hoc analysis was only performed if the one-way ANOVA revealed a significant result.
3. Results and Discussion
3.1. Secondary Structure Analysis
The TIMELINE extension of VMD is an analysis tool that creates a graphical representation of secondary structure changes that occur during MD simulation. It is based on the STRIDE algorithm and maps alterations in secondary structure frame by frame over the simulation’s trajectory [41]. The y-axis of the TIMELINE graph details the amino acid sequence of the studied protein, while the x-axis follows the simulation over its length in picoseconds. The secondary structure each amino acid takes on, through every picosecond of the simulation, is illustrated on the graph using a pre-defined color code. Secondary structure configurations are depicted as follows: turns in teal, extended configurations in bright yellow, isolated β-bridges in dark yellow, α-helices in pink, 3–10 helices in blue, π-helices in red, and coils in white. To be able to effectively interpret the results of the TIMELINE analyses performed in this study, the initial conformation of the Ara h 6 allergen simulated must first be carefully defined.
RCSB PDB structure 1W2Q, the starting structure of the Ara h 6 allergen employed, is 127 residues long with a secondary structure mainly composed of α-helices, coils, and turns. The α-helices range over Cys16–Asp21, Leu25–Gly38, Arg52–Asn65, Cys73–Gln85 and Met94–Cys109 and are six, fourteen, fourteen, thirteen and sixteen residues long, respectively. 1W2Q equally features two isolated β-bridges located in positions Tyr46 and Arg49 and a single 3–10 helix ranging three residues over Pro2–Arg4. An important tertiary structure feature of the Ara h 6 allergen to also consider is the five disulfide bonds, located between residues Cys16–Cys73, Cys28–Cys60, Cys61–Cys109, Cys75–Cys117 and Cys86–Cys126. Having defined the key features of the Ara h 6 allergen’s starting structure, the generated TIMELINE graphs for each thermal treatment, illustrated in Figure 5, Figure 6, Figure 7 and Figure 8, could then be interpreted. To ensure that no important structural changes were overlooked, the TIMELINE graphs were analyzed systematically, first focusing on the more global changes and then finally on the more minute changes occurring within specific regions of the structure through the replicates. In this analysis, the TIMELINE structural changes identified were only considered significant if they were observed in all three replicates (A, B, and C) performed to study a given thermal treatment.

Figure 5.
VMD TIMELINE analysis of the Ara h 6 allergen undergoing thermal treatment at 300 K (turns in teal, extended configurations in bright yellow, isolated β-bridges in dark yellow, α-helices in pink, 3–10 helices in blue, π-helices in red, coils in white): (A) Replicate 1, (B) Replicate 2, and (C) Replicate 3.

Figure 6.
VMD TIMELINE analysis of the Ara h 6 allergen undergoing thermal treatment at 350 K (turns in teal, extended configurations in bright yellow, isolated β-bridges in dark yellow, α-helices in pink, 3–10 helices in blue, π-helices in red, coils in white): (A) Replicate 1, (B) Replicate 2, and (C) Replicate 3.

Figure 7.
VMD TIMELINE analysis of the Ara h 6 allergen undergoing thermal treatment at 400 K (turns in teal, extended configurations in bright yellow, isolated β-bridges in dark yellow, α-helices in pink, 3–10 helices in blue, π-helices in red, coils in white): (A) Replicate 1, (B) Replicate 2, and (C) Replicate 3.
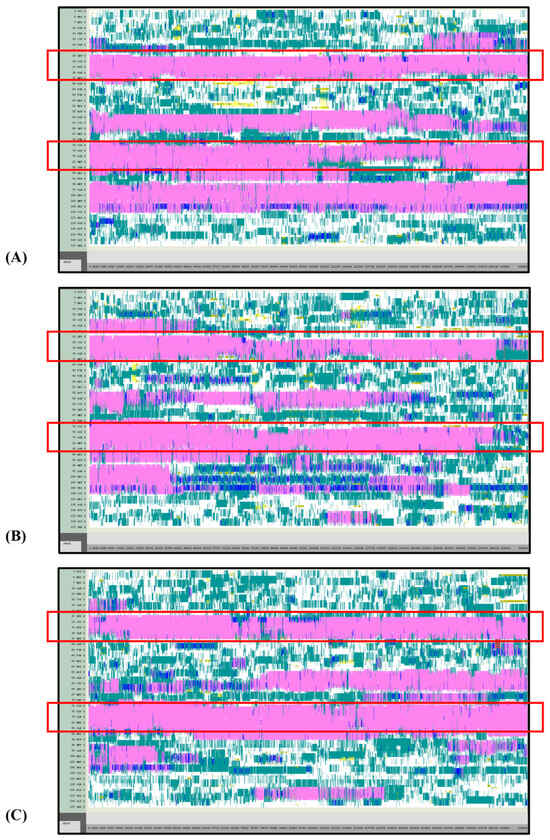
Figure 8.
VMD TIMELINE analysis of the Ara h 6 allergen undergoing thermal treatment at 450 K (turns in teal, extended configurations in bright yellow, isolated β-bridges in dark yellow, α-helices in pink, 3–10 helices in blue, π-helices in red, coils in white): (A) Replicate 1, (B) Replicate 2, and (C) Replicate 3.
Overall, it was found that as the thermal treatment temperature rose from 300 K (Figure 5) to 350 K (Figure 6) to 400 K (Figure 7) and finally to 450 K (Figure 8), the TIMELINE graphs became increasingly chaotic. An increasing proportion of turns, colored in teal, and fewer coils, colored in white, were visible as the treatment temperature increased. The five α-helices of the Ara h 6’s structure were easily distinguished and relatively uniform over the entire length of the 300 K replicates (Figure 5). This uniformity was not observed in the 450 K replicates (Figure 8), as the location of the α-helices in those simulations was slightly more difficult to discern and the α-helices fluctuated more significantly, repeatedly breaking and being reformed. Even though fluctuations in secondary structure were more significant in the simulations performed at higher temperatures, the dominant structural features of the Ara h 6 protein still appear to have been retained. The two α-helices that best resisted and rarely fluctuated were those ranging between Leu25–Gly38 and Cys73–Gln85. Their positions are marked in Figure 5, Figure 6, Figure 7 and Figure 8 by red boxes. These two α-helices maintained their secondary structure, for the most part, through all replicates at the four treatment temperatures. The stability of these two α-helices may be partially attributed to the disulfide bonds, namely Cys16–Cys73, Cys28–Cys60 and Cys75–Cys117, that involve residues in their range. The presence of disulfide bonds in the Ara h 6 structure likely plays a role in stabilizing the protein’s secondary structure elements, namely the α-helices, and appears to make the allergen less vulnerable to denaturation and protein unfolding at high temperatures. Even though fluctuations were observed, the overall stability of the dominant secondary structure features at the core of the Ara h 6 allergen may contribute to its ability to maintain its allergenicity, even after high-temperature food processing treatment.
3.2. Root-Mean-Square Deviation (RMSD)
Backbone-to-backbone root-mean-square deviation (RMSD) is calculated at each timestep of the simulation and measures by what average distance the backbone atoms of a molecule have deviated from their reference structure [42]. The reference structure used in this study, for all replicates, was the minimized, NVT- and NPT-equilibrated structure of the Ara h 6 allergen. In other words, the reference structure used was the Ara h 6 allergen’s structure at the first frame of the simulation, timestep 0 [42]. Peaks in calculated RMSD allowed for the identification of periods in the simulation of important conformational change. Furthermore, a flattening of the RMSD curve toward the end of the simulation suggested the achievement of some degree of structural equilibrium [9]. GROMACS gmx rms function calculates RMSD by “least-square fitting the structure to the reference structure”, according to Equation (1) [43,44] (p. 543).
As indicated in Equation (1), the position of atom i in the reference structure is compared to the position of the same atom i in the heat-stressed structure [44]. This calculation is repeated for each timestep t of the simulation (every 1 ps). In Equation (1), N equals the total number of atom coordinates [44].
From Figure 9 and replicate data presented in Table 1, it was observed that backbone-to-backbone RMSD consistently increased as the treatment temperature rose from 300 K through to 450 K. A higher proportion of large peaks were evident in the 450 K treatment curve (green) of Figure 9 than for any other treatment. The larger and more frequent peaks at 450 K indicate that larger conformational changes in the Ara h 6 structures arose during the 450 K treatment than in any other. The 450 K curve in Figure 9 was, however, the only curve to not flatten toward the end of the 200 ns simulation time. This indicates that the 450 K treated systems did not converge to an equilibrium state in 200 ns and that relevant data may be missing towards the end of these simulations [9]. A longer simulation time may be required for the 450 K treatment temperature.
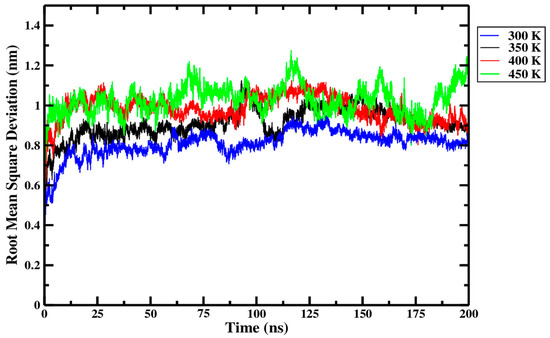
Figure 9.
Average root-mean-square deviation (RMSD) of triplicate 200 ns GROMACS MD simulations of the Ara h 6 allergen undergoing thermal processing at 300, 350, 400 or 450 K.

Table 1.
One-way ANOVA statistical analysis of the effect of processing temperature on the RMSD of the Ara h 6 allergen over a 200 ns GROMACS MD simulation.
As explored above, Figure 9 and data in Table 1 seem to indicate a relationship between increasing thermal treatment temperature and higher RMSD values of the Ara h 6 allergen. To define if the effect of thermal processing temperature on RMSD of the Ara h 6 allergen is statistically significant, a one-way ANOVA test was performed. The one-way ANOVA revealed that there was a statistically significant difference in the RMSD of the Ara h 6 allergen between at least two groups [F(3, 8) = 20.211, p < 0.001]. Eta squared (η2), a measure of effect size, allowed for the determination of the strength of this association and suggested a large effect size (η2 = 0.883, 95% C.I. = [0.469, 0.922]). Tukey’s HSD test for multiple comparisons found that the mean value of RMSD of the Ara h 6 allergen was significantly different between 300 K and 350 K (p = 0.037, 95% C.I. = [−0.197, −0.006]), between 300 K and 400 K (p = 0.002, 95% C.I. = [−0.268, −0.078]), between 300 K and 450 K (p < 0.001, 95% C.I. = [−0.312, −0.121]), and between 350 K and 450 K (p = 0.020, 95% C.I. = [−0.210, −0.020]). There was no statistically significant difference between 350 K and 400 K (p = 0.155) or between 400 K and 450 K (p = 0.497). Assumptions of the one-way ANOVA test were validated to verify the strength of this statistical analysis. The only identified limitation is the significant Shapiro–Wilk test of normality at 350 K (p = 0.017), indicating that the averaged over the simulation length RMSD values for replicate 1 (0.94 ± 0.12), for replicate 2 (0.94 ± 0.11) and replicate 3 (0.84 ± 0.07) at 350 K do not form a normal distribution. The one-way ANOVA is considered robust to violations of normality, but a larger sample size, more than three replicates, would be required in future studies to limit the risk of seeing this type of assumption violation. A larger number of replicates, and thus a larger sample size, would increase the power of this study [45].
3.3. Root-Mean-Square Fluctuation (RMSF)
Cα root-mean-square fluctuation (RMSF) was used, in this study, to locate the more flexible and mobile residues of the Ara h 6 allergen’s structure while under thermal treatment [46]. RMSF can be computed on a per-atom or per-residue basis and measures the distance over which an atom or residue has moved, relative to the atom or residue’s average position during the simulation. GROMACS gmx rmsf function calculates RMSF based on Equation (2) [12]. In this study, RMSF was calculated on a per-residue basis for the Cα atom of each amino acid. In Equation (2), indicates the position of the Cα atom i and indicates the Cα atom i’s average position based on all timesteps of the simulation. The -oq option of the gmx rmsf function uses RMSF values obtained to determine B-factor values and subsequently writes these B-factor values to an output .pdb file that can be displayed visually in VMD [44]. B-factors are calculated based on Equation (3) and are given in units of Å2.
Figure 10 and Figure 11, respectively, summarize the RMSF and B-factor values obtained for the Ara h 6 allergen over triplicate 200 ns GROMACS MD simulations at 300 K, 350 K, 400 K and 450 K. In Figure 10, it can be generally observed that as the processing temperature increased from 300 K through to 450 K, the RMSF values obtained increased, meaning that the residues of the Ara h 6 allergen fluctuated more about their average position during the simulation when exposed to a higher temperature processing treatment. Visually, in Figure 10, it can be observed that the increase in RMSF between 350 K (black curve) and 400 K (red curve), although present, is rather small relative to the increases observed between other temperature groups. Further investigation is required to provide an explanation for this visually smaller increase and to determine if it is statistically significant or not. The B-factor graphs of Figure 11 allow a similar observation as to the effect of processing temperature. The B-factors of the more exposed, less compact regions of the Ara h 6 protein, namely the loop to the left and the tail to the right, are highly variable at all temperatures over all replicates. Interpreting the B-factors of these regions is therefore difficult and not particularly compelling. However, the visible effect that the treatment temperature had on the B-factors of the more compact, center portion of the molecule is of greater interest. For all three replicates, as temperature increased, it can be seen in Figure 11 that the more compact, center portion of the molecule became less uniformly dark red and showed increased color variation as the B-factors in this region rose.
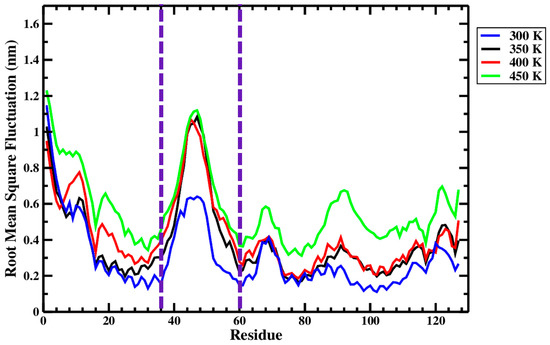
Figure 10.
Average root-mean-square fluctuation (RMSF), on a per-residue (amino acid) basis, of triplicate 200 ns GROMACS MD simulations of the Ara h 6 allergen undergoing thermal processing at 300, 350, 400 or 450 K.
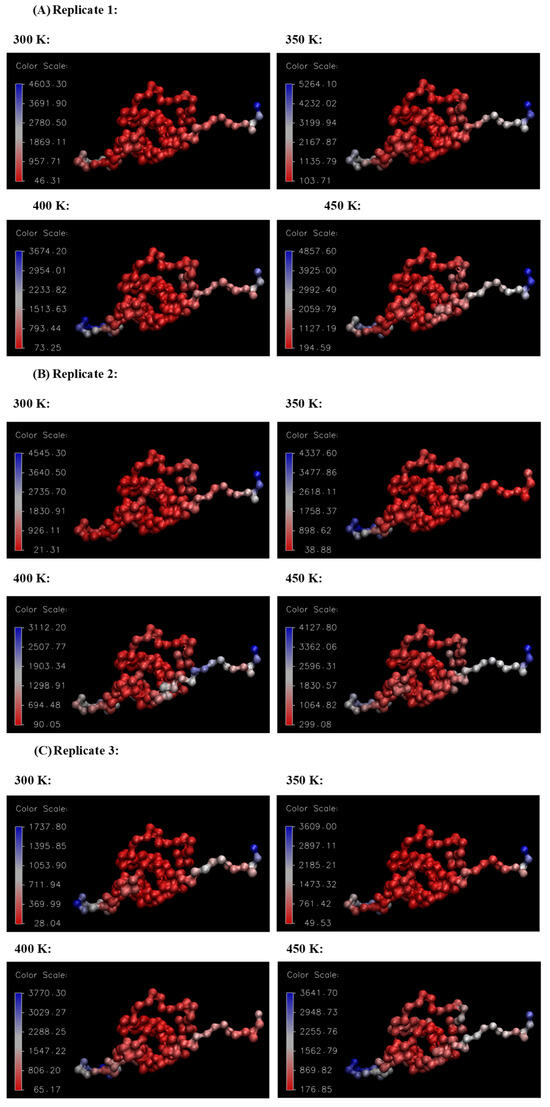
Figure 11.
VMD B-factor analysis, aiding in the visualization of the areas of high atomic fluctuation during the thermal processing of the Ara h 6 allergen. Color scale units are Å2. (A) Replicate 1, (B) Replicate 2, (C) Replicate 3.
Figure 10, as has been explored, provides a general outlook as to how the RMSF of the Ara h 6 allergen is affected by processing temperature. However, Figure 10 equally highlights the sequences of amino acids in the Ara h 6 protein’s structure that are more flexible and mobile under treatment. The peaks of the four treatment curves in Figure 10 follow each other well, often showing peaks in RMSF over the same sequences. The largest peak and most flexible region of the Ara h 6 protein identified under all four thermal treatments is between Ile36–Cys60. The Ile36–Cys60 region is delineated in Figure 10 by two dashed, vertical purple lines. This region may be of particular importance in future studies of the allergenicity of the Ara h 6 protein. As can be seen in Table 2, various linear and conformational IgE-binding epitopes have been identified within the 127-residue sequence of the Ara h 6 allergen, many within or partially within the flexible region Ile36–Cys60 identified above. The increased mobility and flexibility that the epitopes within this Ile36–Cys60 acquire during thermal processing may either increase or decrease their exposure for IgE-binding. An epitope that is more exposed would have an increased risk of triggering an allergic reaction. In order to ascertain how exactly the increased flexibility in the Ile36–Cys60 region of the Ara h 6 allergen affects its allergic potential, experimental work would need to be performed.
Table 2.
Allergen epitopes present in Ara h 6 protein.
3.4. Radius of Gyration (Rg)
The radius of gyration (Rg) is an indicator of the compactness of a protein’s structure [48]. A decrease in Rg suggests a protein’s structure is becoming more compact, whereas an increase in Rg suggests a protein’s structure is becoming more spread out [49]. More precisely, Rg is a measure of “the distribution of atoms in [a protein molecule] with respect to its center of mass” [12] (p. 6). GROMACS gmx gyrate function calculates the Rg of the protein, based on Equation (4) [44]. In Equation (4), “ is the mass of atom i and the position of atom i with respect to the center of mass of the [protein] molecule” [44] (p. 542).
From Figure 12 and within the replicate data presented in Table 3, it can be seen that only very minute differences in Rg were observed across the different processing temperatures. Rg is highest at 300 K, decreases through to 400 K, and then rises slightly at 450 K. The most marked difference in Rg, visible in Figure 12, was a decrease observed as the treatment temperature rose from 300 K (blue curve) to 400 K (red curve). The relatively higher Rg at 300 K suggests that the Ara h 6 allergen retained its more extended initial configuration during this treatment. The noticeable drop in Rg as the treatment temperature rose from 300 K to 400 K suggests that the Ara h 6 allergen, during thermal processing, initially tends to compact, not unfold. The observed tendency of the Ara h 6 allergen structure to compact and resist denaturation is further evidence of its structural stability. The thermal stability of the Ara h 6 allergen was also observed experimentally by Lehmann et al. [7]. As compaction of the Ara h 6 allergen was detected in this analysis, it is important to highlight a potential confounding factor, the CHARMM36m force field. The CHARMM36m has been found, in certain studies, to over-stabilize helices and over-compact protein structures [50]. GROMACS MD simulations performed using a different force field could help better define the contribution, if any, of the CHARMM36m force field to the compaction of the Ara h 6 allergen observed in this analysis. The slight rise in Rg observed between 400 K and 450 K is difficult to interpret in this experiment. In order to interpret this deviation to the overall trend in Rg observed between 300 K, 350 K, and 400 K, it would be necessary to further investigate the effect that temperatures exceeding 450 K have on the Rg of the Ara h 6 peanut protein. Studying this would provide insight into whether this rise in Rg observed in this experiment at 450 K is a short spike or the beginning of a new trend wherein the Rg of the Ara h 6 peanut protein increases at higher treatment temperatures. Further experimentation at higher treatment temperatures, such as 500 K and 550 K, is necessary to elucidate this potential effect.
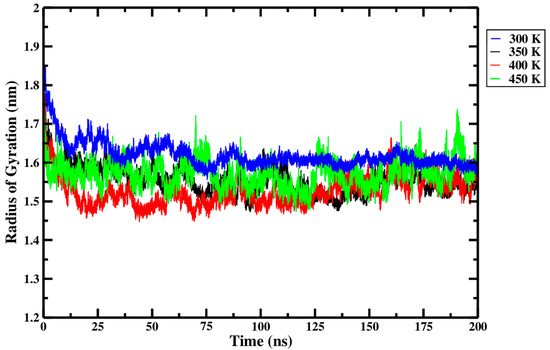
Figure 12.
Average radius of gyration (Rg) of triplicate 200 ns GROMACS MD simulations of the Ara h 6 allergen undergoing thermal processing at 300, 350, 400 or 450 K.
Table 3.
One-way ANOVA statistical analysis of the effect of processing temperature on the Rg of the Ara h 6 allergen over a 200 ns GROMACS MD simulation.
A one-way ANOVA was performed to validate the observations described above and to further study the effect of thermal processing temperature on the Rg of the Ara h 6 allergen. The one-way ANOVA revealed that there was a statistically significant difference in Rg of the Ara h 6 allergen between at least two thermal processing temperature groups [F(3, 8) = 4.479, p = 0.040]. Eta squared (η2), a measure of effect size, allowed for the determination of the strength of this association and suggested a large effect size (η2 = 0.627, 95% C.I. = [0.000, 0.753]). However, it is important to note the large confidence interval of η2, which includes 0.000, suggests that this may be an overestimate. Tukey’s HSD test for multiple comparisons found that the mean value of Rg of the Ara h 6 allergen was significantly different between 300 K and 400 K (p = 0.030, 95% C.I. = [0.009, 0.175]). This supports the observations made above. Tukey’s HSD test, however, located no statistically significant difference between 300 K and 350 K (p = 0.137), between 300 K and 450 K (p = 0.313), between 350 K and 400 K (p = 0.710), between 350 K and 450 K (p = 0.922), or between 400 K and 450 K (p = 0.381). For this reason, variations between these thermal treatment groups were not addressed in this analysis. Assumptions of the one-way ANOVA test performed were validated, and no violations were noted, as seen by the results presented in Table 3.
3.5. Intra-Peptide Hydrogen Bonds
Hydrogen bonds exist when a hydrogen atom that is covalently bonded to an electronegative atom (a donor D) forms a weak bond with another electronegative atom (an acceptor A) [51]. It has been suggested that intra-peptide hydrogen bonds may play a role in stabilizing secondary protein structure [52]. Thus, the loss of intra-peptide hydrogen bonds due to external stress application may promote protein unfolding [52]. GROMACS gmx hbond function, used to determine the presence of hydrogen bonds between the protein and itself (protein–protein), is based on the following two criteria. The first criterion that allows GROMACS to determine if a bond in a molecule is a hydrogen bond is a distance r between the donor D and the acceptor A electronegative atoms of less than or equal to 0.35 nm [44]. The second criterion is that the angle α formed between an imaginary line drawn between the donor D and acceptor A atoms and the covalent bond between the donor D electronegative atom and the hydrogen atom must be less than 30 degrees [44].
A one-way ANOVA was performed to determine the effect of thermal processing temperature on the total number of intra-peptide hydrogen bonds in the Ara h 6 allergen. The one-way ANOVA revealed that there was not a statistically significant difference in the number of intra-peptide hydrogen bonds in the Ara h 6 allergen between at least two groups [F(3, 8) = 3.327, p = 0.077]. Even though the one-way ANOVA revealed no statistical significance between the variables, assumptions of the one-way ANOVA test were still explored to determine its validity. The only identified limitations to this statistical analysis were the significant results of the Shapiro–Wilk test of normality obtained at 400 K (p = 0.028) and 450 K (p = 0.002). These results indicated that the replicate data for the 400 K group (replicate 1 (88.33 ± 7.77), replicate 2 (88.44 ± 8.25) and replicate 3 (81.94 ± 8.85)) and the replicate data for the 450 K group (replicate 1 (84.12 ± 7.91), replicate 2 (80.26 ± 9.78), and replicate 3 (84.13 ± 8.50)) did not form normal distributions. The one-way ANOVA is considered robust to violations of normality; however, as was suggested beforehand when a similar issue arose in the analysis of the effect of RMSD, a larger sample size of more than three GROMACS MD replicates would be suggested to optimize future studies and limit the risk of this type of assumption violation.
Even though the Shapiro–Wilk test of normality failed in two circumstances, the conclusion of the statistical analysis performed above, wherein there is no significance between the total number of intra-peptide hydrogen bonds in the Ara h 6 allergen and processing temperature, is still meaningful, as it is supported by the data presented in Figure 13 and Table 4. The average total number of intra-peptide hydrogen bonds of the three replicates, presented in the final row of Table 4 and calculated for each thermal treatment group, suggests that the total number of intra-peptide hydrogen bonds of the Ara h 6 allergen changed by no more than a maximum of seven hydrogen bonds, regardless of the processing temperature. Furthermore, the highly overlapping curves in Figure 13 suggest that only very minor variations in hydrogen bonding within the Ara h 6 protein occurred during thermal processing. All this put together suggests that the magnitude of hydrogen bonding in the Ara h 6 allergen may remain largely unchanged during thermal processing. This likely contributes to the observed general stability of the secondary structures of the Ara h 6 protein.
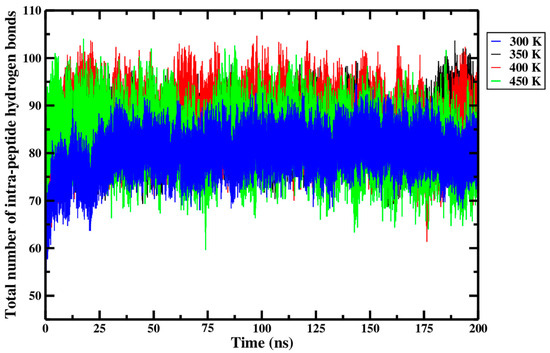
Figure 13.
Average total number of intra-peptide hydrogen bonds of triplicate 200 ns GROMACS MD simulations of the Ara h 6 allergen undergoing thermal processing at 300, 350, 400 or 450 K.
Table 4.
One-way ANOVA statistical analysis of the effect of processing temperature on the total number of intra-peptide hydrogen bonds of the Ara h 6 allergen over a 200 ns GROMACS MD simulation.
3.6. Solvent Accessible Surface Area (SASA)
Solvent accessible surface area (SASA) of a protein is, as its name indicates, a measure of the surface area of the protein accessible to the solvent surrounding it. As a protein’s secondary structure fluctuates under stress, the surface area of the protein accessible to the solvent may vary along with it [43]. Investigating how SASA evolves may allow deeper insight into how a protein’s conformation changes under different external stresses [43]. GROMACS gmx sasa function applies the double cubic lattice method (DCLM) approach, defined by Eisenhaber et al. [53], to calculate SASA.
From Figure 14 and within the replicate data presented in Table 5, it can be seen that only small differences in SASA were observed across the different processing temperatures. The SASA of the Ara h 6 allergen was highest at 300 K, then dropped as the temperature rose to 350 K and again as it rose to 400 K, only to rise slightly at 450 K. In Figure 14, it can be seen that over more than half of the simulation trajectory, the 300 K (blue curve) and 450 K (green curve) overlap. However, in Figure 14, the 350 K (black) and 400 K (red) curves are consistently below the 300 K curve (blue). This suggests a significant drop in the SASA of the Ara h 6 allergen as the temperature rose from 300 K to 350 K and 400 K. A one-way ANOVA was performed to determine the statistical significance of these observed differences and to qualify the effect that thermal processing temperature has on the SASA of the Ara h 6 allergen. The one-way ANOVA revealed that there was a statistically significant difference in SASA of the Ara h 6 allergen between at least two groups [F(3, 8) = 9.169, p = 0.006]. Eta squared (η2), a measure of effect size, allowed for the determination of the strength of this association and suggested a large effect size (η2 = 0.775, 95% C.I. = [0.164, 0.850]). Tukey’s HSD test for multiple comparisons found that the mean value of SASA of the Ara h 6 allergen was significantly different between 300 K and 350 K (p = 0.018, 95% C.I. = [1.032, 10.208]) and between 300 K and 400 K (p = 0.007, 95% C.I. = [2.117, 11.292]). However, there was no statistically significant difference detected between 300 K and 450 K (p = 0.412), between 350 K and 400 K (p = 0.871), between 350 K and 450 K (p = 0.181), or between 400 K and 450 K (p = 0.063). Assumptions of the one-way ANOVA test performed were validated, and no violations were noted. This statistical analysis supports qualitative observations made within the physical data, presented in Figure 14 and Table 5. Similarly to what was observed in the analysis of the results for the Rg, a slight rise in SASA was observed between 400 K and 450 K. This deviation in the overall trend in SASA observed between 300 K, 350 K and 400 K is difficult to interpret. Without data supporting the effect of thermal treatments exceeding 450 K, it is impossible to determine if this rise in SASA is an isolated peak or the beginning of a new overall trend, wherein the SASA begins to increase. This demands further investigation.
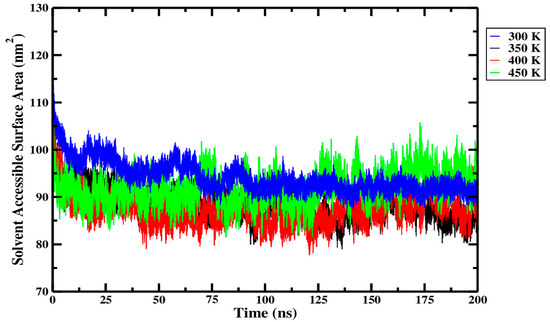
Figure 14.
Average solvent accessible surface area (SASA) of triplicate 200 ns GROMACS MD simulations of the Ara h 6 allergen undergoing thermal processing at 300, 350, 400 or 450 K.
Table 5.
One-way ANOVA statistical analysis of the effect of processing temperature on SASA of the Ara h 6 allergen over a 200 ns GROMACS MD simulation.
4. Conclusions
Before consumption, peanuts (Arachis hypogaea) are often thermally processed. Peanuts contain numerous known allergens, including the Ara h 6 protein, whose structure contains various linear and conformational IgE-binding epitopes. Understanding how thermal processing affects the structure of the Ara h 6 protein is a crucial first step in the process of discovering methods to attenuate its allergenicity. Thus, the aim of this study was to explore, using long-timescale 200 ns GROMACS MD simulations, the structural changes that occur in the Ara h 6 allergen during thermal processing. After a thorough analysis of the results, it was concluded that the Ara h 6 allergen’s structure is highly thermostable. Pronounced fluctuations in the Ara h 6 allergen’s secondary structure, RMSD and RMSF were observed during higher temperature processing treatments. However, concurrent decreases in Rg and SASA as treatment temperature rose from 300 K to 400 K suggested that these observed fluctuations in the structure were not due to the denaturation or unfolding of the Ara h 6 protein but were more likely attributed to a compaction of the protein’s structure. The statistical significance of these results was confirmed by one-way ANOVA. Two weaknesses of this study, important to acknowledge, are the small sample size explored and the risk, given the chosen CHARMM36m force field, of the overestimation of molecular compaction. Future work must be performed to determine if the CHARMM36m force field, utilized in this study, is over-stabilizing the Ara h 6 allergen relative to other force fields. Future studies should also consider increasing the number of replicate GROMACS MD simulations performed to a minimum of five in order to increase the power of the study. In this study, between 400 K and 450 K, a slight rise in Rg and SASA was observed. This result deviated from the general trend observed through 300 K to 400 K, wherein Rg and SASA consistently decreased. In order to elucidate whether this rise in Rg and SASA at 450 K is significant and the beginning of a different overall trend in the response of the structure of the Ara h 6 peanut protein to thermal treatment, even higher treatment temperatures, such as 500 K and 550 K, should be examined in future works.
Author Contributions
Conceptualization, A.S. and V.R.; methodology, A.S.; software, A.S.; validation, A.S.; formal analysis, A.S.; data curation, A.S.; writing—original draft preparation, A.S.; writing—review and editing, A.S. and V.R.; visualization, A.S.; supervision, V.R.; project administration, V.R.; funding acquisition, V.R. All authors have read and agreed to the published version of the manuscript.
Funding
The authors would like to acknowledge the financial support of the Natural Sciences and Engineering Research Council of Canada (NSERC) [RGPIN-2014-04190] for the research program executed by the Department of Bioresource Engineering.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors on request.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Iqbal, A.; Shah, F.; Hamayun, M.; Ahmad, A.; Hussain, A.; Waqas, M.; Kang, S.M.; Lee, I.J. Allergens of Arachis hypogaea the effect of processing on their detection by ELISA. Food Nutr. Res. 2016, 60, 28945. [Google Scholar] [CrossRef][Green Version]
- Beyer, K.; Morrow, E.; Li, X.M.; Bardina, L.; Bannon, G.A.; Burks, A.W.; Sampson, H.A. Effects of cooking methods on peanut allergenicity. J. Allergy Clin. Immunol. 2001, 107, 1077–1081. [Google Scholar] [CrossRef] [PubMed]
- Arya, S.S.; Salve, A.R.; Chauhan, S. Peanuts as functional food: A review. J. Food Sci. Technol. 2016, 53, 31–41. [Google Scholar] [CrossRef]
- Mueller, G.A.; Maleki, S.J.; Pedersen, L.C. The molecular basis of peanut allergy. Curr. Allergy Asthma Rep. 2014, 14, 429. [Google Scholar] [CrossRef] [PubMed]
- Kulis, M.; Chen, X.; Lew, J.; Wang, Q.; Patel, O.P.; Zhuang, Y.; Murray, K.S.; Duncan, M.W.; Porterfield, H.S.; Burks, A.W.; et al. The 2s albumin allergens of Arachis hypogaea, ara h 2 and ara h 6, are the major elicitors of anaphylaxis and can effectively desensitize peanut-allergic mice. Clin. Exp. Allergy J. Br. Soc. Allergy Clin. Immunol. 2012, 42, 326–336. [Google Scholar] [CrossRef]
- Chen, X.; Wang, Q.; El-Mezayen, R.; Zhuang, Y.; Dreskin, S.C. Ara h 2 and Ara h 6 have similar allergenic activity and are substantially redundant. Int. Arch. Allergy Immunol. 2013, 160, 251–258. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, K.; Schweimer, K.; Reese, G.; Randow, S.; Suhr, M.; Becker, W.M.; Vieths, S.; Rösch, P. Structure and stability of 2S albumin-type peanut allergens: Implications for the severity of peanut allergic reactions. Biochem. J. 2006, 395, 463–472. [Google Scholar] [CrossRef]
- Otsu, K.; Guo, R.; Dreskin, S.C. Epitope analysis of Ara h 2 and Ara h 6: Characteristic patterns of IgE-binding fingerprints among individuals with similar clinical histories. Clin. Exp. Allergy J. Br. Soc. Allergy Clin. Immunol. 2015, 45, 471–484. [Google Scholar] [CrossRef] [PubMed]
- Hollingsworth, L.R.; Brown, A.M.; Bevan, D.R. Utilizing computational techniques to accelerate discovery in peanut allergenicity: A case study. In Proceedings of the Practice and Experience in Advanced Research Computing 2017 on Sustainability, Success and Impact, New Orleans, LA, USA, 9–13 July 2017; pp. 1–4. [Google Scholar] [CrossRef]
- Vanga, S.K.; Singh, A.; Raghavan, V. Effect of thermal and electric field treatment on the conformation of ara h 6 peanut protein allergen. Innov. Food Sci. Emerg. Technol. 2015, 30, 79–88. [Google Scholar] [CrossRef]
- Vagadia, B.H.; Vanga, S.K.; Singh, A.; Raghavan, V. Effects of thermal and electric fields on soybean trypsin inhibitor protein: A molecular modelling study. Innov. Food Sci. Emerg. Technol. 2016, 35, 9–20. [Google Scholar] [CrossRef]
- Saxena, R.; Vanga, S.K.; Raghavan, V. Effect of thermal and microwave processing on secondary structure of bovine β-lactoglobulin: A molecular modeling study. J. Food Biochem. 2019, 43, e12898. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Vanga, S.K.; Raghavan, V. Structural responses of kiwifruit allergen act d 2 to thermal and electric field stresses based on molecular dynamics simulations and experiments. Food Funct. 2020, 11, 1373–1384. [Google Scholar] [CrossRef]
- Zhu, Y.; Wang, J.; Vanga, S.K.; Raghavan, V. Visualizing structural changes of egg avidin to thermal and electric field stresses by molecular dynamics simulation. LWT 2021, 151, 112139. [Google Scholar] [CrossRef]
- Lemkul, J.A. From Proteins to Perturbed Hamiltonians: A Suite of Tutorials for the GROMACS-2018 Molecular Simulation Package. Living J. Comput. Mol. Sci. 2018, 1, 5068. [Google Scholar] [CrossRef]
- Bekker, H.; Berendsen, H.J.C.; Dijkstra, E.J.; Achterop, S.; van Drunen, R.; van der Spoel, D.; Sijbers, A.; Keegstra, H.; Reitsma, B.; Renardus, M.K.R. Gromacs: A parallel computer for molecular dynamics simulations. Phys. Comput. 1993, 92, 252–256. [Google Scholar]
- Berendsen, H.J.C.; van der Spoel, D.; van Drunen, R. Gromacs: A message-passing parallel molecular dynamics implementation. Comput. Phys. Commun. 1995, 91, 43–56. [Google Scholar] [CrossRef]
- van der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J.C. Gromacs: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. Gromacs: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Goddard, T.D.; Huang, C.C.; Meng, E.C.; Pettersen, E.F.; Couch, G.S.; Morris, J.H.; Ferrin, T.E. UCSF ChimeraX: Meeting modern challenges in visualization and analysis. Protein Sci. Publ. Protein Soc. 2018, 27, 14–25. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Meng, E.C.; Couch, G.S.; Croll, T.I.; Morris, J.H.; Ferrin, T.E. UCSF ChimeraX: Structure visualization for researchers, educators, and developers. Protein Sci. Publ. Protein Soc. 2021, 30, 70–82. [Google Scholar] [CrossRef]
- Jo, S.; Kim, T.; Iyer, V.G.; Im, W. Charmm-gui: A web-based graphical user interface for charmm. J. Comput. Chem. 2008, 29, 1859–1865. [Google Scholar] [CrossRef] [PubMed]
- Brooks, B.R.; Brooks, C.L.; Mackerell, A.D.; Nilsson, L.; Petrella, R.J.; Roux, B.; Won, Y.; Archontis, G.; Bartels, C.; Boresch, S.; et al. Charmm: The biomolecular simulation program. J. Comput. Chem. 2009, 30, 1545–1614. [Google Scholar] [CrossRef] [PubMed]
- Jo, S.; Cheng, X.; Islam, S.M.; Huang, L.; Rui, H.; Zhu, A.; Lee, H.S.; Qi, Y.; Han, W.; Vanommeslaeghe, K.; et al. Charmm-gui pdb manipulator for advanced modeling and simulations of proteins containing non-standard residues. Adv. Protein Chem. Struct. Biol. 2014, 96, 235–265. [Google Scholar] [CrossRef]
- Lee, J.; Cheng, X.; Swails, J.M.; Yeom, M.S.; Eastman, P.K.; Lemkul, J.A.; Wei, S.; Buckner, J.; Jeong, J.C.; Qi, Y.; et al. Charmm-gui input generator for namd, gromacs, amber, openmm, and charmm/openmm simulations using the charmm36 additive force field. J. Chem. Theory Comput. 2016, 12, 405–413. [Google Scholar] [CrossRef]
- Park, S.-J.; Kern, N.; Brown, T.; Lee, J.; Im, W. Charmm-gui pdb manipulator: Various pdb structural modifications for biomolecular modeling and simulation. J. Mol. Biol. 2023, 435, 167995. [Google Scholar] [CrossRef]
- Huang, J.; Rauscher, S.; Nawrocki, G.; Ran, T.; Feig, M.; de Groot, B.L.; Grubmüller, H.; MacKerell, A.D. Charmm36m: An improved force field for folded and intrinsically disordered proteins. Nat. Methods 2016, 14, 71–73. [Google Scholar] [CrossRef] [PubMed]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007, 126, 014101. [Google Scholar] [CrossRef]
- Smith, A.; Dong, X.; Raghavan, V. An Overview of Molecular Dynamics Simulation for Food Products and Processes. Processes 2022, 10, 119. [Google Scholar] [CrossRef]
- McDaniel, K.A.; White, B.L.; Dean, L.L.; Sanders, T.H.; Davis, J.P. Compositional and Mechanical Properties of Peanuts Roasted to Equivalent Colors using Different Time/Temperature Combinations. J. Food Sci. 2012, 77, C1293–C1299. [Google Scholar] [CrossRef]
- Shi, X.; Dean, L.O.; Davis, J.P.; Sandeep, K.P.; Sanders, T.H. The effects of different dry roast parameters on peanut quality using an industrial belt-type roaster simulator. Food Chem. 2018, 240, 974–979. [Google Scholar] [CrossRef]
- Parrinello, M.; Rahman, A. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle mesh ewald: An N·log(N) method for ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. Lincs: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Grace Development Team. Grace User’s Guide (for Grace-5.1.25); Weizmann Institute of Science, 2015; Available online: https://plasma-gate.weizmann.ac.il/Grace/ (accessed on 28 August 2023).
- Frishman, D.; Argos, P. Knowledge-based protein secondary structure assignment. Proteins 1995, 23, 566–579. [Google Scholar] [CrossRef]
- Levene, H. Robust Tests for Equality of Variances. In Contributions to Probability and Statistics; Olkin, I., Ed.; Stanford University Press: Palo Alto, CA, USA, 1960; pp. 278–292. [Google Scholar]
- Shapiro, S.S.; Wilk, M.B. An Analysis of Variance Test for Normality (Complete Samples). Biometrika 1965, 52, 591–611. [Google Scholar] [CrossRef]
- Heinig, M.; Frishman, D. Stride: A web server for secondary structure assignment from known atomic coordinates of proteins. Nucleic Acids Res. 2004, 32, 502. [Google Scholar] [CrossRef]
- Martínez, L. Automatic identification of mobile and rigid substructures in molecular dynamics simulations and fractional structural fluctuation analysis. PLoS ONE 2015, 10, e0119264. [Google Scholar] [CrossRef]
- Singh, A.; Munshi, S.; Raghavan, V. Effect of external electric field stress on gliadin protein conformation. Proteomes 2013, 1, 25–39. [Google Scholar] [CrossRef]
- Abraham, M.; Alekseenko, A.; Bergh, C.; Blau, C.; Briand, E.; Doijade, M.; Fleischmann, S.; Gapsys, V.; Garg, G.; Gorelov, S.; et al. GROMACS 2023.2 Manual (2023.2). Zenodo. 2023. Available online: https://zenodo.org/records/8134388 (accessed on 26 January 2025).
- Uttley, J. Power analysis, sample size, and assessment of statistical assumptions—Improving the evidential value of lighting research. Leukos 2019, 15, 143–162. [Google Scholar] [CrossRef]
- Tomasiak, L.; Karch, R.; Schreiner, W. Conformational flexibility of a free and tcr-bound pmhc-i protein investigated by long-term molecular dynamics simulations. BMC Immunol. 2022, 23 (Suppl. S1), 36. [Google Scholar] [CrossRef]
- Mishra, A.; Jain, A.; Arora, N. Mapping B-cell epitopes of major and minor peanut allergens and identifying residues contributing to IgE binding. J. Sci. Food Agric. 2016, 96, 539–547. [Google Scholar] [CrossRef] [PubMed]
- Lobanov, M.Y.; Bogatyreva, N.S.; Galzitskaya, O.V. Radius of gyration as an indicator of protein structure compactness. Mol. Biol. 2008, 42, 623–628. [Google Scholar] [CrossRef]
- Ogunlana, A.T.; Boyenle, I.D.; Ojo, T.O.; Quadri, B.O.; Elegbeleye, O.E.; Ogbonna, H.N.; Ayoola, S.O.; Badmus, I.O.; Manica, A.K.; Joshua, K.I.; et al. Structure-based computational design of novel covalent binders for the treatment of sickle cell disease. J. Mol. Graph. Model. 2023, 124, 108549. [Google Scholar] [CrossRef] [PubMed]
- Gil Pineda, L.I.; Milko, L.N.; He, Y. Performance of charmm36m with modified water model in simulating intrinsically disordered proteins: A case study. Biophys. Rep. 2020, 6, 80–87. [Google Scholar] [CrossRef]
- Hubbard, R.E.; Haider, M.K. Hydrogen bonds in proteins: Role and strength. In Encyclopedia of Life Sciences (ELS); John Wiley & Sons, Ltd.: Chichester, UK, 2010. [Google Scholar] [CrossRef]
- Budi, A.; Legge, S.; Treutlein, H.; Yarovsky, I. Effect of external stresses on protein conformation: A computer modelling study. Eur. Biophys. J. EBJ 2004, 33, 121–129. [Google Scholar] [CrossRef]
- Eisenhaber, F.; Lijnzaad, P.; Argos, P.; Sander, C.; Scharf, M. The double cubic lattice method: Efficient approaches to numerical integration of surface area and volume and to dot surface contouring of molecular assemblies. J. Comput. Chem. 1995, 16, 273–284. [Google Scholar] [CrossRef]














Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).