Abstract
It is essential to use highly antigenic epitope areas, since the development of peptide vaccines heavily relies on the precise design of epitope regions that can elicit a strong immune response. Choosing epitope regions experimentally for the production of the SARS-CoV-2 vaccine can be time-consuming, costly, and labor-intensive. Scientists have created in silico prediction techniques based on machine learning to find these regions, to cut down the number of candidate epitopes that might be tested in experiments, and, as a result, to lessen the time-consuming process of their mapping. However, the tools and approaches involved continue to have low accuracy. In this work, we propose a hybrid deep learning model based on a convolutional neural network (CNN) and long short-term memory (LSTM) for the classification of peptides into epitopes or non-epitopes. Numerous transfer learning strategies were utilized, and the fine-tuned method gave the best result, with an AUC of 0.979, an f1 score of 0.902, and 95.1% accuracy, which was far better than the performance of the model trained from scratch. The experimental results obtained show that this model has superior performance when compared to other methods trained on IEDB datasets. Using bioinformatics tools such as ToxinPred, VaxiJen, and AllerTop2.0, the toxicities, antigenicities, and allergenicities, respectively, of the predicted epitopes were determined. In silico cloning and codon optimization were used to successfully express the vaccine in E. coli. This work will help scientists choose the best epitope for the development of the COVID-19 vaccine, reducing cost and labor and thereby accelerating vaccine production.
1. Introduction
The contagious disease called coronavirus disease 2019 (COVID-19) is brought on by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) [1]. The virus, which was first discovered in Wuhan in December 2019, has since spread around the world, causing the deaths of millions and having catastrophic effects on the economy and society. The necessity for effective vaccinations is therefore extremely important [2]. Several strategies have been put up by researchers to create SARS-CoV-2 vaccines [3,4]. Growing pathogens is the basis of the conventional vaccine development method, which makes it exceedingly time-consuming to isolate, inactivate, and inject the disease-causing virus [5]. This method typically takes over a year to produce effective vaccinations, and as a result, it does very little to stop the disease’s spread [6,7].
The current COVID-19 control measures, which do not yet include a single particular antiviral drug for SARS-CoV-2, include early diagnosis, reporting, isolation, and supportive therapies [8]. Elderly persons and those with weakened immune systems tend to have more severe illnesses and are more likely to perish because their cells’ ability to resist infection and repair themselves is decreased by a compromised immune system [9]. The immune system has two distinct response mechanisms: innate and adaptive. Innate immunity swiftly intervenes and initiates early immune responses when it comes into contact with pathogens [10]. On the other hand, adaptive immunity may be developed in several ways, including through disease exposure or externally delivered serum and vaccinations. Adaptive immune systems contain memories that allow them to recall previous infections; as such, antigen-specific responses are produced by adaptive immune systems [11]. Adaptive immune responses come in both humoral and cellular forms. A humoral response utilizes B-cells to produce antibodies that can target an antigen when exposed to it. To create a vaccine, the highly immunogenic portions of the protein of a pathogenic organism must first be identified. These areas are referred to as B- and T-cell epitopes and are in charge of triggering immune responses [12,13].
SARS-CoV-2 has a large 26–32 kb RNA genome that encodes several structural and non-structural proteins, such as Spike (S), Envelope (E), Membrane (M), and Nucleocapsid (N), which are crucial for triggering immunological responses [14,15,16,17]. Therefore, an epitope peptide vaccine made of viral proteins S, M, N, and E is highly required to control disease spread and another SARS virus in the future. B-cell epitopes can be linear or conformational. While conformational epitopes are made up of amino acids that are connected during the folding of a protein, linear epitopes are produced by a sequence of the amino acids of the protein [13,18]. One study [19] identified the best epitopes for an epitopic vaccination to prevent SARS-CoV-2 infection. To identify and describe potential B and T-cell epitopes for the creation of the epitopic vaccine, immunoinformatics were used. The SARS-CoV-2 spike glycoprotein was selected as the target because it creates the virus’ distinctive crown and protrudes from the viral membrane. Multiple servers and pieces of software built on the immunoinformatic platform were used to explore the spike glycoprotein’s protein sequence. The focus of this study is on linear epitope prediction due to the limited number of available datasets for conformational epitopes. Conventional approaches for creating vaccines against deadly diseases have proven to be exceedingly time- and money-consuming. Using in silico techniques, vaccine candidates for earlier viruses (Zika, Ebola, HPV, and MERS) have been successfully designed [20,21].
In a study, immunoinformatics-based techniques were used to find possible immunodominant SARS-CoV-2 epitopes, which may be relevant to creating COVID-19 vaccines. In total, 25 epitopes that were 100% similar to experimentally verified SARS-CoV epitopes and 15 putative immunogenic areas from three SARS-CoV-2 proteins were found. Analysis was carried out to test the suitability of the epitopes as a vaccine [22]. Similarly, immunoinformatics tools were used to create a multi-epitope vaccine that could be employed for COVID-19 prevention as well as treatment. B-cell, CTL, and HTL epitopes were combined to create this multi-epitope vaccine. Using online tools, additional research was done to predict and evaluate the vaccine structure and efficacy [23]. To provide a list of possibly immunogenic and antigenic peptide epitopes that might aid in vaccine creation, several immunoinformatics methods were integrated. Spike proteins’ S1 and S2 domains were examined, and two vaccine constructions, with T- and B-cell epitopes, were given priority. Using linkers and adjuvants, prioritized epitopes were then modeled, and corresponding 3D models were built to assess their physiochemical characteristics and potential interactions with ACE2 and HLA superfamily alleles [24].
Machine learning algorithms’ architecture automatically identifies patterns in data, which is perfect for data-driven sciences, such as genomics [25,26]. The usage of DL frameworks in medical imaging has been widespread [27,28,29,30,31]. To create computer models that can more accurately predict the existence of linear B-cell epitopes from an amino acid sequence for vaccine production against a pathogenic organism, in silico techniques have been frequently employed [32]. There are several programs available and cited in the literature that employ machine learning methods to predict linear B-cell epitopes, including BepiPred-2.0 [33], BCPred [34], EpiDope [35], ABCPred [36], and SVMTrip [37]. In addition, the Lbtope tool and SVM, K-nearest neighbor models [38], and genetic algorithms [39] have been used in vaccine design for B-cell or T-cell epitopes. Most of these machine learning models rely on features related to amino acid sequences; therefore, the portrayed effectiveness of such models is ineffective.
The web server BepiPred-2.0 is used to predict B-cell epitopes from antigen sequences. The data utilized had 11,834 positive and 18,722 negative epitopes, and it was taken from the immune epitope database (IEDB) [40]. Since epitopes are rarely found outside of peptides consisting of five to twenty-five amino acids, the peptides within this range were eliminated. The random forest (RF) regression technique with fivefold cross-validation was utilized as the training approach; the method was reported to have an AUC of 0.62 on test datasets [33]. BCPred, another method to predict linear B-cell epitopes, employed SVM that utilized a radial-based kernel with five kernel modifications and fivefold cross-validation. The proposed technique eventually achieved an AUC of around 0.76 [34]. In another study, deep neural networks were used by the application EpiDope to locate B-cell epitopes in specific protein sequences composed of ELMo DNN and biLSTM. Each of the 30,556 protein sequences in the dataset, which was taken from the IEDB, had experimentally validated epitopes or non-epitopes. A bidirectional LSTM (long short-term memory) layer was linked to a vector of length 10 that was used to encode each amino acid in the ELMo DNN branch. Tenfold cross-validation was employed for validation, achieving an AUC of 0.67 [35]. ABCPred employed a recurrent neural network (RNN) to predict linear B-cell epitopes. For each residue, sliding windows containing 10 to 20 amino acids were employed to determine its attributes. Fivefold cross-validation was utilized for these tests, yielding an accuracy of 66% [36]. The SVMTriP approach, which combines SVM with tri-peptide similarity and propensity scores, is used to predict linear antigenic B-cell epitopes. Our dataset was taken from the IEDB and consisted of 65,456 positive epitopes [40]. Using fivefold cross-validation, it obtained an AUC value of 0.702. The authors of [41] created a strategy for predicting B-cell linear epitopes that was based on the design of a fuzzy-ARTMAP neural network. This was trained on 15 attributes, including an amino acid ratio scale and a set of 14 physicochemical scales, using a linear averaging approach. Fivefold cross-validation procedures were employed with datasets taken from the IEDB to train and validate the knowledge of models and were shown to achieve an AUC of 0.7831 on test data, which is a good performance. To take into account the properties of a whole antigen protein in combination with the target sequence, [42] presents a deep learning approach based on short-term memory with an attention mechanism. In experimental epitope location prediction with data taken from the immune epitope database, the suggested technique outperformed the standard method, with an AUC of 0.822. The SMOTE technique produced more accurate predictions than other methods when evaluated for the datasets balanced using it. It was shown that once the SARS-CoV and B-cell datasets used for training were balanced, the epitope prediction success of the models generally rose, with an accuracy of 0.914.
There are very few deep learning methods available for forecasting B-cell epitopes, and even though some of the models are good at predicting linear B-cell epitopes, based on their performances, they still have some difficulties with making good predictions, which makes it necessary to develop better models. This research suggests a novel and effective hybrid transfer learning strategy, for forecasting SARS-CoV-2 epitopes, that integrates data preprocessing of physiochemical characteristics and sequence-based features. This study’s goal is to provide a better method of identifying the epitope areas that could be candidates for vaccines. The list of abbreviations is presented in Table 1. The contributions made by this study are as follows:
- We proposed CNN-LSTM, a hybrid architecture that combines a CNN with bidirectional LSTM (BiLSTM), to predict B-cell epitopes given a peptide sequence that may be employed for vaccine development. The idea behind this hybrid architecture is to employ the CNN for feature extraction and LSTM for modeling feature relationships in order of appearance.
- To address the issue of limited sample sizes and enhance model performance, we put forth a transfer learning technique. In particular, we first pre-trained the proposed CNN-LSTM using the B-cell dataset. The pre-trained CNN-LSTM was then adjusted to predict epitopes using SARS-CoV datasets.
- To forecast epitopes from the SARS-CoV-2 datasets, we looked into three transfer learning strategies—fine-tuned, frozen CNN, and frozen LSTM—using information learnt from the previous model.
- With the use of the bioinformatics tools AllerTop, VaxiJen, ToxinPred, Jcat, and Snapgene, we investigated epitopes discovered with transfer learning techniques.

Table 1.
List of abbreviations.
2. Materials and Methods
2.1. Datasets
The Kaggle database provided the datasets utilized in this study, which were made available to the general public (https://www.kaggle.com/datasets/futurecorporation/epitope-prediction, accessed on 13 March 2023). This database includes SARS-CoV, B-cell, and SARS-CoV-2 datasets. The B-cell datasets consisted of 14,387 samples, of which 10,485 were non-epitopes (negative) and 3902 were epitopes (positive). The SARS-CoV dataset consisted of 520 samples; 380 were non-epitopes and 140 were epitopes. The data consisted of ten features, both structural and chemical. The protein sequences and peptide sequences were in categorical form but were later converted to numerical form using their sequence lengths so that each protein sequence or peptide sequence would have a value that corresponded to the number of its categorical letters, while chou_fasman (beta turn), kolaskar_tongaonkar (antigenicity), Parker (hydrophobicity), Emini (relative surface accessibility), stability, isoelectric_point, aromaticity, and hydrophobicity were numerical, as previously described in [43]; see Figure 1 for the data distribution. The SARS-CoV-2 dataset lacked label information and comprised 20,312 peptides isolated from the virus’ spike protein. The SARS-CoV-2 dataset served as a test set because it was unlabeled, and it consisted of 20,312 samples. The fine-tuned model was utilized to predict the B-cell epitopes in the SARS-CoV-2 datasets.
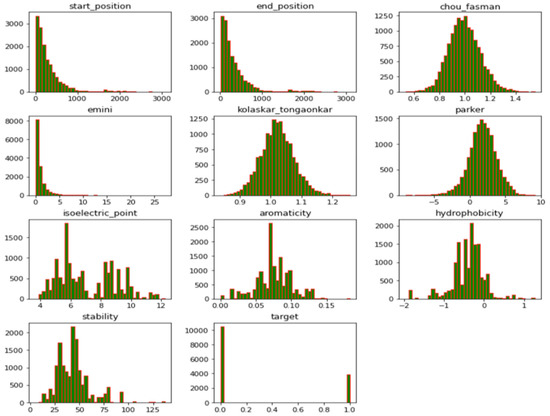
Figure 1.
Data distribution of features in the datasets showing different variables.
2.2. Models
Overview of the Method Proposed
The presented approach combined a CNN and LSTM. To identify sequence patterns, the convolutional module stage scanned the sequence using a set of 1D convolutional filters. The next RNN step was used to learn intricate high-level correlations by taking the orientations and spatial interactions between the motifs into account. The B-cell datasets were used to train this model, which was then enhanced using transfer learning strategies with SARS-CoV datasets. The improved model was then used to forecast epitopes from the SARS-CoV-2 datasets that may be exploited for vaccine development. The toxicologies, allergenicities, and antigenicities of the anticipated epitopes were examined. The whole process of the study is presented in Figure 2.
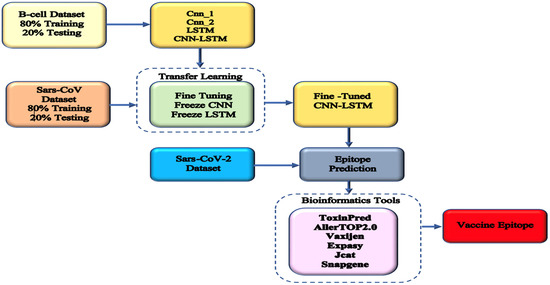
Figure 2.
Overview of the proposed method. Three sets of data were used: B-cell for developing different methods, SARS-CoV for transfer learning, and SARS-CoV-2 for predicting COVID-19 epitopes. Bioinformatic tools were used to screen for the best epitope for vaccine production.
A deep, feed-forward artificial neural network known as a convolutional neural network (CNN) can record hierarchical spatial representations without the need for time-consuming human feature engineering [44]. In addition, deep neural network variants include recurrent neural networks (RNNs) [45]. The internal state of an RNN is updated as it scans an input sequence, unlike a CNN, which is not. RNN is frequently used in the field of NLP because of its internal memory, which enables it to record interactions between elements along a sequence [46]. Information that has to be preserved for a long period can be sent by a memory cell without being discarded. RNN architecture [47] can be applied to handle series data. In light of the problem of vanishing gradients, which occur when backpropagation occurs in training and causes the gradient to become smaller, the RNN is not suited to long-range series data. An RNN model called LSTM [48] will handle long-range range series data by employing memory cells and gates. In this work, bi-directional LSTM [49], which combines forward and backward LSTM models, was used to merge the data acquired from the series in correspondence to opposite contexts.
First, the input features are fed into the input layer, and an embedding layer is followed to map the input features in a higher dimensional space. The weight matrix that makes up the embedding layer is modified during training to reduce error, since it is manually tweaked along with the model’s hyperparameters. This method results in superior model performance, since the weight matrix may be adjusted to enhance model performance. This differs from conventional encoding, which maintains the numerical values for encoded features during training.
For the CNN model, two levels of convolution layers were used. The first layer was made up of a 1D convolution layer (conv 1) that used 256 convolution kernels of size 9 to extract the important local characteristics from the gRNA sequence. To the output of each convolution layer, a Rectified Linear Unit (ReLU) with an activation function was applied [50]. After that, a pooling layer received the output to conduct average pooling. A 128-dimensional LSTM layer was the third layer. This LSTM layer was added because it is effective at strengthening the relevance between sequence feature attributes. To create our final feature representation, which contained both forward and backward information, the outputs of two concurrent LSTMs were combined. The collected features were then dropped from the model at the rate of 0.3 for regularization to prevent overfitting [51]. Lastly, to forecast the epitopes, the outputs of the features were then passed into a dense layer for classification with softmax. The batch normalization layer was incorporated into the model before the CNN layer to improve the model’s performance. To overcome the gradient vanishing problem and further speed up training, batch normalization forcefully changes the values of the input of every neuron into a normal distribution with a mean of 0 and a variance equal to 1. This ensures that the input value of each layer of the network is distributed uniformly. Similarly, to prevent overfitting, apart from adding a dropout layer, early stopping was used with a patience of 3, which implies that the model should stop training when the loss or accuracy does not improve after 3 epochs.
Different models were trained by changing some layers to obtain varieties of architecture to select the best model. For instance, we tried the CNN alone, with one and two CNN layers (Cnn_1 and Cnn_2), then the single LSTM, the LSTM with Cnn_1, and the LSTM with Cnn_2. Hyperparameter tuning was carried out first to select the models utilized in this work. We first used different epoch amounts, 10, 100, and 1000, and decided to work with 100 epochs. Other hyperparameters included batch normalization and dropout. Models with and without batch normalization were trained, and better performance was obtained after batch normalization. Similarly, the addition of the dropout layer in the model gave better performance.
Finally, after selecting the best model for this task, we performed transfer learning using the best model, which was the CNN-LSTM. Since the CNN-LSTM model was already trained using the B-cell dataset, the architecture and weights of this model were utilized for the following steps: For the transfer learning models, we tried different methods, such as freezing the CNN and in some cases freezing the LSTM or fine-tuning the pre-trained model. This was carried out to improve the model’s performance, especially when dealing with a small size of data. The final pre-trained model was utilized to forecast whether the targets in the SARS-CoV-2 datasets were epitopes or not. The recognized epitopes were further analyzed using the necessary bioinformatics tools: ToxinPred [52] for verifying toxicity; AllerTOP2.0 [53], a tool for checking for possible allergens in epitopes; and VaxiJen [54] to check the ability to recognize antigens from the epitopes predicted.
2.3. Evaluation Metrics
In order to assess the model’s performance, accuracy was used. This important model performance indicator measures the proportion of accurate predictions to all data samples. Therefore, with accuracy, we can see how well the model was able to make correct predictions in the entire dataset. In addition, the F1 score was utilized to evaluate the model because it could also be used to check the model’s accuracy. The difference is that accuracy is based on correct predictions in the entire dataset while the F1 score is based on accuracy in each class, which is why the F1 score combined the means of recall and precision [55,56].
The performance of the model could be ascertained by measuring the accuracy, the area under the ROC curve (AUC), and the F1 score. Accuracy determines how well a model can make correct predictions [57]. It is given as:
The F1 score [58] provides a score within the range of (0, 1), meaning that if a model is well-trained, it will give a score that is close to 1. It is used to try to find a balance between precision and recall. The F1 score can be calculated using:
The AUC [59] is the area under the plot of the false positive rate and the sensitivity, known as the receiver operating characteristics (ROCs). It is widely used to determine the correctness of predictions in test data. It gives a value within (0, 1), and the best models will have AUCs close to 1.
3. Results
3.1. Performance Comparison between Different Architectures Developed with the B-Cell Datasets
First, we looked at convolutional models without LSTM, that is, Cnn_1 and Cnn_2. The Cnn_1 model, having one layer of convolution, showed average performance, but there was a small increase in performance with Cnn_2, which had two layers of convolution, as shown in Figure 3. Next, the LSTM alone gave a better performance, with an accuracy of 0.77 and an AUC of 0.78, than Cnn_1, which had an accuracy of 0.73 and an AUC of 0.76, and Cnn_2, with an accuracy of 0.74 and an AUC of 0.762; this might be because LSTM is good for understanding sequence dependencies that CNNs do not. Since CNNs are good for feature extraction when combined with LSTM, we obtained far better model performance. In addition, we tried LSTM with Cnn_1 and Cnn_2 and observed that the LSTM with Cnn_2 gave the best performance, with a 0.779 accuracy and an AUC of 0.81, while the LSTM with Cnn_1 had an accuracy of 0.764 and an AUC of 0.785. Detailed results are presented in Table 2. The training accuracy for all the models is included to show the ability of each model to make good generalizations in the test sets.
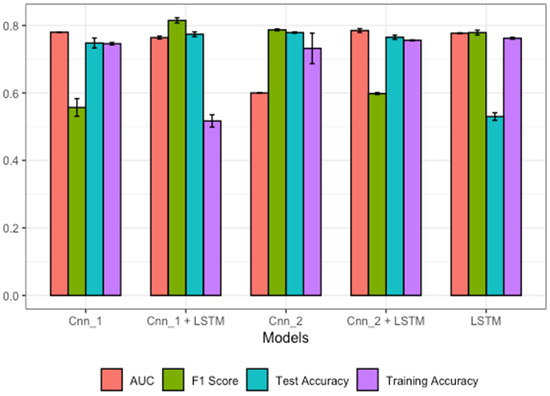
Figure 3.
Performances of different model architectures using the B-cell dataset.
Table 2.
Model performance comparison for various architectures.
3.2. Performances of Different Transfer Learning Architectures
The results obtained using the transferring knowledge strategy were far better than in the first method, where the models were trained with the B-cell data. This implies that at first, we used the B-cell data to train methods for the prediction task; then, we transferred the knowledge that was learnt from those models to the new models. As can be seen in Figure 3, the models that were fine-tuned after addition of two more dense layers during re-training gave better performance. Here, we froze the weight of the CNN-LSTM, and two dense layers were added to train the model with SARS-CoV data. This gave an accuracy of 0.951 and an AUC of 0.979, which was the best performance. In the second scenario, we froze the weights of Cnn_1 and Cnn_2 and trained the model using the new SARS-CoV data. It was observed that the performance declined from 0.951 to 0.928 in accuracy, which means that the presence of convolution in the updated model was significant to model performance. Finally, the LSTM was frozen and the weight of the model was updated using the SARS-CoV for training. Similarly, there was a decrease in performance from 0.951 to 0.943, meaning that the presence of the LSTM in the fine-tuned method highly increased its performance. In addition, the F1 scores obtained with this technique for all the models are better than those when the models were built from the beginning using the B-cell data. Detailed results can be seen in Table 3 and Figure 4; training accuracy for all the models is included to show the ability of each model to make a good generalization on the test sets.
Table 3.
Performances of transfer learning techniques on different model architectures.
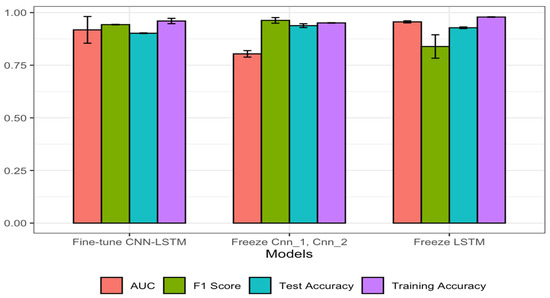
Figure 4.
Performances of different transfer learning strategies using the SARS-CoV dataset.
3.3. Prediction of Epitopes in SARS-CoV-2 for Possible Vaccine Development
It is highly time-consuming and expensive to design and test a vaccine using the conventional method; therefore, the use of epitope prediction methods can lower the cost and time of developing a vaccine. In this work, we utilized SARS-CoV-2 data for the epitope forecast. It consisted of 20,312 unlabeled peptide samples that needed to be classified as either epitopes or not. After training the CNN-LSTM with B-cell data, we applied different transfer learning solutions to improve the model by updating the learning weights using the SARS-CoV data, and finally, the best accuracy, of 0.951, was obtained with the fine-tuned CNN-LSTM for the classification task; therefore, it was utilized to make predictions of the SARS-CoV-2 data into epitopes or not. According to the model’s forecast, the last 5000 samples consisted of 18 epitopes and 4982 non-epitopes, as shown in the Figure 5. After the prediction, the last 18 epitope samples were selected for further analysis using the bioinformatic tools mentioned above; see Table 4 for more details.
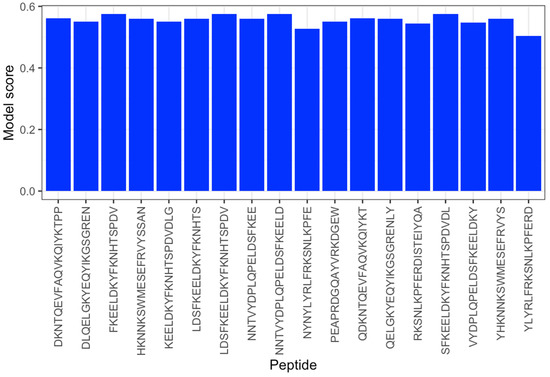
Figure 5.
Scores of epitopes detected with the fine-tuned CNN-LSTM method.
Table 4.
The epitopes detected in the samples of the SARS-CoV-2 datasets with fine-tuned CNN-LSTM.
Toxicity Determination Using ToxinPred
The determination of the toxicity status of an epitope is required before it can be chosen for vaccine development. Here, the ToxinPred tool was chosen to analyze the epitopes selected by our fine-tuned CNN-LSTM. The ToxinPred [52] tool was created using a support vector machine, and a score of <0.0 is regarded as non-toxic. The ToxinPred score depends on some physiochemical properties of an epitope, such as molecular weight, hydrophilic nature, and possible mutations. The results can be seen in Table 5 below. It can be seen that all the peptide sequences that were shown to be epitopes by our fine-tuned CNN-LSTM tend to be non-toxic and would not be able to generate any mutation. The ability to predict a peptide or protein’s toxicity before its synthesis as a vaccine is crucial to reducing the time and cost spent developing peptide- or protein-based drugs. Similarly, in Table 6, we utilized ToxinPred to analyze the peptides that were classified as non-epitopes by our model, and they were predicted to be non-toxic; only one peptide was found to be toxic, with an SVM score of 0.1.
Table 5.
Epitope toxicity analysis using the ToxinPred tool.
Table 6.
Non-epitope toxicity analysis using the ToxinPred tool.
3.4. Optimization of Codons and In Silico Cloning
The Java Codon Adaptation Tool (JCat) was used to check the amount of protein that would be expressed in E. coli. (Strain K12) host to optimize the codon usage for the vaccine constructs [60]. The length of the codon sequence that was optimized was 3843 nucleotides. The average GC content of the adapted sequence was 51.73%, and the projected codon optimization index (CAI) value of 1.0 suggested strong expression in the E. coli host. Finally, using SnapGene software, the recombinant plasmid sequence was created by inserting the modified codon sequences into the plasmid carrier, pCC1BAC, as shown in Figure 6. On the left is the step for the cloning, and the right side shows the final cloned vector, which was obtained using the Snapgene free trial software (https://www.snapgene.com/free-trial/, accessed on 3 March 2023). The vaccine sequence was inserted between the ApaLI and StuI restriction sites. The black region on the cloned pCC1BAC shows the vector backbone while the red is the codon sequence of the vaccine obtained from Jcat. This analysis suggests that our vaccine sequence would be highly expressed in the host and, as such, would lead to the production of antibodies against COVID-19. Finally, the instability index, obtained with Expasy, was 31.38, with a half-life of >10 h in E. coli in vivo and 30 h in mammalian reticulocytes in vitro; this makes the vaccine sequence stable (https://web.expasy.org/protparam/, accessed on 3 March 2023).
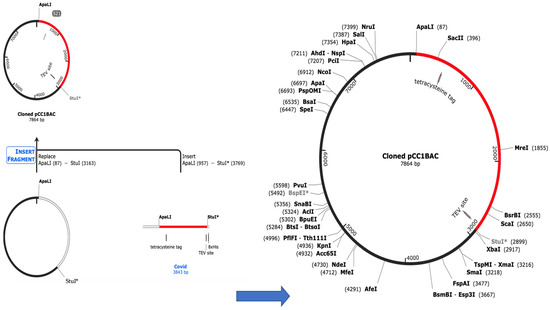
Figure 6.
Cloning of codon sequences into the pCC1BAC vector.
3.5. Determination of Allergenicity and Antigenicity
Allergenicity determination was important because our target was not to create a vaccine that humans would be allergic to. In addition, a good vaccine needs to be antigenic, meaning it will be able to generate an immune response in humans to fight the SARS-CoV-2 virus. The allergenicity was determined using AllerTop2.0 [53], and twelve epitopes were considered non-allergens while four were allergens. The possible vaccine candidate could therefore be chosen from the 12 non-allergens, shown in Table 7. Out of the 12 non-allergens, one epitope was found to be antigenic and the remaining 11 non-antigenic using Vaxijen2.0 [54]. The non-epitopes predicted by our model were found to be allergens and non-antigenic, except for one. This shows that the model was able to correctly predict the non-epitopes in Table 8, since they would generate allergic reactions and would not be able to induce the production of antibodies, even though most of them are non-toxic.
Table 7.
Screening of epitopes for antigenicity and allergenicity.
Table 8.
Screening of non-epitopes for antigenicity and allergenicity.
3.6. Immune Response Simulation with C-IMMSIM Server
The possible immunological response that the vaccine candidate could cause was assessed using the C-ImmSim server [61]. We demonstrated, using the immune simulation study, that the suggested vaccine candidate might potentially elicit an immune response against the virus. Due to a strong immunological response after vaccination, as shown in Figure 7A, leading to the production of immunoglobulins IgM and IgG, the active B-cell population also seemed to be very high (Figure 7D). Helper T lymphocytes (Th) and cytotoxic T lymphocytes (Tc) both expanded in number in a similar way (Figure 7C,E), and the number of Tc kept increasing over time. The level of interferon (IFN-γ) was sustained until the fifteenth day (Figure 7B). IFN-γ controls the immune response to viruses and bacteria, and is mostly produced following NK and T-cell activation.
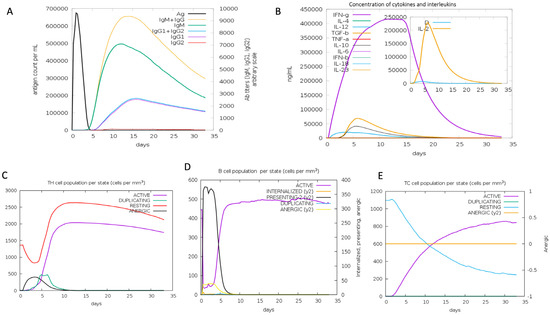
Figure 7.
Immune response simulation using C-IMMSISM.
4. Discussion
In this work, a CNN-LSTM method was pre-trained using a B-cell dataset; the pre-trained CNN-LSTM was then adjusted using SARS-CoV datasets, and that knowledge was transferred to a new, fine-tuned CNN-LSTM model. It could be seen that combining the CNN with LSTM was a good strategy, since there was an increase in performance from using either the CNN or LSTM alone. The first CNN-LSTM model was trained using the B-cell dataset. In the second stage of the knowledge transfer technique, the model was fine-tuned using SARS-CoV datasets. It could be observed that there was an 18% improvement in the accuracy of the model. There are a limited number of machine learning tools for epitope determination. Additionally, the accuracy of some methods, when compared to our CNN-LSTM performance for the epitope prediction task, is low; our method gave a better performance even when compared to the best method so far. These methods were recently developed using the same datasets utilized in this study, so they were chosen for fair comparison in the prediction of SARS-CoV epitopes. The ensemble utilized by [62] was a layered ensemble, with XGBoost on the outer layer and random forest regression with gradient boosting at the inner layer, and gave an AUC of 0.923 and an accuracy of 87.79%. In another study [42], the attention method and LSTM were paired for this task by combining protein with chemical and structural features, during which they achieved an accuracy of 79% and an AUC of 0.822. In another study, a Bayesian neural network was used for this task and was able to achieve an accuracy of 85% [63]. It can be seen from Table 9 that the random forest model developed in [43] was able to make good predictions, although there is a need for improvement in the model’s performance. In all the methods used previously, to the best of our knowledge, none have attempted the transfer learning strategy that was used in this study. This technique was able to significantly improve performance to an accuracy of 95.1% and an AUC of 0.979, which are better than all the results obtained in other methods. In this study, the performance of the proposed method was compared with that of state-of-the-art methods, and it gave outstanding results, with 95.1% accuracy. The benchmark deep learning model was an attention–LSTM-based model and was able to achieve 79% accuracy. Therefore, there was about a 16% increase in performance.
Table 9.
Results of other methods compared to the proposed CNN-LSTM.
In the next stage, we tried to analyze the predicted epitopes to screen the best ones for vaccine development. Here, tools such as Toxinpred were utilized for determination of toxicity. When a peptide sequence is provided to the tool, it will return a score, and when that score is less than 0.0, then the peptide sequence is non-toxic. Other parameters, such as molecular weight and hydrophilicity, can be seen with the tool. It has been found that peptides with lower molecular weights tend to be non-toxic [52], and hydrophilicity is quite important as well, for generating the process of the immune response [64]. A good vaccine must not cause allergies; hence, the prediction of vaccine allergenicity is crucial. We used the AllerTOP2.0 service, which assesses peptide allergenicity. Among the predicted epitopes, twelve were considered non-allergens, while four were allergens, according to the AllerTOP2.0 [53]. Finally, to choose the best sequence for vaccine development, it is important to determine antigenicity because antigenic peptides will generate high immune responses in humans. For this task, the VaxiJen tool was used to predict antigenicity [54].
In order to check the expression of the vaccine sequence, a codon adaptation tool, Jcat, was used to optimize the codon usage of the designed vaccine. A codon optimization index (CAI) value of 1.0 was obtained, which means that the vaccine would be highly expressed in E. coli. It is important to have the vaccine expressed in the host organism in other to elicit an immune response. It was successfully cloned into the pCC1BAC vector using the snapgene tool. Further analysis with the Expasy tool revealed that the vaccine would have a half-life of 30 h in mammalian reticulocytes and would be stable. Finally, the immune response simulation showed that the vaccine sequence, when injected, would produce high numbers of B-cells and T-cells as well as a high amount of IFN-γ, which would assist other immune cells in killing the virus.
Even though fine-tuned CNN-LSTM has enhanced epitope prediction performance and has been turned into a powerful method, there are still several interesting directions to explore. Three issues will be the focus of our next approach. First, investigating more deep-learning-based frameworks and utilizing techniques for the best hyperparameter search may result in improved performance. Secondly, it is important to consider peptide sequence information in the feature space apart from the length that was considered in this work. Finally, we will explore explainable AI to understand the features that are utilized by this model for decision-making. This study will assist scientists in designing a suitable vaccine for COVID-19, which can be validated experimentally, in a short period.
5. Conclusions
In this work, we have introduced a transfer learning epitope prediction model with superior performance. This type of method has not been considered anywhere for this task. This method has shown that combing a CNN and LSTM into a single architecture will go a long way in improving model performance. Similarly, to boost performance, especially when data is very limited, the proposed method is highly efficient. This can be seen from the results obtained when the CNN-LSTM was fine-tuned using SARS-CoV data, generating almost an 18% increase in accuracy (95.1% accuracy). The fine-tuned model was utilized to predict epitopes from SARS-CoV-2 data. When our proposed method was compared to those available, it was discovered that ours outperformed competing techniques. To choose the best epitope for vaccine development, other important parameters were checked using bioinformatic tools. Allergenicity, toxicity, and antigenicity analyses were carried out; stability, expression, cloning, and immune simulation also gave insight into the vaccine’s immune response. Therefore, the findings of this research can help in successful COVID-19 vaccine development, saving time and costs.
Author Contributions
Conceptualization, Z.S.A. and A.S.M.; methodology, Z.S.A. and A.S.M.; software, Z.S.A.; validation, H.M., D.U.O. and A.S.M.; data curation, Z.S.A.; writing—original draft preparation, Z.S.A. and A.S.M.; writing—review and editing, Z.S.A., H.M. and A.S.M.; visualization, H.M.; supervision, D.U.O.; project administration, D.U.O.; funding acquisition, H.M. All authors have read and agreed to the published version of the manuscript.
Funding
Princess Nourah bint Abdulrahman University Researchers Supporting Project number (PNURSP2023R137), Princess Nourah bint Abdulrahman University, Riyadh, Saudi Arabia.
Data Availability Statement
Available on request.
Acknowledgments
We would like to acknowledge the Operational Research Center in Healthcare, Near East University, and Princess Nourah bint Abdulrahman University for their support throughout this project.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Zhou, P.; Yang, X.-L.; Wang, X.-G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.-R.; Zhu, Y.; Li, B.; Huang, C.-L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef]
- Shang, W.; Yang, Y.; Rao, Y.; Rao, X. The outbreak of SARS-CoV-2 pneumonia calls for viral vaccines. NPJ Vaccines 2020, 5, 18. [Google Scholar] [CrossRef]
- Callaway, E. The race for coronavirus vaccines: A graphical guide. Nature 2020, 580, 576–577. [Google Scholar] [CrossRef]
- Alcorta-Nuñez, F.; Pérez-Ibave, D.C.; Burciaga-Flores, C.H.; Garza, M.; González-Escamilla, M.; Rodríguez-Niño, P.; González-Guerrero, J.F.; Alcorta-Garza, A.; Vidal-Gutiérrez, O.; Ramírez-Correa, G.A.; et al. SARS-CoV-2 Neutralizing Antibodies in Mexican Population: A Five Vaccine Comparison. Diagnostics 2023, 13, 1194. [Google Scholar] [CrossRef]
- Gandon, S.; Mackinnon, M.J.; Nee, S.; Read, A.F. Imperfect vaccines and the evolution of pathogen virulence. Nature 2001, 414, 751–756. [Google Scholar] [CrossRef]
- Ferreira, R.G.; Gordon, N.F.; Stock, R.; Petrides, D. Adenoviral Vector COVID-19 Vaccines: Process and Cost Analysis. Processes 2021, 9, 1430. [Google Scholar] [CrossRef]
- Kim, Y.C.; Dema, B.; Reyes-Sandoval, A. COVID-19 vaccines: Breaking record times to first-in-human trials. NPJ Vaccines 2020, 5, 19–21. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Liu, Q.; Guo, D. Emerging coronaviruses: Genome structure, replication, and pathogenesis. J. Med. Virol. 2020, 92, 418–423. [Google Scholar] [CrossRef]
- Yang, X.; Yu, Y.; Xu, J.; Shu, H.; Xia, J.; Liu, H.; Wu, Y.; Zhang, L.; Yu, Z.; Fang, M.; et al. Clinical course and outcomes of critically ill patients with SARS-CoV-2 pneumonia in Wuhan, China: A single-centered, retrospective, observational study. Lancet Respir. Med. 2020, 8, 475–481. [Google Scholar] [CrossRef] [PubMed]
- Medzhitov, R.; Janeway, C., Jr. Innate immune recognition: Mechanisms and pathways. Immunol. Rev. 2000, 173, 89–97. [Google Scholar] [CrossRef]
- Cooper, M.D.; Alder, M.N. The Evolution of Adaptive Immune Systems. Cell 2006, 124, 815–822. [Google Scholar] [CrossRef]
- Kringelum, J.V.; Nielsen, M.; Padkjær, S.B.; Lund, O. Structural analysis of B-cell epitopes in antibody:protein complexes. Mol. Immunol. 2013, 53, 24–34. [Google Scholar] [CrossRef]
- Ansari, H.R.; Raghava, G.P. Identification of conformational B-cell Epitopes in an antigen from its primary sequence. Immunome Res. 2010, 6, 6–9. [Google Scholar] [CrossRef]
- Kang, S.; Yang, M.; Hong, Z.; Zhang, L.; Huang, Z.; Chen, X.; He, S.; Zhou, Z.; Zhou, Z.; Chen, Q.; et al. Crystal structure of SARS-CoV-2 nucleocapsid protein RNA binding domain reveals potential unique drug targeting sites. Acta Pharm. Sin. B 2020, 10, 1228–1238. [Google Scholar] [CrossRef]
- Wrapp, D.; Wang, N.; Corbett, K.S.; Goldsmith, J.A.; Hsieh, C.-L.; Abiona, O.; Graham, B.S.; McLellan, J.S. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 2020, 367, 1260–1263. [Google Scholar] [CrossRef]
- Westerbeck, J.W.; Machamer, C.E. The Infectious Bronchitis Coronavirus Envelope Protein Alters Golgi pH To Protect the Spike Protein and Promote the Release of Infectious Virus. J. Virol. 2019, 93, e00015-19. [Google Scholar] [CrossRef]
- Yuan, P.; Huang, S.; Yang, Z.; Xie, L.; Wang, K.; Yang, Y.; Ran, L.; Yu, Q.; Song, Z. UBXN1 interacts with the S1 protein of transmissible gastroenteritis coronavirus and plays a role in viral replication. Veter Res. 2019, 50, 28. [Google Scholar] [CrossRef]
- Van Regenmortel, M.H.V. Chapter 1 What Is a B-Cell Epitope? In Methods in Molecular Biology, Epitope Mapping Protocols; Ulrich, R., Schutkowsk, W., Eds.; Humana Press: Totowa, NJ, USA, 2009; Volume 524. [Google Scholar] [CrossRef]
- Bhattacharya, M.; Sharma, A.R.; Patra, P.; Ghosh, P.; Sharma, G.; Patra, B.C.; Lee, S.; Chakraborty, C. Development of epitope-based peptide vaccine against novel coronavirus 2019 (SARS-COV-2): Immunoinformatics approach. J. Med. Virol. 2020, 92, 618–631. [Google Scholar] [CrossRef]
- Yazdani, Z.; Rafiei, A.; Yazdani, M.; Valadan, R. Design an Efficient Multi-Epitope Peptide Vaccine Candidate against SARS-CoV-2: An in silico Analysis. Infect. Drug Resist. 2020, 13, 3007–3022. [Google Scholar] [CrossRef]
- Bukhari, S.N.H.; Jain, A.; Haq, E.; Mehbodniya, A.; Webber, J. Ensemble Machine Learning Model to Predict SARS-CoV-2 T-Cell Epitopes as Potential Vaccine Targets. Diagnostics 2021, 11, 1990. [Google Scholar] [CrossRef]
- Mukherjee, S.; Tworowski, D.; Detroja, R.; Mukherjee, S.B.; Frenkel-Morgenstern, M. Immunoinformatics and Structural Analysis for Identification of Immunodominant Epitopes in SARS-CoV-2 as Potential Vaccine Targets. Vaccines 2020, 8, 290. [Google Scholar] [CrossRef] [PubMed]
- Dong, R.; Chu, Z.; Yu, F.; Zha, Y. Contriving Multi-Epitope Subunit of Vaccine for COVID-19: Immunoinformatics Approaches. Front. Immunol. 2020, 11, 1784. [Google Scholar] [CrossRef] [PubMed]
- Naz, A.; Shahid, F.; Butt, T.T.; Awan, F.M.; Ali, A.; Malik, A. Designing Multi-Epitope Vaccines to Combat Emerging Coronavirus Disease 2019 (COVID-19) by Employing Immuno-Informatics Approach. Front. Immunol. 2020, 11, 1663. [Google Scholar] [CrossRef]
- Eraslan, G.; Avsec, Ž.; Gagneur, J.; Theis, F.J. Deep learning: New computational modelling techniques for genomics. Nat. Rev. Genet. 2019, 20, 389–403. [Google Scholar] [CrossRef] [PubMed]
- Ameen, Z.S.; Ozsoz, M.; Mubarak, A.S.; Al Turjman, F.; Serte, S. C-SVR Crispr: Prediction of CRISPR/Cas12 guideRNA activity using deep learning models. Alex. Eng. J. 2021, 60, 3501–3508. [Google Scholar] [CrossRef]
- Ameen, Z.S.; Mubarak, A.S.; Altrjman, C.; Alturjman, S.; Abdulkadir, R.A. Explainable Residual Network for Tuberculosis Classification in the IoT Era. In Proceedings of the 2021 International Conference on Forthcoming Networks and Sustainability in AIoT Era (FoNeS-AIoT), Nicosia, Turkey, 27–28 December 2021; pp. 9–12. [Google Scholar] [CrossRef]
- Ozsoz, M.; Mubarak, A.; Said, Z.; Aliyu, R.; Al Turjman, F.; Serte, S. Deep learning-based feature extraction coupled with multi-class SVM for COVID-19 detection in the IoT era. Int. J. Nanotechnol. 2021, 1, 1–18. [Google Scholar] [CrossRef]
- Wang, D.; Hu, B.; Hu, C.; Zhu, F.; Liu, X.; Zhang, J.; Wang, B.; Xiang, H.; Cheng, Z.; Xiong, Y.; et al. Clinical characteristics of 138 hospitalized patients with 2019 novel coronavirus–infected pneumonia in Wuhan, China. JAMA 2020, 323, 1061–1069. [Google Scholar] [CrossRef]
- Mubarak, A.S.; Serte, S.; Al-Turjman, F.; Ameen, Z.S.; Ozsoz, M. Local binary pattern and deep learning feature extraction fusion for COVID-19 detection on computed tomography images. Expert Syst. 2021, 39, e12842. [Google Scholar] [CrossRef]
- Alhazmi, W.; Turki, T. Applying Deep Transfer Learning to Assess the Impact of Imaging Modalities on Colon Cancer Detection. Diagnostics 2023, 13, 1721. [Google Scholar] [CrossRef]
- Sun, P.; Guo, S.; Sun, J.; Tan, L.; Lu, C.; Ma, Z. Advances in In-silico B-cell Epitope Prediction. Curr. Top. Med. Chem. 2019, 19, 105–115. [Google Scholar] [CrossRef]
- Jespersen, M.C.; Peters, B.; Nielsen, M.; Marcatili, P. BepiPred-2.0: Improving sequence-based B-cell epitope prediction using conformational epitopes. Nucleic Acids Res. 2017, 45, 24–29. [Google Scholar] [CrossRef]
- El-Manzalawy, Y.; Dobbs, D.; Honavar, V. Predicting linear B-cell epitopes using string kernels. J. Mol. Recognit. 2008, 21, 243–255. [Google Scholar] [CrossRef] [PubMed]
- Collatz, M.; Mock, F.; Barth, E.; Hölzer, M.; Sachse, K.; Marz, M. EpiDope: A deep neural network for linear B-cell epitope prediction. Bioinformatics 2020, 37, 448–455. [Google Scholar] [CrossRef] [PubMed]
- Saha, S.; Raghava, G.P.S. Prediction of continuous B-cell epitopes in an antigen using recurrent neural network. Proteins Struct. Funct. Bioinform. 2006, 65, 40–48. [Google Scholar] [CrossRef] [PubMed]
- Yao, B.; Zhang, L.; Liang, S.; Zhang, C. SVMTriP: A Method to Predict Antigenic Epitopes Using Support Vector Machine to Integrate Tri-Peptide Similarity and Propensity. PLoS ONE 2012, 7, e45152. [Google Scholar] [CrossRef]
- Singh, H.; Ansari, H.R.; Raghava, G.P.S. Improved Method for Linear B-Cell Epitope Prediction Using Antigen’s Primary Sequence. PLoS ONE 2013, 8, e62216. [Google Scholar] [CrossRef]
- Fischer, W.; Perkins, S.; Theiler, J.; Bhattacharya, T.; Yusim, K.; Funkhouser, R.; Kuiken, C.; Haynes, B.; Letvin, N.L.; Walker, B.D.; et al. Polyvalent vaccines for optimal coverage of potential T-cell epitopes in global HIV-1 variants. Nat. Med. 2006, 13, 100–106. [Google Scholar] [CrossRef]
- Vita, R.; Zarebski, L.; Greenbaum, J.A.; Emami, H.; Hoof, I.; Salimi, N.; Damle, R.; Sette, A.; Peters, B. The Immune Epitope Database 2.0. Nucleic Acids Res. 2009, 38, D854–D862. [Google Scholar] [CrossRef]
- La Marca, A.F.; Lopes, R.D.S.; Lotufo, A.D.P.; Bartholomeu, D.C.; Minussi, C.R. BepFAMN: A Method for Linear B-Cell Epitope Predictions Based on Fuzzy-ARTMAP Artificial Neural Network. Sensors 2022, 22, 4027. [Google Scholar] [CrossRef]
- Noumi, T.; Lnoue, S.; Fujita, H.; Sadamitsu, K.; Sakaguchi, M.; Tenma, A.; Nakagami, H. Epitope Prediction of Antigen Protein using Attention-Based LSTM Network. Inf. Process. 2020, 29, 321–327. [Google Scholar] [CrossRef]
- Cihan, P.; Ozger, Z.B. A new approach for determining SARS-CoV-2 epitopes using machine learning-based in silico methods. Comput. Biol. Chem. 2022, 98, 107688. [Google Scholar] [CrossRef]
- LeCun, Y.; Boser, B.; Denker, J.; Henderson, D.; Howard, R.; Hubbard, W.; Jackel, L. Handwritten Digit Recognition with a Back-Propagation Network. In Advances in Neural Information Processing Systems; Morgan-Kaufmann: Denver, CO, USA, 1989; pp. 396–404. [Google Scholar]
- Graves, A.; Mohamed, A.; Hinton, G. Speech recognition with deep recurrent neural networks. In Proceedings of the 2013 IEEE International Conference on Acoustics, Speech and Signal Processing, Vancouver, BC, Canada, 26–31 May 2013; pp. 6645–6649. [Google Scholar] [CrossRef]
- Yang, Z.; Dai, Z.; Yang, Y.; Carbonell, J.; Salakhutdinov, R.; Le, Q.V. XLNet: Generalized Autoregressive Pretraining for Language Understanding. In Proceedings of the 33rd International Conference on Neural Information Processing Systems, Vancouver, BC, Canada, 8–14 December 2019; pp. 1–18. [Google Scholar]
- Rumelhart, D.E.; Hinton, G.E.; Williams, R.J. Learning representations by back-propagating errors. Nature 1986, 323, 533–536. [Google Scholar] [CrossRef]
- Hochreiter, S.; Schmidhuber, J. Long short-term memory. Neural Comput. 1997, 9, 1735–1780. [Google Scholar] [CrossRef]
- Graves, A.; Schmidhuber, J. Framewise phoneme classification with bidirectional LSTM and other neural network architectures. Neural Netw. 2005, 18, 602–610. [Google Scholar] [CrossRef] [PubMed]
- Levine, S.; Krizhevsky, A. Learning Hand-Eye Coordination for Robotic Grasping with Deep Learning and Large-Scale Data Collection. Springer Proc. Adv. Robot. 2017, 1, 173–184. [Google Scholar] [CrossRef]
- Srivastava, N.; Hinton, G.R.; Krizhevsky, A.; Sutskever, I.; Salakhutdinov, R. Dropout: A Simple Way to Prevent Neural Networks from Over fitting. J. Mach. Learn. Res. 2014, 15, 1929–1958. [Google Scholar] [CrossRef]
- Gupta, S.; Kapoor, P.; Chaudhary, K.; Gautam, A.; Kumar, R.; Raghava, G.P.S.; Open Source Drug Discovery Consortium. In Silico Approach for Predicting Toxicity of Peptides and Proteins. PLoS ONE 2013, 8, e73957. [Google Scholar] [CrossRef] [PubMed]
- Dimitrov, I.; Bangov, I.; Flower, D.R.; Doytchinova, I. AllerTOP v.2—A server for in silico prediction of allergens. J. Mol. Model. 2014, 20, 2278. [Google Scholar] [CrossRef]
- Doytchinova, I.A.; Flower, D.R. VaxiJen: A server for prediction of protective antigens, tumour antigens and subunit vaccines. BMC Bioinform. 2007, 8, 4. [Google Scholar] [CrossRef]
- Lever, J.; Krzywinski, M.; Altman, N. Classification evaluation. Nat. Methods 2016, 13, 603–604. [Google Scholar] [CrossRef]
- Gao, X.W.; James-Reynolds, C.; Currie, E. Analysis of tuberculosis severity levels from CT pulmonary images based on enhanced residual deep learning architecture. Neurocomputing 2019, 392, 233–244. [Google Scholar] [CrossRef]
- Baldi, P.; Brunak, S.; Chauvin, Y.; Andersen, C.A.F.; Nielsen, H. Assessing the accuracy of prediction algorithms for classification: An overview. Bioinformatics 2000, 16, 412–424. [Google Scholar] [CrossRef] [PubMed]
- Powers, D.M.W. Evaluation: From Precision, Recall and F-Measure to ROC, Informedness, Markedness and Correlation. J. Mach. Learn. Technol. 2021, 2, 37–63. [Google Scholar]
- Bradley, A.P. The use of the area under the ROC curve in the evaluation of machine learning algorithms. Pattern Recognit. 1997, 30, 1145–1159. [Google Scholar] [CrossRef]
- Grote, A.; Hiller, K.; Scheer, M.; Münch, R.; Nörtemann, B.; Hempel, D.C.; Jahn, D. JCat: A novel tool to adapt codon usage of a target gene to its potential expression host. Nucleic Acids Res. 2005, 33, 526–531. [Google Scholar] [CrossRef]
- Rapin, N.; Lund, O.; Bernaschi, M.; Castiglione, F. Computational Immunology Meets Bioinformatics: The Use of Prediction Tools for Molecular Binding in the Simulation of the Immune System. PLoS ONE 2010, 5, e9862. [Google Scholar] [CrossRef] [PubMed]
- Jain, N.; Jhunthra, S.; Garg, H.; Gupta, V.; Mohan, S.; Ahmadian, A.; Salahshour, S.; Ferrara, M. Prediction modelling of COVID using machine learning methods from B-cell dataset. Results Phys. 2021, 21, 103813. [Google Scholar] [CrossRef]
- Ghoshal, B.; Swift, S.; Tucker, A. Uncertainty Estimation in SARS-CoV-2 B-Cell Epitope Prediction for Vaccine Development. In Proceedings of the Artificial Intelligence in Medicine: 19th International Conference on Artificial Intelligence in Medicine, AIME 2021, Virtual Event, 15–18 June 2021. [Google Scholar] [CrossRef]
- Pooja, K.; Rani, S.; Kanwate, B.; Pal, G.K. Physico-chemical, Sensory and Toxicity Characteristics of Dipeptidyl Peptidase-IV Inhibitory Peptides from Rice Bran-derived Globulin Using Computational Approaches. Int. J. Pept. Res. Ther. 2017, 23, 519–529. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).