
Understanding Cd2+ Adsorption Mechanism on Montmorillonite Surfaces by Combining DFT and MD


Abstract
:1. Introduction
2. Model and Calculation
2.1. Density Functional Theory Calculations
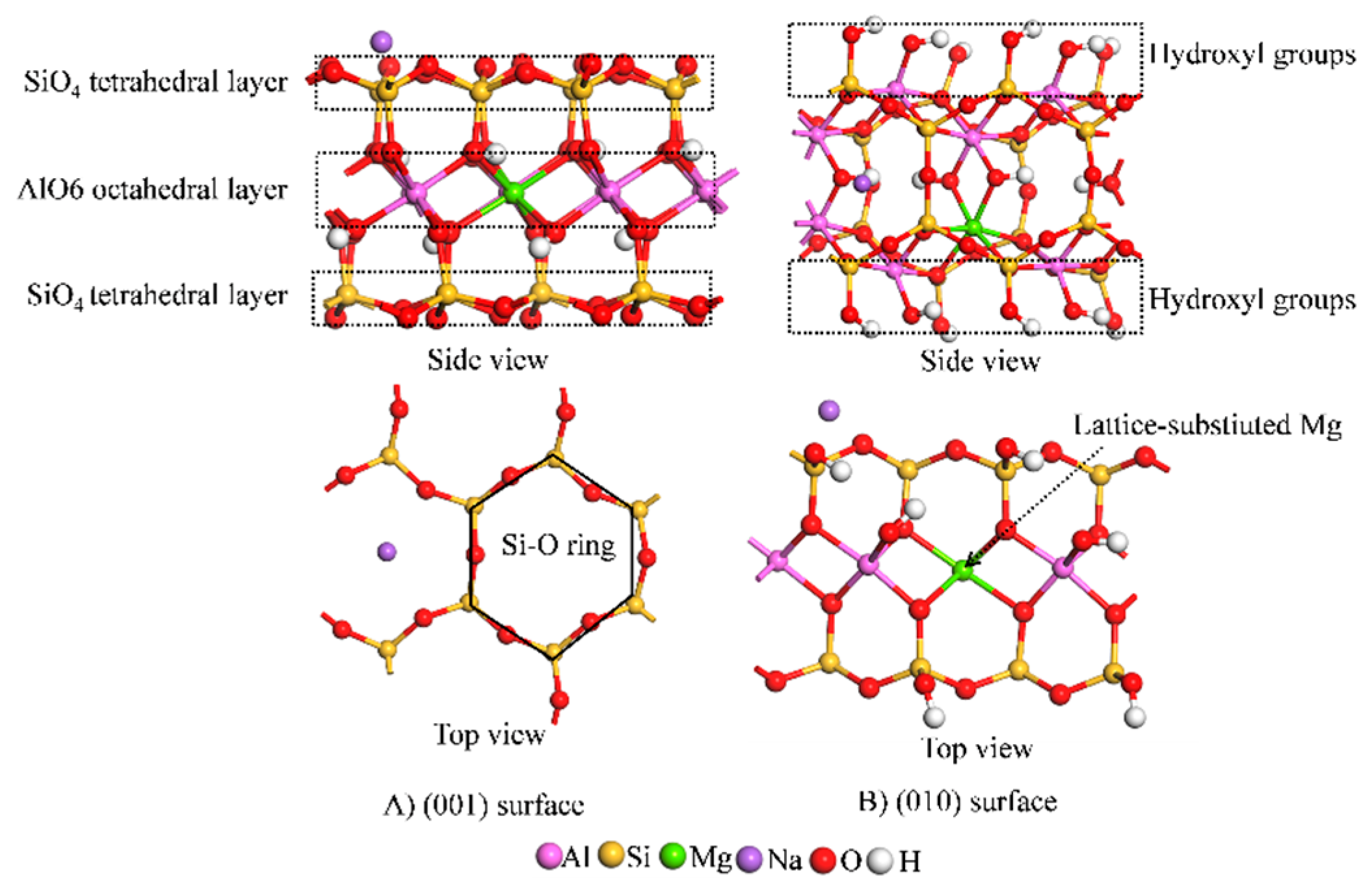
2.1.1. Model Construction
2.1.2. Calculation Method
2.2. Molecular Dynamics Calculation
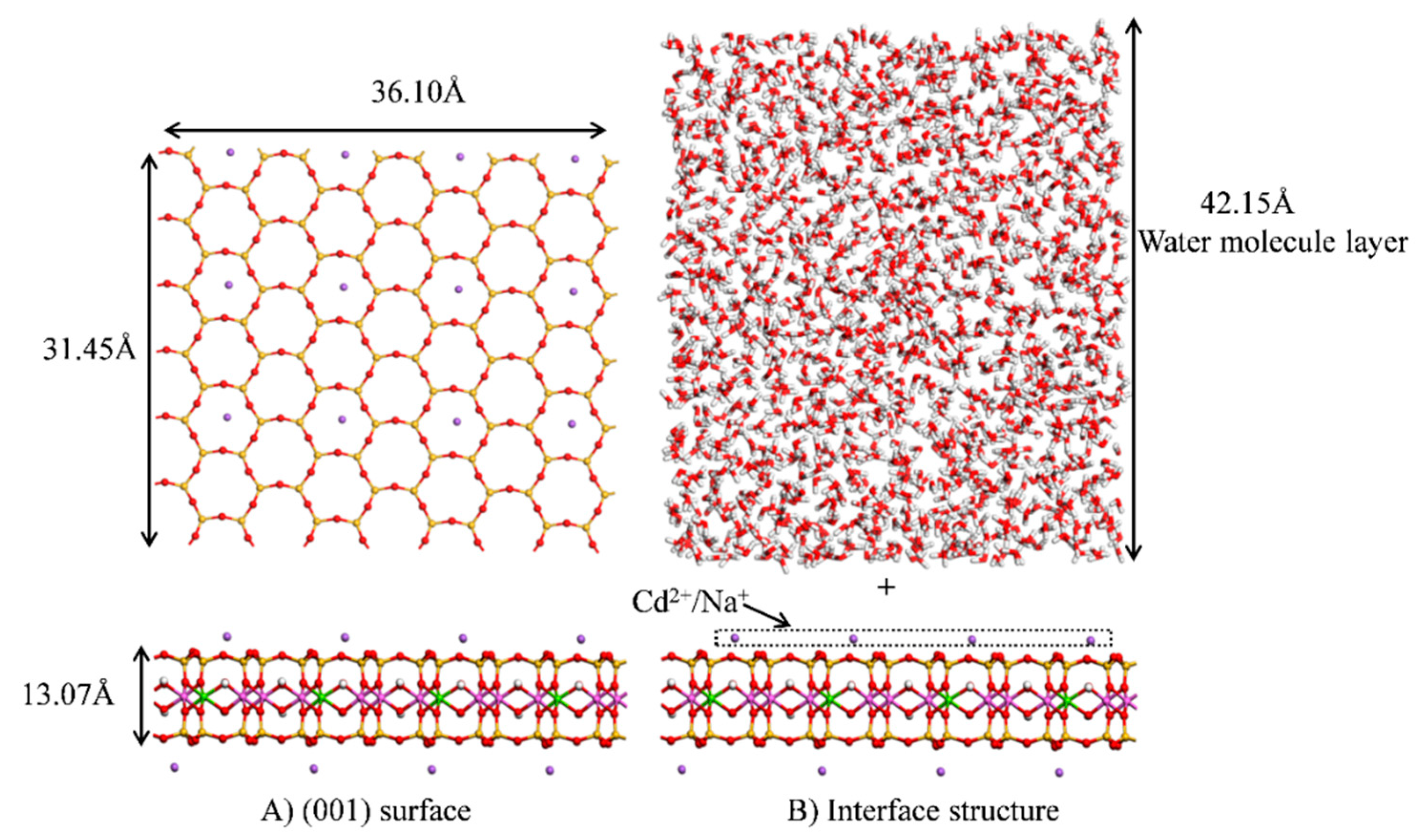
2.2.1. Construction of the Model
2.2.2. MD Simulations
3. Discussion
3.1. DFT
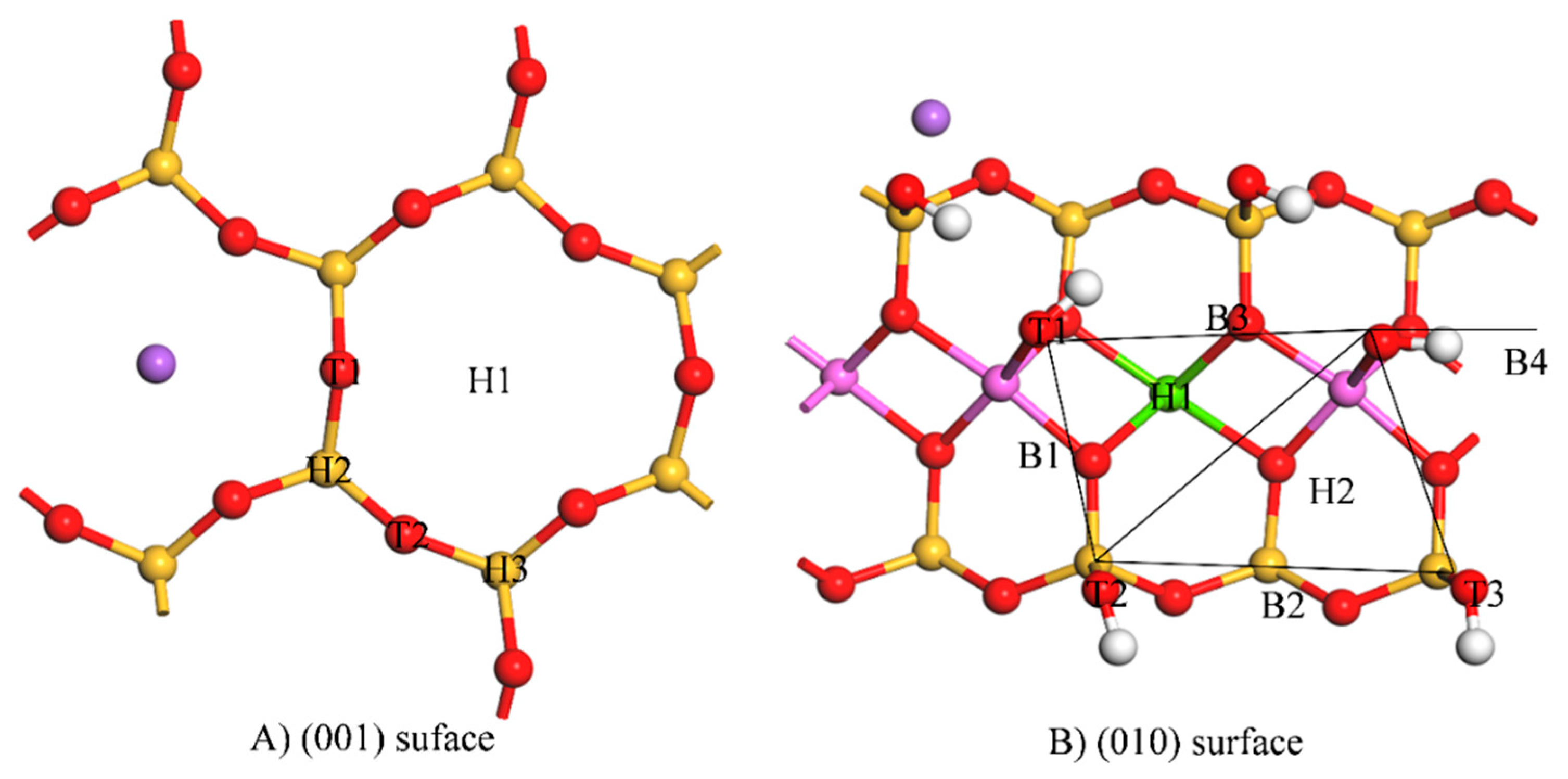
3.1.1. Initial Adsorption Position and Adsorption Energy
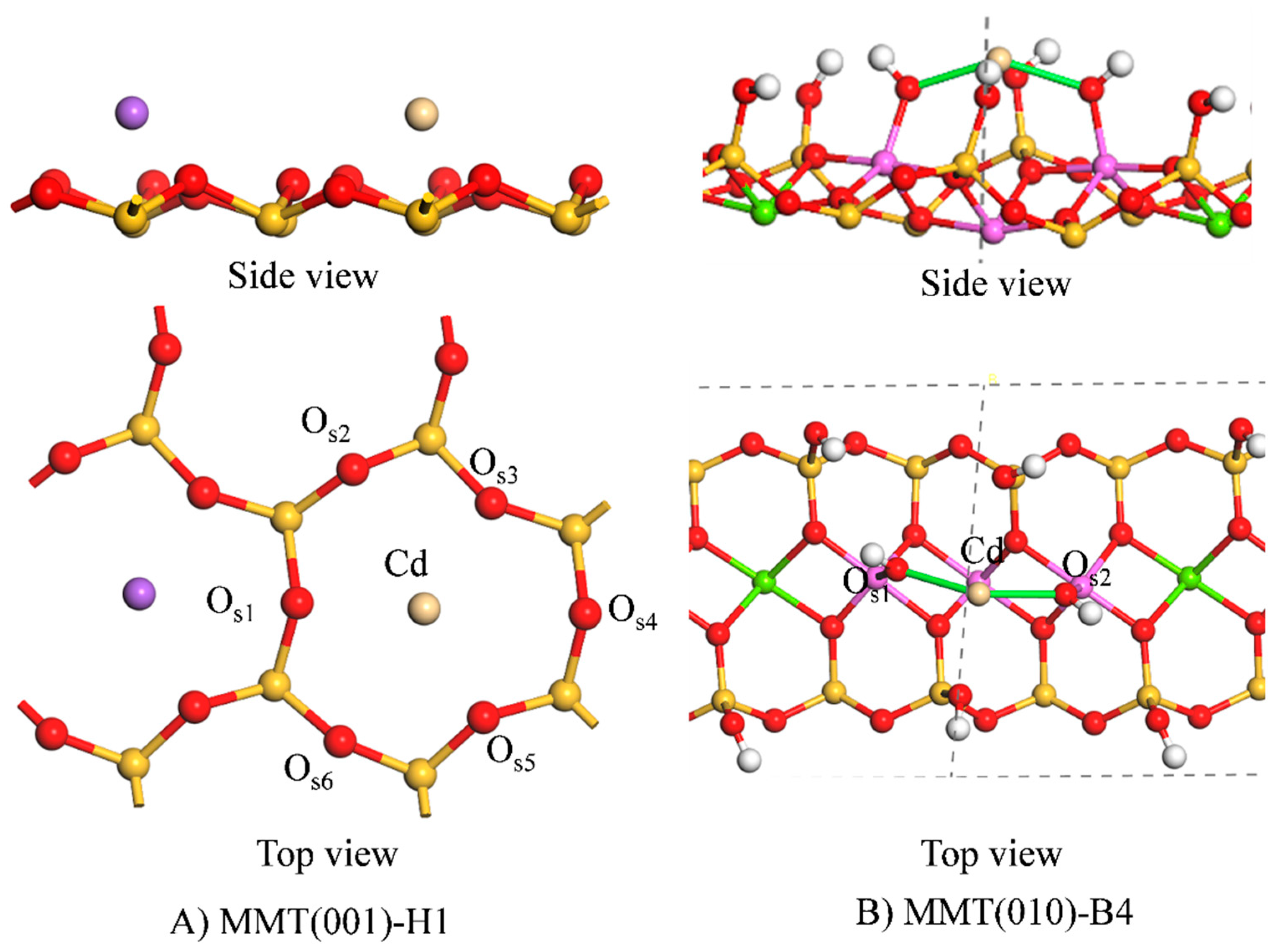
3.1.2. Adsorption Configuration
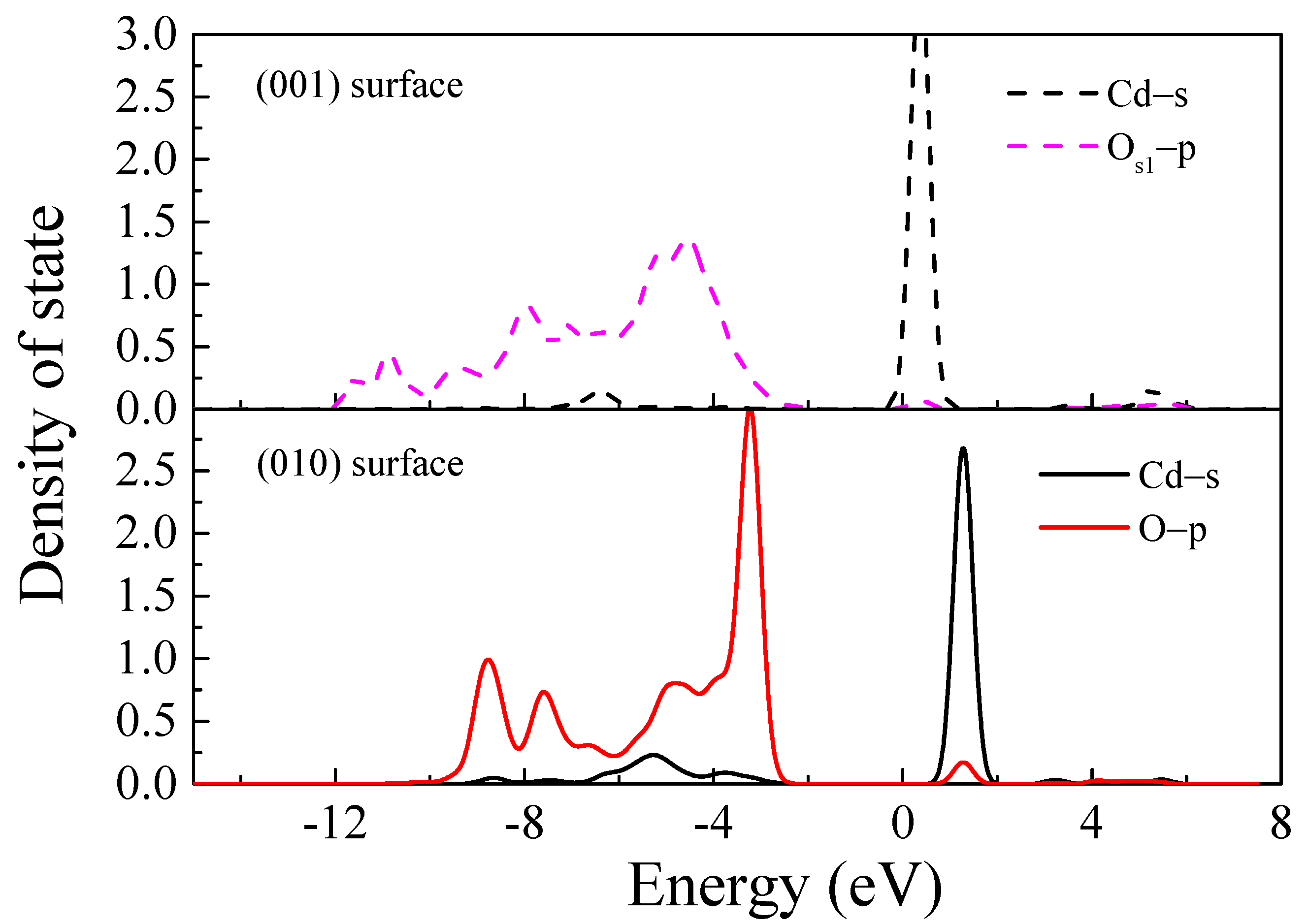
3.1.3. Local State Density Analysis
3.2. MD Simulations
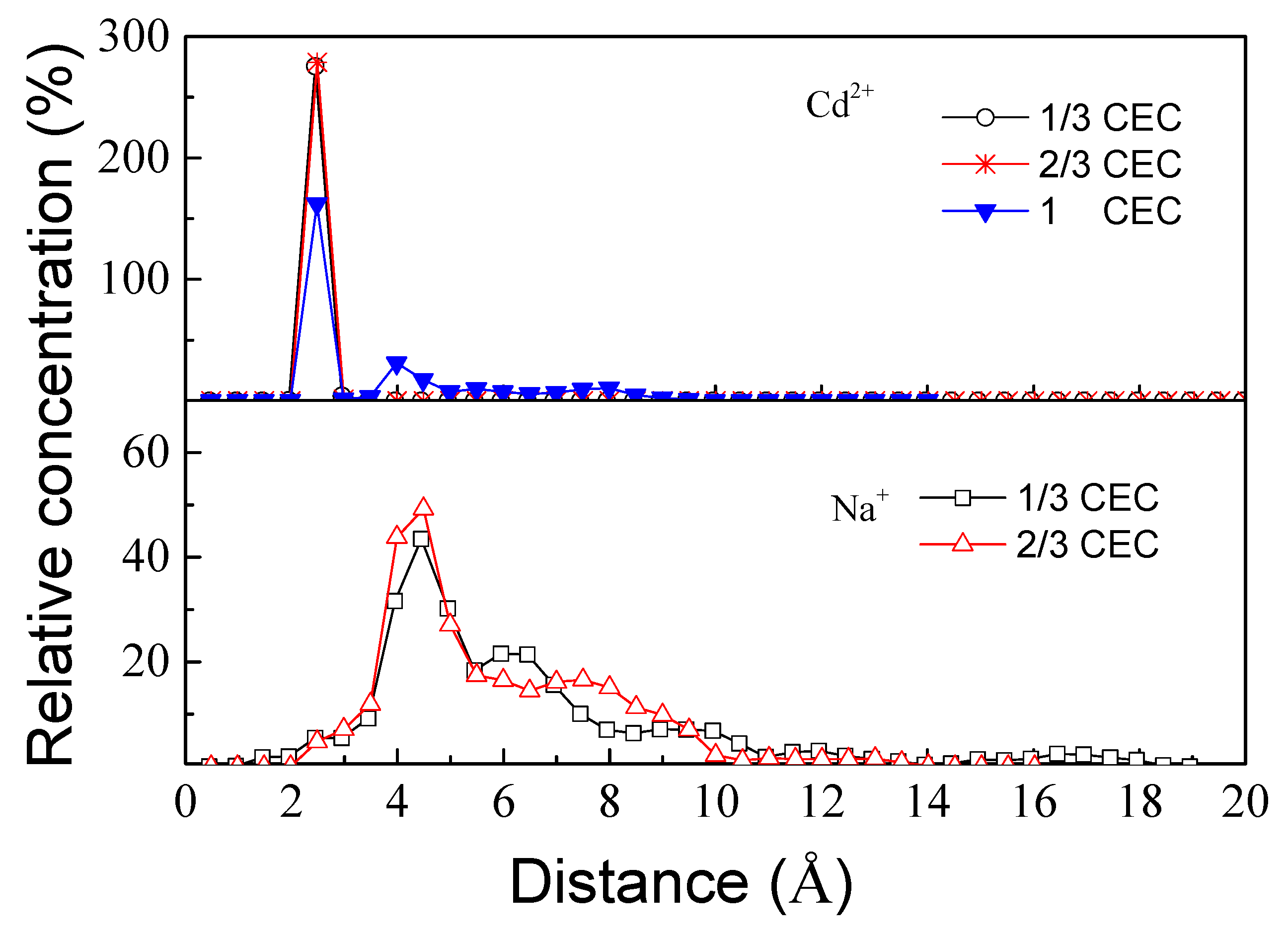
3.2.1. Relative Concentration Distribution
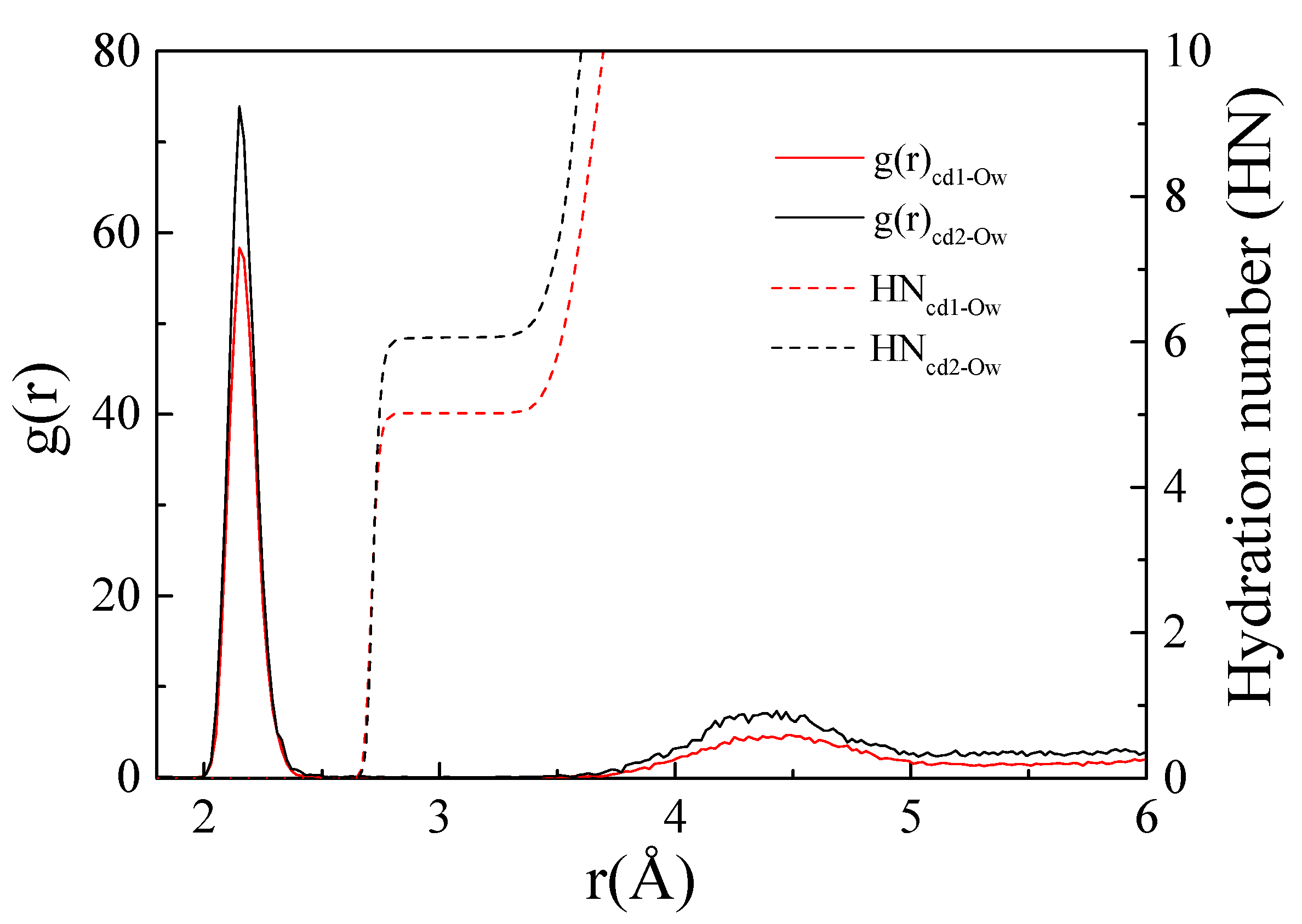
3.2.2. Radial Distribution
3.2.3. Diffusion Coefficient
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Gupta, S.S.; Bhattacharyya, K.G. Removal of Cd(II) from aqueous solution by kaolinite, montmorillonite and their poly(oxo zirconium) and tetrabutylammonium derivatives. J. Hazard. Mater. 2006, 128, 247–257. [Google Scholar] [CrossRef]
- Uddin, M.K. A review on the adsorption of heavy metals by clay minerals, with special focus on the past decade. Chem. Eng. J. 2017, 308, 438–462. [Google Scholar] [CrossRef]
- Hu, C.; Hu, H.; Song, M.; Tan, J. Preparation, characterization, and Cd(II) sorption of/on cysteine-montmorillonite composites synthesized at various pH. Environ. Sci. Pollut. Res. 2020, 27, 10599–10606. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Yuan, Y.; Zhao, Y.; Rao, F. Effect of layer charges on exfoliation of montmorillonite in aqueous solutions. Colloids Surf. A 2018, 548, 92–97. [Google Scholar] [CrossRef]
- Wang, W.; Zhao, Y.; Yi, H.; Chen, T.; Kang, S.; Zhang, T.; Rao, F.; Song, S. Pb(ΙΙ) removal from water using porous hydrogel of chitosan-2D montmorillonite. Int. J. Biol. Macromol. 2019, 128, 85–93. [Google Scholar] [CrossRef] [PubMed]
- Juan, Y.; Chaobing, D.; Zhen, Y.; Wang, X.; Xu, G. Effective removal of lead ions from aqueous solution using nano Illite/Smectite clay: Isotherm, kinetic, and thermodynamic modeling of adsorption. Water 2018, 10, 210. [Google Scholar]
- Chen, Y.; Liu, Y.; Li, Y.; Li Zhao, L.; Chen, Y.; Li, H.; Liu, Y.; Li, L.; Xu, F.; Li, M. Functional wastepaper-montmorillonite composite aerogel for Cd2+ adsorption. Environ. Sci. Pollut. R. 2020, 27, 38644–38653. [Google Scholar] [CrossRef]
- Peng, C.; Min, F.; Liu, L.; Chen, J. The adsorption of CaOH+ on (001) basal and (010) edge surface of Na-montmorillonite: A DFT study. Surf. Interface Anal. 2017, 49, 267–277. [Google Scholar] [CrossRef]
- Chen, J.; Li, Y. Orbital symmetry matching study on the interactions of flotation reagents with mineral surfaces. Miner. Eng. 2022, 179, 107469. [Google Scholar] [CrossRef]
- Chen, J. The interaction of flotation reagents with metal ions in mineral surfaces. Miner. Eng. 2021, 171, 107067. [Google Scholar] [CrossRef]
- Peng, C.; Wang, G.; Zhang, C.; Qin, L. Molecular dynamics simulation of NH4+-smectite interlayer hydration: Influence of layer charge density and location. J. Mol. Liq. 2021, 336, 116232. [Google Scholar] [CrossRef]
- Peng, C.; Wang, G.; Qin, L.; Luo, S. Molecular dynamics simulation of NH4+-montmorillonite interlayer hydration: Structure, energetics, and dynamics. Appl. Clay Sci. 2020, 195, 105657. [Google Scholar] [CrossRef]
- Wardle, R.; Brindley, G. The Crystal structures of pyrophyllite, 1Tc, and of its dehydroxylate. Am. Mineral. 1972, 57, 732–750. [Google Scholar]
- Delley, B. An all-electron numerical method for solving the localdensity functional for polyatomic molecules. J. Chem. Phys. 1990, 92, 508–517. [Google Scholar] [CrossRef]
- Delley, B. From molecules to solids with the DMol3 approach. J. Chem. Phys. 2000, 113, 7756–7764. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [Green Version]
- Grimme, S. Semiempirical hybrid density functional with perturbative second-order correlation. J. Chem. Phys. 2006, 124, 034108. [Google Scholar] [CrossRef] [Green Version]
- Pack, J.D. Special points for Brillon-zone integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar]
- Kai, C.; Zh, B. A new method for quantifying cation exchange capacity in clay minerals. Appl. Clay Sci. 2018, 161, 444–455. [Google Scholar]
- Kobayashi, K.; Liang, Y.; Murata, S.; Matsuoka, T.; Takahashi, S.; Nishi, N.; Sakka, T. Ion distribution and hydration structure in the stern layer on muscovite surface. Langmuir 2017, 33, 3892–3899. [Google Scholar] [CrossRef]
- Kirkpatrick, R.; Kalinichev, A.; Bowers, G.M.; Yazaydin, A.O. NMR and computational molecular modeling studies of mineral surfaces and interlayer galleries: A review. Am. Mineral. 2015, 100, 1341–1354. [Google Scholar] [CrossRef]
- Cygan, R.; Liang, J.; Kalinichev, A. Molecular models of hydroxide, oxyhydroxide, and clay phases and the development of a general force field. J. Phys. Chem. B 2004, 108, 1255–1266. [Google Scholar] [CrossRef]
- Peng, C.; Min, F.; Liu, L.; Chen, J. A periodic DFT study of adsorption of water on sodium-montmorillonite (001) basal and (010) edge surface. Appl. Surf. Sci. 2016, 387, 308–316. [Google Scholar] [CrossRef]
- Mignon, P.; Ugliengo, P.; Sodupe, M. Ab initio molecular dynamics study of the hydration of Li+, Na+ and K+ in a montmorillonite model. Influence of isomorphic substitution. Phys. Chem. Chem. Phys. 2010, 12, 688–697. [Google Scholar] [CrossRef]
- Mohammed, A.M. Hydration of Cd(II): Molecular dynamics study. Chem. Soc. Ethiop. 2008, 22, 423–432. [Google Scholar] [CrossRef] [Green Version]
- Abollino, O.; Aceto, M.; Malandrino, M.; Sarzanini, C.; Mentasti, E. Adsorption of heavy metals on Na-montmorillonite. Effect of pH and organic substances. Water Res. 2003, 37, 1619–1627. [Google Scholar] [CrossRef]
- Liu, X.; Cheng, J.; Sprik, M.; Lu, X. Surface acidity of 2:1-type dioctahedral clay minerals from first principles molecular dynamics simulations. Geochim. Cosmochim. Acta 2014, 140, 410–417. [Google Scholar] [CrossRef]
- Peng, C.; Zhong, Y.; Wang, G.; Min, F.; Qin, L. Atomic-level insights into the adsorption of rare earth Y(OH)3−nn (n = 1–3) ion. Appl. Surf. Sci. 2019, 469, 357–367. [Google Scholar] [CrossRef]
- Ramadugu, S.K.; Mason, S.E. DFT study antimony(V) oxyanion adsorption on a-A2O3(1102). J. Phys. Chem. 2015, 119, 18149–18159. [Google Scholar]
- Du, J.; Fan, L.; Wang, Q.; Min, F. Adsorption of Cr(OH)n(3−n)+ (n=1–3) on illite (001) and (010) surfaces: A DFT study. Processes 2021, 9, 2048–2059. [Google Scholar] [CrossRef]
- Xia, S.; Yang, Y.; Yu, L. DFT study on structures and stabilities of hydration and hydrolysis Cd(Ⅱ). Riodical Ocean. Univ. China 2009, 39, 115–118. [Google Scholar]
- Du, J.; Min, F.; Zhang, M.; Peng, C. Study on hydration of illite in K+, Na+, Ca2+, Mg2+, and Al3+ electrolyte solutions. Z. Phys. Chem. 2019, 233, 721–735. [Google Scholar] [CrossRef]
- Yi, H.; Zhang, X.; Zhao, Y.; Liu, L. Molecular dynamics simulations of hydration shell on montmorillonite (001) in water. Surf. Interface Anal. 2016, 48, 976–980. [Google Scholar] [CrossRef]
- Chen, J.; Min, F.; Liu, L.; Liu, C. Mechanism research on surface hydration of kaolinite insights from DFT and MD simulations. Appl. Surf. Sci. 2019, 476, 6–15. [Google Scholar] [CrossRef]
- Loganathan, N.; Yazaydin, A.O.; Bowers, G.M.; Kalinichev, A.G.; Kirkpatrick, R.J. Structure, energetics, and dynamics of Cs+ and H2O in hectorite: Molecular dynamics simulations with an unconstrained substrate surface. J. Phys. Chem. C 2016, 120, 10298–10310. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| (001) Surface | Initial Position | Final Position | Adsorption Energy (kJ/mol) | Initial Position | Final Position | Adsorption Energy (kJ/mol) |
|---|---|---|---|---|---|---|
| H1 | H1 | −88.74 | T1 | H1 | −81.65 | |
| H2 | H1 | −75.61 | T2 | H1 | −80.87 | |
| H3 | H3 | −44.11 | - | |||
| (010) surface | H1 | H1 | −254.67 | B4 | B4 | −283.55 |
| H2 | H2 | −189.56 | T1 | T1 | −183.79 | |
| B1 | B1 | −183.79 | T2 | B2 | −183.55 | |
| B2 | B2 | −178.53 | T3 | B2 | −183.12 | |
| B3 | H1 | −254.67 | - |
| Configuration | Cd-Os1 /Å | Cd-Os2 /Å | Cd-Os3 /Å | Cd-Os4 /Å | Cd-Os5 /Å | Cd-Os6 /Å | Cd-Place
/Å | Before
Na-Place/Å | After
Na-Place/Å |
|---|---|---|---|---|---|---|---|---|---|
| MMT(001)-H1 | 2.612 | 3.064 | 2.668 | 3.151 | 2.533 | 3.154 | 1.1 | 0.7 | 1.3 |
| MMT(010)-B4 | 2.209 | 2.210 | - | - | - | - | - | - | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Du, J.; Wang, Q.; Chen, J. Understanding Cd2+ Adsorption Mechanism on Montmorillonite Surfaces by Combining DFT and MD. Processes 2022, 10, 1381. https://doi.org/10.3390/pr10071381
Du J, Wang Q, Chen J. Understanding Cd2+ Adsorption Mechanism on Montmorillonite Surfaces by Combining DFT and MD. Processes. 2022; 10(7):1381. https://doi.org/10.3390/pr10071381
Chicago/Turabian StyleDu, Jia, Qinghe Wang, and Jun Chen. 2022. "Understanding Cd2+ Adsorption Mechanism on Montmorillonite Surfaces by Combining DFT and MD" Processes 10, no. 7: 1381. https://doi.org/10.3390/pr10071381
APA StyleDu, J., Wang, Q., & Chen, J. (2022). Understanding Cd2+ Adsorption Mechanism on Montmorillonite Surfaces by Combining DFT and MD. Processes, 10(7), 1381. https://doi.org/10.3390/pr10071381