
Highly Efficient Regioselective Acylation of Dihydromyricetin Catalyzed by Lipase in Nonaqueous Solvents




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Enzymatic Acylation of DHM
2.3. Procedure for HPLC Analysis
2.4. Purification of DHM Derivatives
2.5. Structural Identification of DHM Derivatives
2.6. Optimization of Reaction Conditions
2.7. Determination of Octanol–Water Partition Coefficient (Log P)
2.8. Inhibition of Lecithin Peroxidation
2.9. Statistical Analysis
3. Results and Discussion
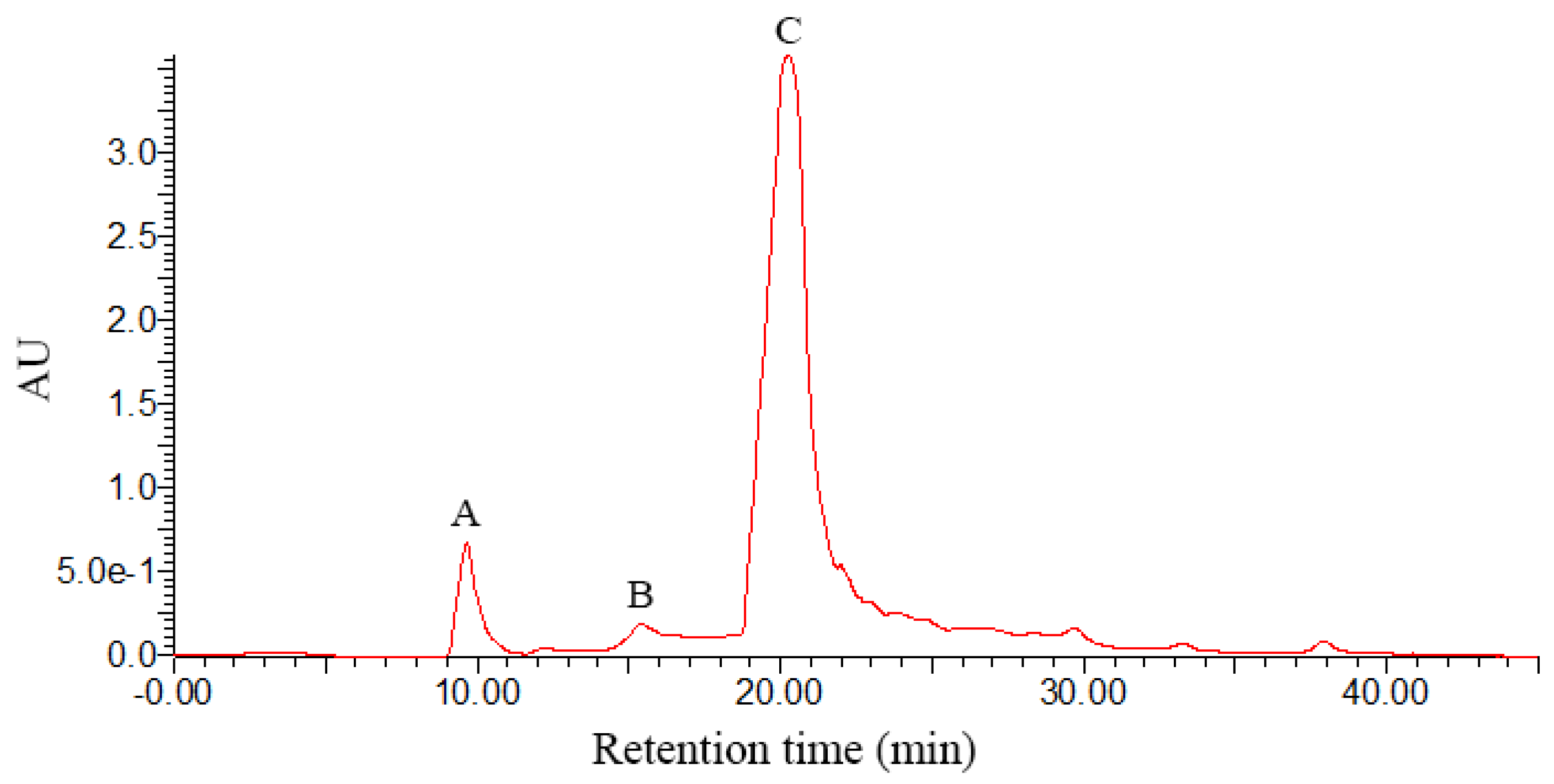
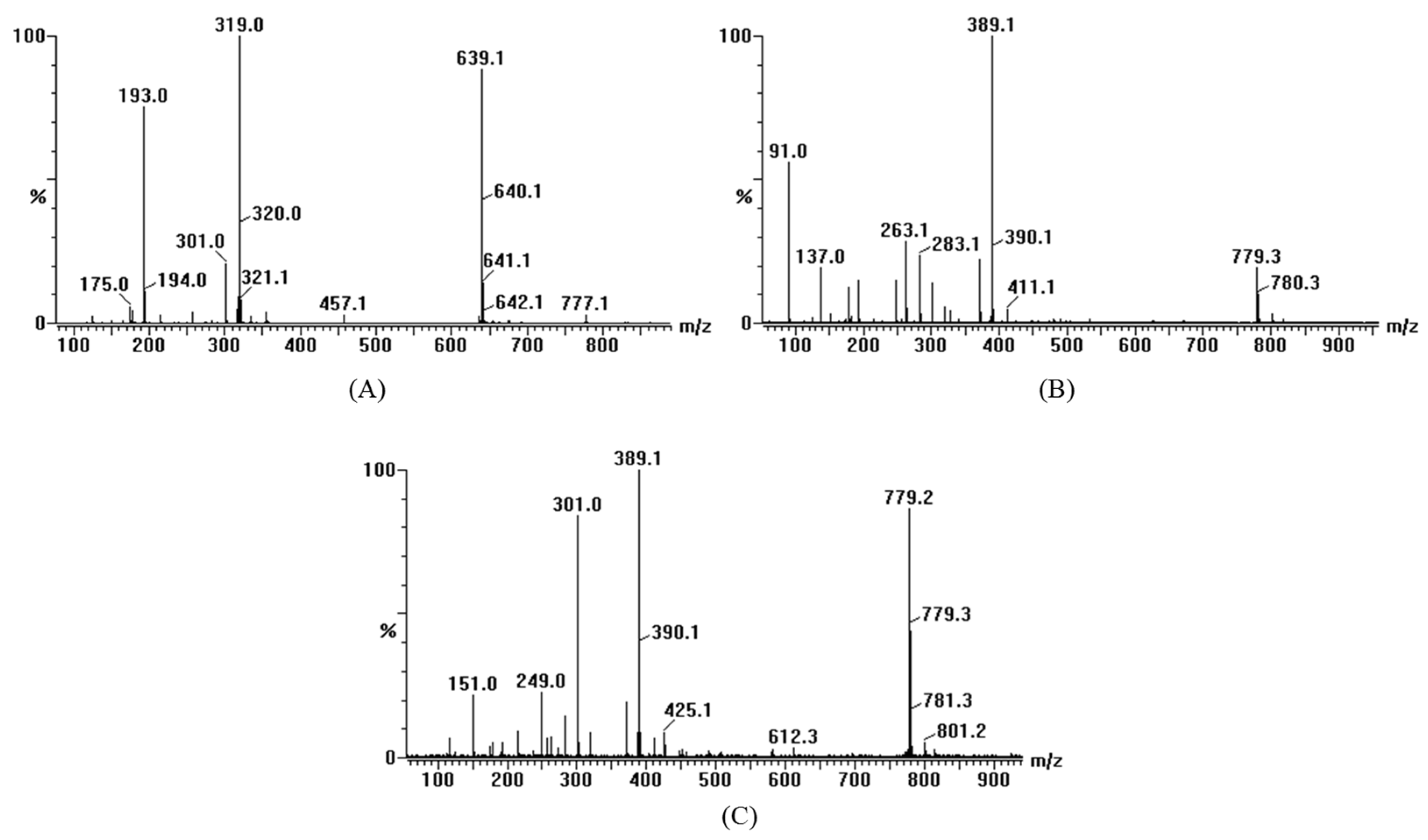
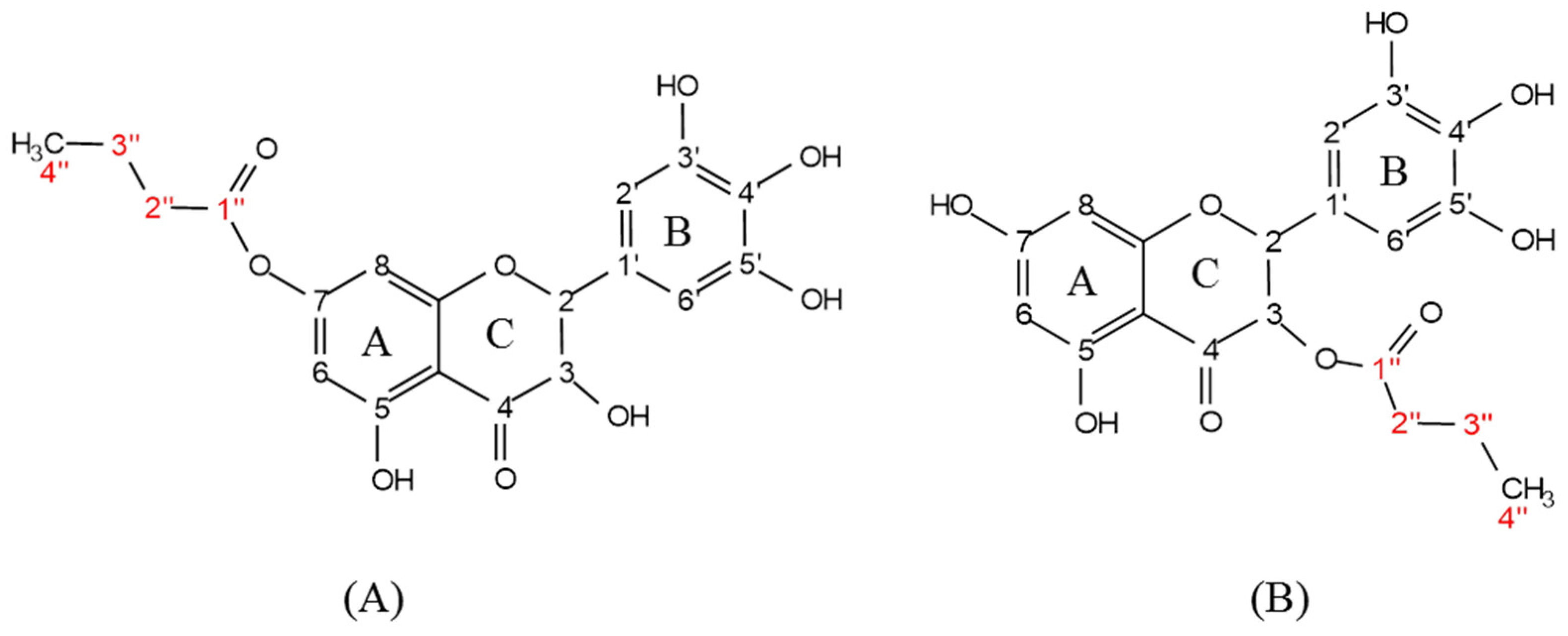
3.1. Structural Identification of DHM Derivatives
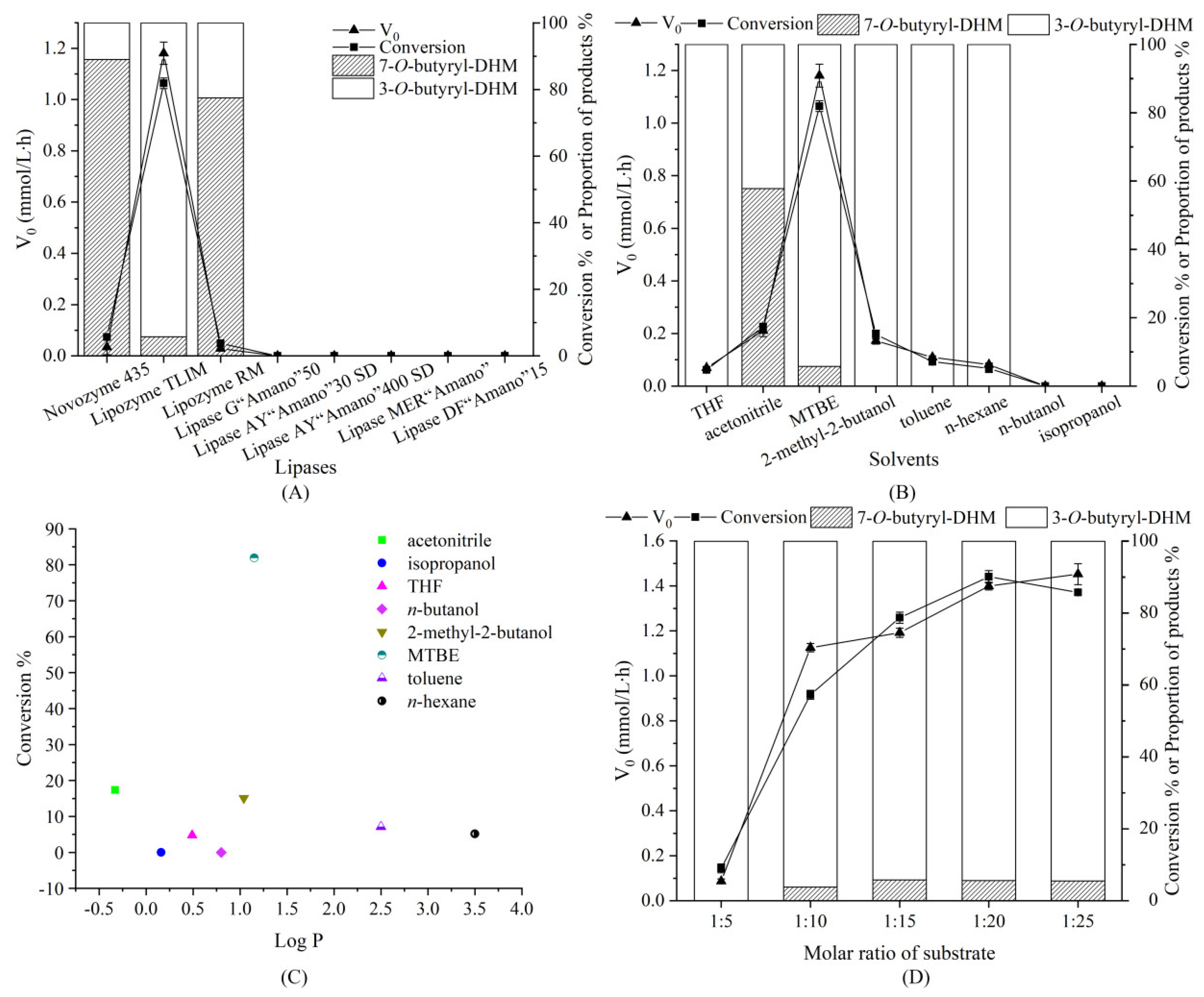
3.2. Effects of Lipase
3.3. Effect of Solvent System
3.4. Effect of Substrate Molar Ratio
3.5. Effect of Lipase Dosage
3.6. Effects of Reaction Temperature, Stirrer Speed, and Reaction Time
3.7. Effect of Acyl Donor
3.8. Determination of Octanol–Water Partition Coefficient (log P)
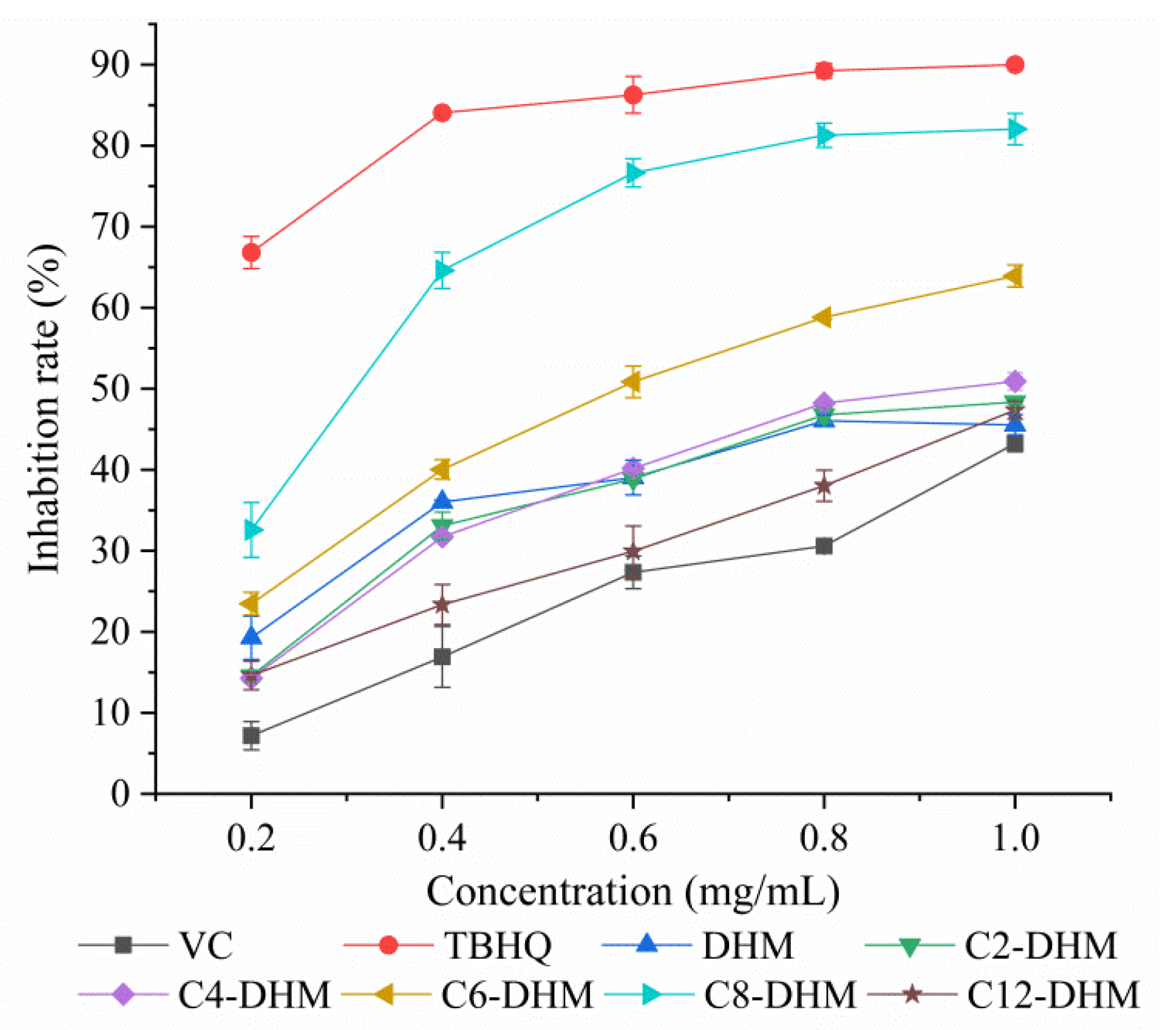
3.9. Inhibition of Lecithin Peroxidation
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Liu, D.; Mao, Y.Q.; Ding, L.J.; Zeng, X.A. Dihydromyricetin: A review on identification and quantification methods, biological activities, chemical stability, metabolism and approaches to enhance its bioavailability. Trends Food Sci. Technol. 2019, 91, 586–597. [Google Scholar] [CrossRef] [PubMed]
- Cao, S.L.; Deng, X.; Xu, P.; Huang, Z.X.; Zhou, J.; Li, X.H.; Zong, M.H.; Lou, W.Y. Highly efficient enzymatic acylation of dihydromyricetin by the immobilized lipase with deep eutectic solvents as cosolvent. J. Agric. Food Chem. 2017, 65, 2084–2088. [Google Scholar] [CrossRef] [PubMed]
- Tong, H.H.; Zhang, X.J.; Tan, L.F.; Jin, R.M.; Huang, S.L.; Li, X. Multitarget and promising role of dihydromyricetin in the treatment of metabolic diseases. Eur. J. Pharmacol. 2020, 870, 172888. [Google Scholar] [CrossRef]
- Liu, B.G.; Du, J.Q.; Zeng, J.; Chen, C.G.; Niu, S.Y. Characterization and antioxidant activity of dihydromyricetin–lecithin complex. Eur. Food Res. Technol. 2009, 230, 325–331. [Google Scholar] [CrossRef]
- Plaza, M.; Pozzo, T.; Liu, J.Y.; Gulshan Ara, K.Z.; Turner, C.; Karlsson, E.N. Substituent effects on in vitro antioxidizing properties, stability, and solubility in flavonoids. J. Agricutural Food Chem. 2014, 62, 3321–3333. [Google Scholar] [CrossRef] [PubMed]
- Saik, A.Y.H.; Lim, Y.Y.; Stanslas, J.; Choo, W.S. Lipase-catalyzed acylation of quercetin with cinnamic acid. Biocatal. Biotransform. 2016, 34, 33–43. [Google Scholar] [CrossRef]
- Chebil, L.; Antihoni, J.; Huweau, C.; Gerardin, C.; Engasser, J.M.; Ghoul, M. Enzymatic acylation of flavonoids: Effect of the nature of the substrate, origin of lipase, and operating conditions on conversion yield and regioselectivity. J. Agric. Food Chem. 2007, 55, 9496–9502. [Google Scholar] [CrossRef]
- Yu, M.; Liu, B.; Zhong, F.; Wan, Q.; Zhu, S.; Huang, D.; Li, Y. Interactions between caffeic acid and corn starch with varying amylose content and their effects on starch digestion. Food Hydrocoll. 2021, 114, 106544. [Google Scholar] [CrossRef]
- Chebil, L.; Humeau, C.; Falcimaigne, A.; Engasser, J.M.; Ghoul, M. Enzymatic acylation of flavonoids. Process Biochem. 2006, 41, 2237–2251. [Google Scholar] [CrossRef]
- Dong, W.X.; Chen, D.J.; Chen, Z.Y.; Sun, H.Y.; Xu, Z.M. Antioxidant capacity differences between the major flavonoids in cherry (Prunus pseudocerasus) in vitro and in vivo models. LWT Food Sci. Technol. 2021, 141, 110938. [Google Scholar] [CrossRef]
- Li, W.; Wu, H.; Liu, B.G.; Hou, X.D.; Wan, D.J.; Lou, W.Y.; Zhao, J. Highly efficient and regioselective synthesis of dihydromyricetin esters by immobilized lipase. J. Biotechnol. 2015, 199, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Meng, N.; Chen, S.W.; Li, Y. Study of acetylated EGCG synthesis by enzymatic transesterfication in orgnic media. Arab. J. Chem. 2020, 13, 8824–8834. [Google Scholar] [CrossRef]
- Wang, S.; Li, Y.; Meng, X.Y.; Chen, S.W.; Huang, D.J.; Xia, Y.M.; Zhu, S. Antioxidant activities of chlorogenic acid derivatives with different acyl donor chain lengths and their stabilities during in vitro simulated gastrointestinal digestion. Food Chem. 2021, 357, 129904. [Google Scholar] [CrossRef] [PubMed]
- Mavi, A.; Lawrence, G.D.; Kordali, S.; Yildirim, A.L.I. Inhibition of Iron-Fructose-Phosphate-Induced lipid peroxidation in lecithin liposome and linoleic acid emulsion systems by some edible plants. J. Food Biochem. 2011, 35, 833–844. [Google Scholar] [CrossRef]
- Yadav, G.; Pawar, S.V. Synergism between microwave irradiation and enzyme catalysis in transesterification of ethyl-3-phenylpropanoate with n-butanol. Bioresour. Technol. 2012, 109, 1–6. [Google Scholar] [CrossRef]
- Zare, M.; Golmakani, M.T.; Niakousari, M. Lipase synthesis of isoamyl acetate using different acyl donors: Comparison of novel esterification techniques. LWT Food Sci. Technol. 2019, 101, 214–219. [Google Scholar] [CrossRef]
- Hazarika, S.; Goswami, P.; Dutta, N.N. Lipase catalysed transesterification of 2-O-benzylglycerol with vinyl acetate: Solvent effect. Chem. Eng. J. 2003, 94, 1–10. [Google Scholar] [CrossRef]
- Lu, J.; Nie, K.; Wang, F.; Tan, T.W. Immobilized lipase Candida sp. 99–125 catalyzed methanolysis of glycerol trioleate: Solvent effect. Bioresour. Technol. 2008, 99, 6070–6074. [Google Scholar] [CrossRef]
- Laane, C.; Boeren, S.; Vos, K.; Veeger, C. Rules for optimization of biocatalysis in organic solvents. Biotechnol. Bioeng. 1987, 30, 81–87. [Google Scholar] [CrossRef]
- Yadav, G.; Hude, M.P.; Talpade, A.D. Microwave assisted process intensification of lipase catalyzed transesterification of 1,2 propanediol with dimethyl carbonate for the green synthesis of propylene carbonate: Novelties of kinetics and mechanism of consecutive reactions. Chem. Eng. J. 2015, 281, 199–208. [Google Scholar] [CrossRef]
- Jahangiri, A.; Moller, A.H.; Danielsen, M.; Madsen, B.; Joernsgaard, B.; Vaerbak, S.; Adlercreutz, P.; Dalsgaard, T.K. Hydrophilization of bixin by lipase-catalyzed transesterification with sorbitol. Food Chem. 2018, 268, 203–209. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Wang, S.; Chen, S.W.; Xia, Y.M.; Li, Y. Lipase-catalyzed highly regioselective synthesis of acylated chlorogenic acid. Food Biosci. 2020, 37, 100706. [Google Scholar] [CrossRef]
- Cao, Z.D.; Zhang, R.K.; Hu, X.R.; Sha, J.; Jiang, G.L.; Li, Y.; Li, T.; Ren, B.Z. Thermodynamic modelling, Hansen solubility parameter and solvent effect of oxaprozin in thirteen pure solvents at different temperatures. J. Chem. Thermodyn. 2020, 151, 106239. [Google Scholar] [CrossRef]
- Zheng, M.; Chen, J.; Chen, G.Q.; Farajtabar, A.; Zhao, H. Solubility modelling and solvent effect for domperidone in twelve green solvents. J. Mol. Liq. 2018, 261, 50–56. [Google Scholar] [CrossRef]
- Lorentz, C.; Dulac, A.; Pencreac’h, G.; Ergan, F.; Richomme, P.; Soultani-Vigneron, S. Lipase-catalyzed synthesis of two new antioxidants: 4-O- and 3-O-palmitoyl chlorogenic acids. Biotechnol. Lett. 2010, 32, 1955–1960. [Google Scholar] [CrossRef]
- Bhavsar, K.V.; Yadav, G.D. Process intensification by microwave irradiation in immobilized-lipase catalysis in solvent-free synthesis of ethyl valerate. Mol. Catal. 2018, 461, 34–39. [Google Scholar] [CrossRef]
- Wang, Z.; Zhang, Y.X.; Zheng, L.; Cui, X.Y.; Huang, H.; Geng, X.; Xie, X.N. Regioselective acylation of resveratrol catalyzed by lipase under microwave. Green Chem. Lett. Rev. 2018, 11, 312–317. [Google Scholar] [CrossRef] [Green Version]
- Ziaullah, K.S.; Rupasinghe, H.P. Sonochemical enzyme-catalyzed regioselective acylation of flavonoid glycosides. Bioorgnic Chem. 2016, 65, 17–25. [Google Scholar] [CrossRef]
- Lue, B.M.; Guo, Z.; Xu, X.B. Effect of room temperature ionic liquid structure on the enzymatic acylation of flavonoids. Process Biochem. 2010, 45, 1375–1382. [Google Scholar] [CrossRef]
- De Souza, R.O.; Antunes, O.A.; Kroutil, W.; Kappe, C.O. Kinetic resolution of rac-1-phenylethanol with immobilized lipases: A critical comparison of microwave and conventional heating protocols. J. Org. Chem. 2009, 74, 6157–6162. [Google Scholar] [CrossRef]
- Ardhaoui, M.; Falcimaigne, A.; Ognier, S.; Engasser, J.M.; Moussou, P.; Pauly, G.; Ghoul, M. Effect of acyl donor chain length and substitutions pattern on the enzymatic acylation of flavonoids. J. Biotechnol. 2004, 110, 265–271. [Google Scholar] [CrossRef] [PubMed]
- Deng, X.; Cao, S.L.; LI, N.; Wu, H.; Smith, T.J.; Zong, M.H.; Lou, W.Y. A magnetic biocatalyst based on mussel-inspired polydopamine and its acylation of dihydromyricetin. Chin. J. Catal. 2016, 37, 584–595. [Google Scholar] [CrossRef]
- Enaud, E.; Humeau, C.; Piffaut, B.; Girardin, M. Enzymatic synthesis of new aromatic esters of phloridzin. J. Mol. Catal. B Enzym. 2004, 27, 1–6. [Google Scholar] [CrossRef]
- Bayrasy, C.; Chabi, B.; Laguerre, M.; Lecomte, J.; Jublanc, E.; Villeneuve, P.; Wrutniak-Cabello, C.; Cabello, G. Boosting antioxidants by lipophilization: A strategy to increase cell uptake and target mitochondria. Pharm. Res. 2013, 30, 1979–1989. [Google Scholar] [CrossRef] [PubMed]
- Oh, W.Y.; Shahidi, F. Antioxidant activity of resveratrol ester derivatives in food and biological model systems. Food Chem. 2018, 261, 267–273. [Google Scholar] [CrossRef]
- Laguerre, M.; Lopez Giraldo, L.J.; Lecomte, J.; Figueroa-Espinoza, M.C.; Barea, B.; Weiss, J.; Decker, E.A.; Villeneuve, P. Relationship between hydrophobicity and antioxidant ability of “phenolipids” in emulsion: A parabolic effect of the chain length of rosmarinate esters. J. Agricutural Food Chem. 2010, 58, 2869–2876. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhu, S.; Du, B.; Huang, D.; Xia, Y.; Chen, S.; Li, Y. Highly Efficient Regioselective Acylation of Dihydromyricetin Catalyzed by Lipase in Nonaqueous Solvents. Processes 2022, 10, 1368. https://doi.org/10.3390/pr10071368
Zhu S, Du B, Huang D, Xia Y, Chen S, Li Y. Highly Efficient Regioselective Acylation of Dihydromyricetin Catalyzed by Lipase in Nonaqueous Solvents. Processes. 2022; 10(7):1368. https://doi.org/10.3390/pr10071368
Chicago/Turabian StyleZhu, Song, Baoshuang Du, Dejian Huang, Yongmei Xia, Shangwei Chen, and Yue Li. 2022. "Highly Efficient Regioselective Acylation of Dihydromyricetin Catalyzed by Lipase in Nonaqueous Solvents" Processes 10, no. 7: 1368. https://doi.org/10.3390/pr10071368
APA StyleZhu, S., Du, B., Huang, D., Xia, Y., Chen, S., & Li, Y. (2022). Highly Efficient Regioselective Acylation of Dihydromyricetin Catalyzed by Lipase in Nonaqueous Solvents. Processes, 10(7), 1368. https://doi.org/10.3390/pr10071368