
Explicit-pH Coarse-Grained Molecular Dynamics Simulations Enable Insights into Restructuring of Intestinal Colloidal Aggregates with Permeation Enhancers

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
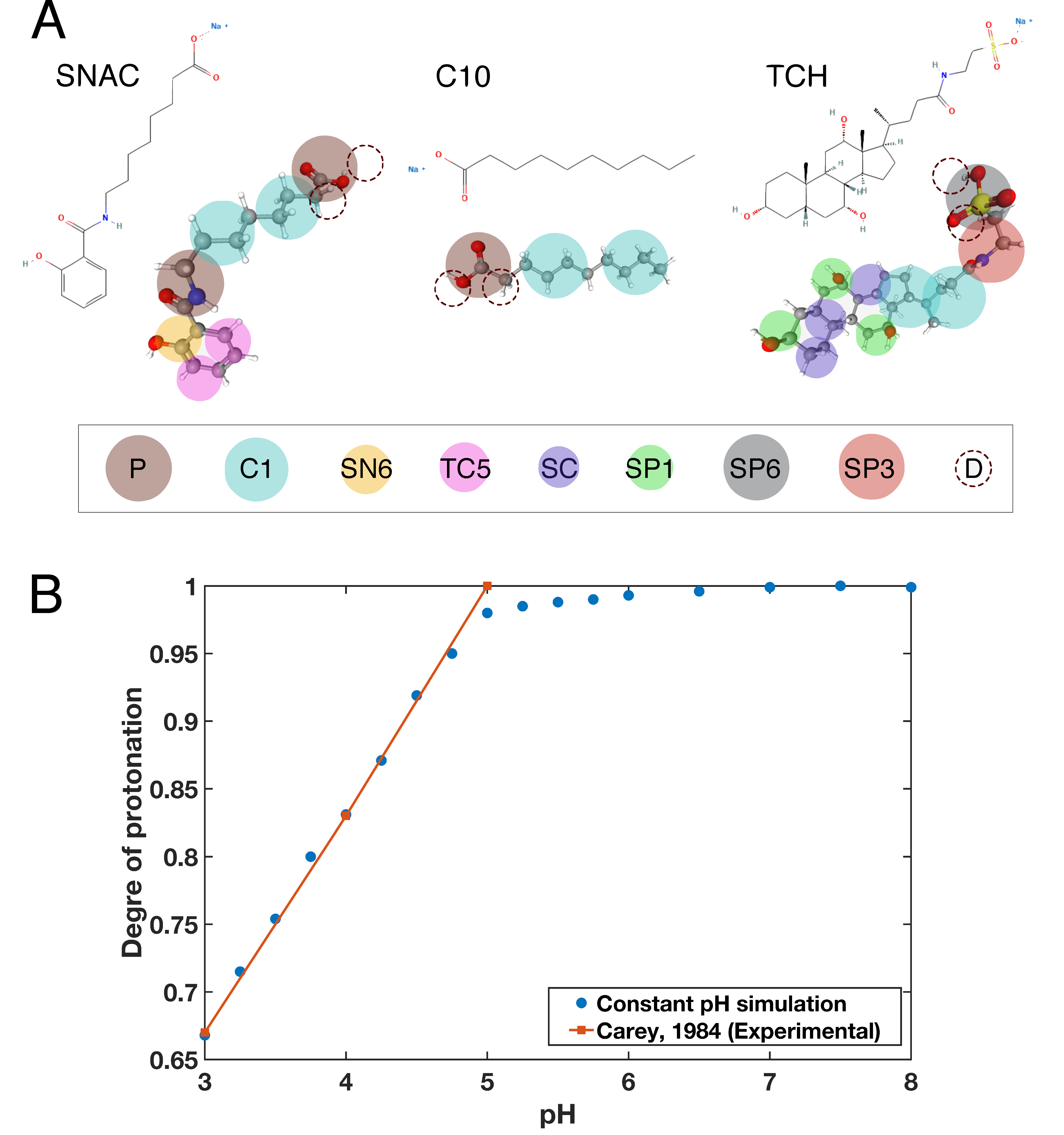
2.1. Parameterization of Taurocholate
2.2. Titratable Martini Model
2.3. Parameterization of the Permeation Enhancers and Taurocholate Titratable Beads
2.4. Simulation Setups
2.5. Analysis
3. Results and Discussion
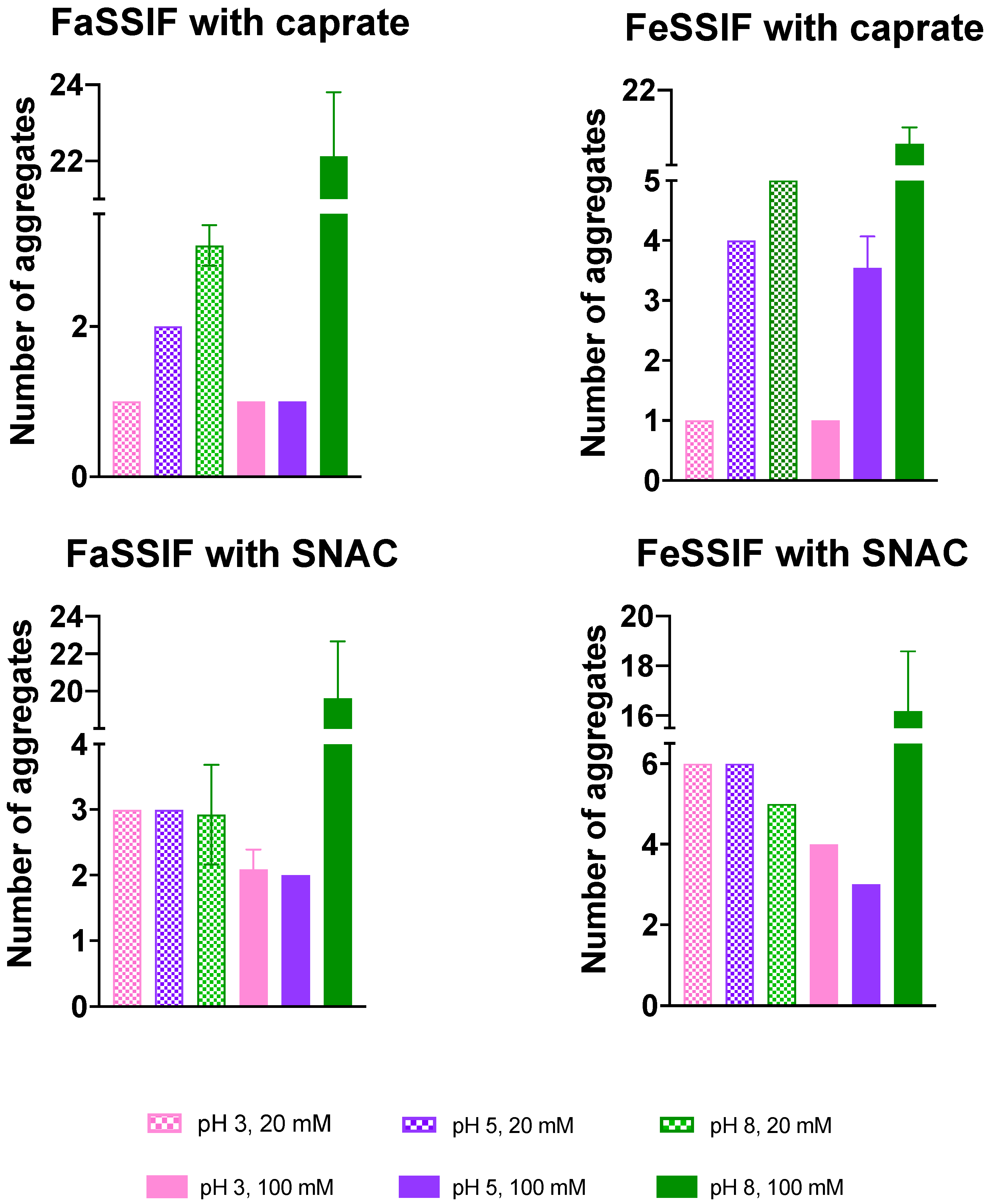
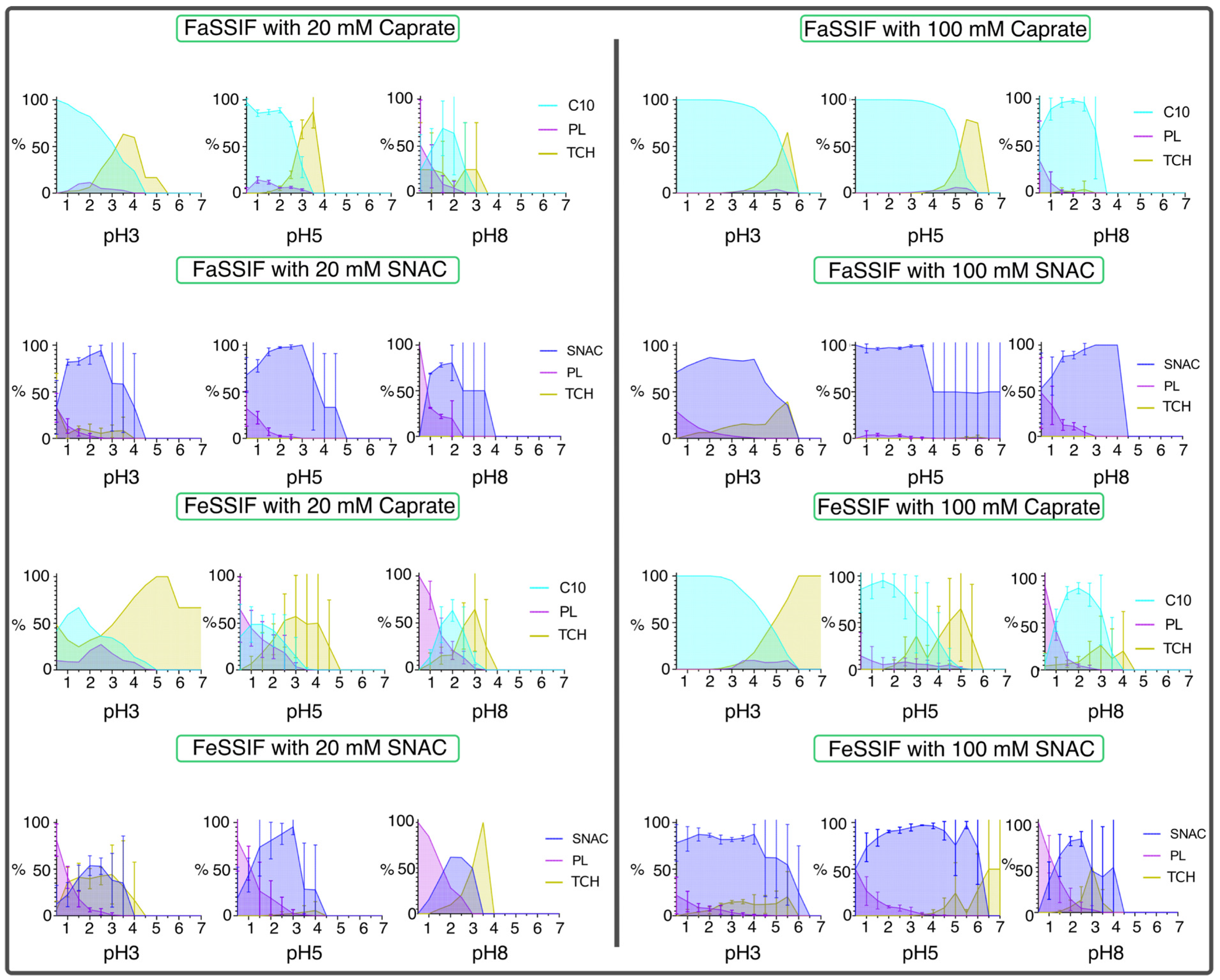
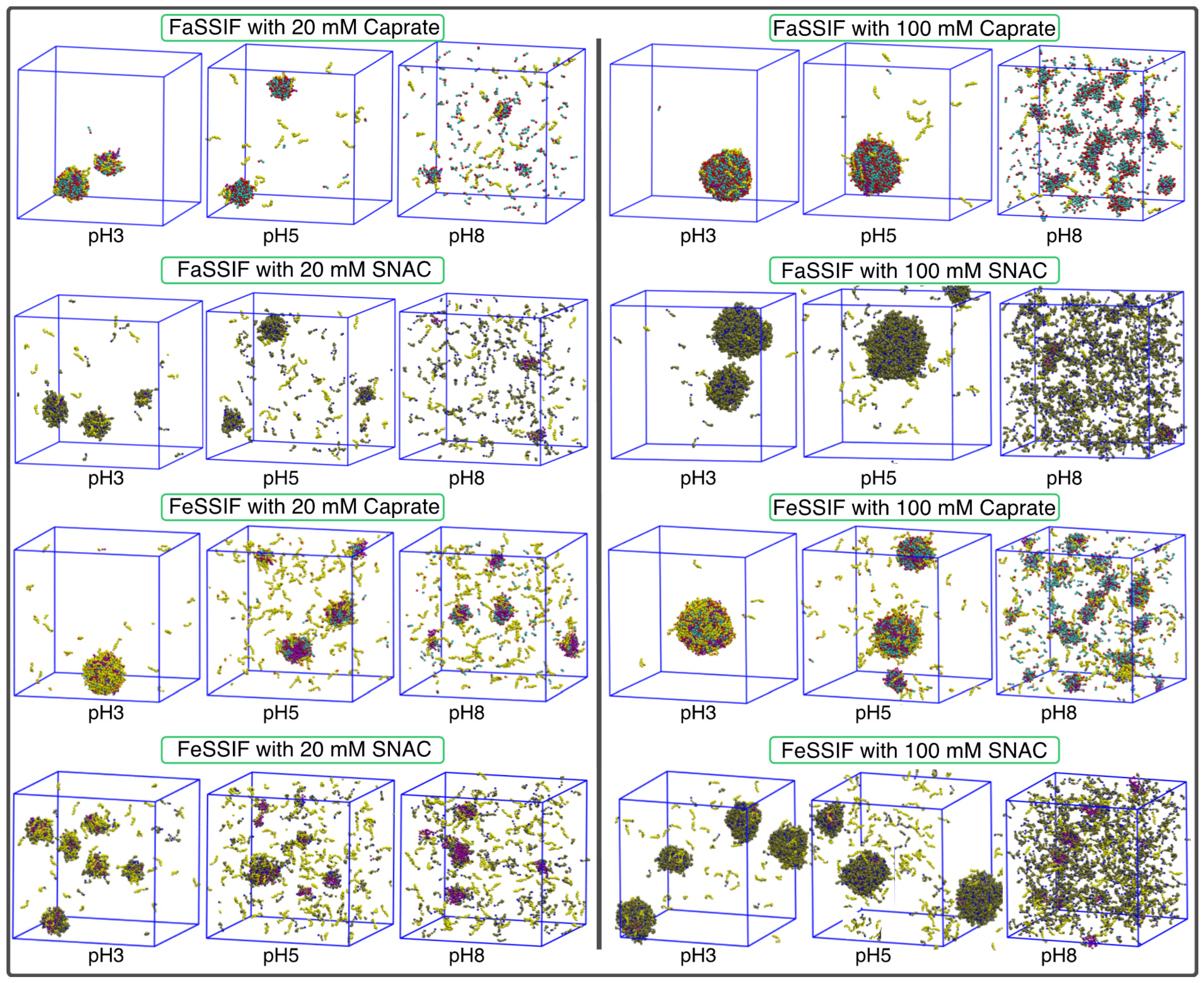
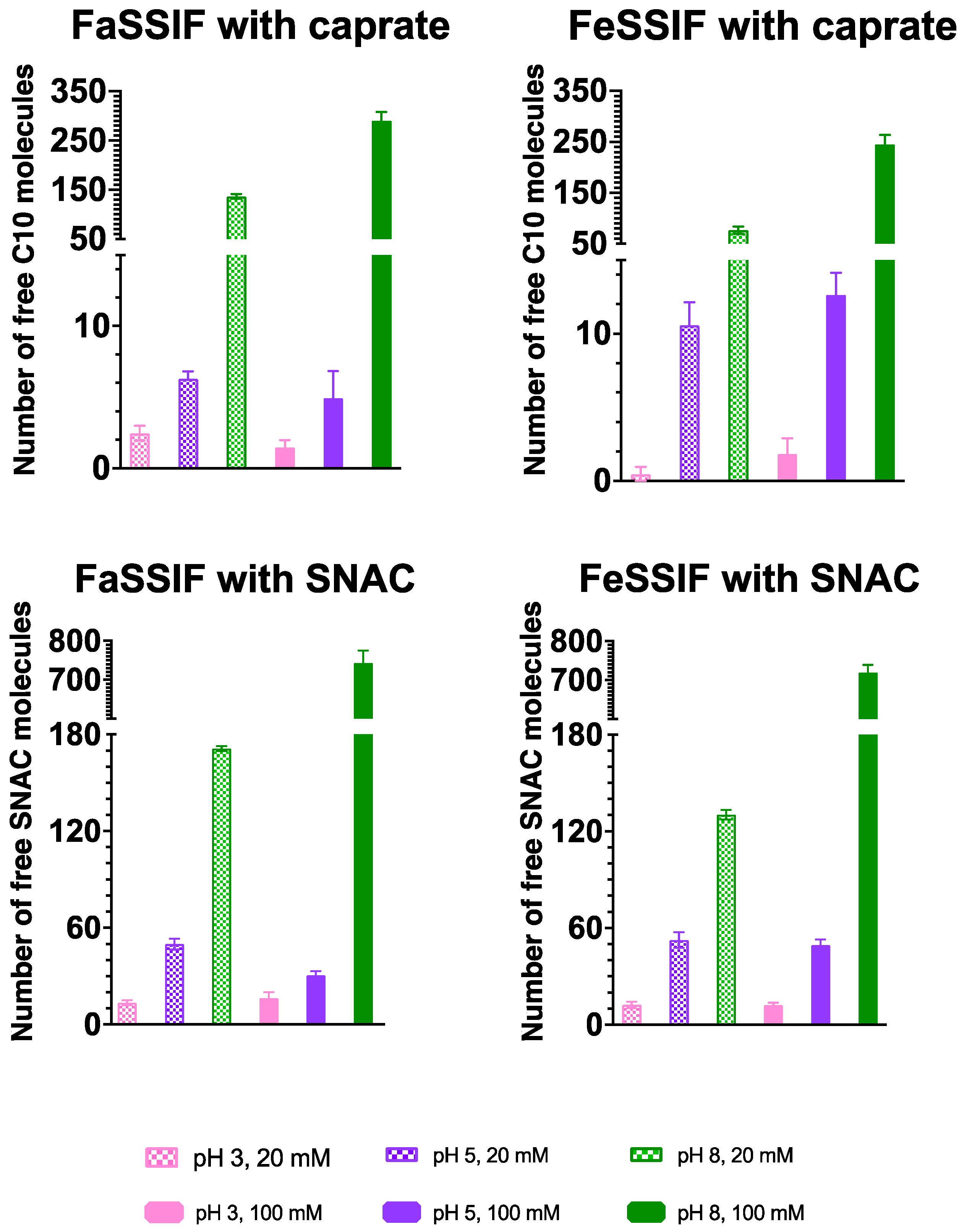
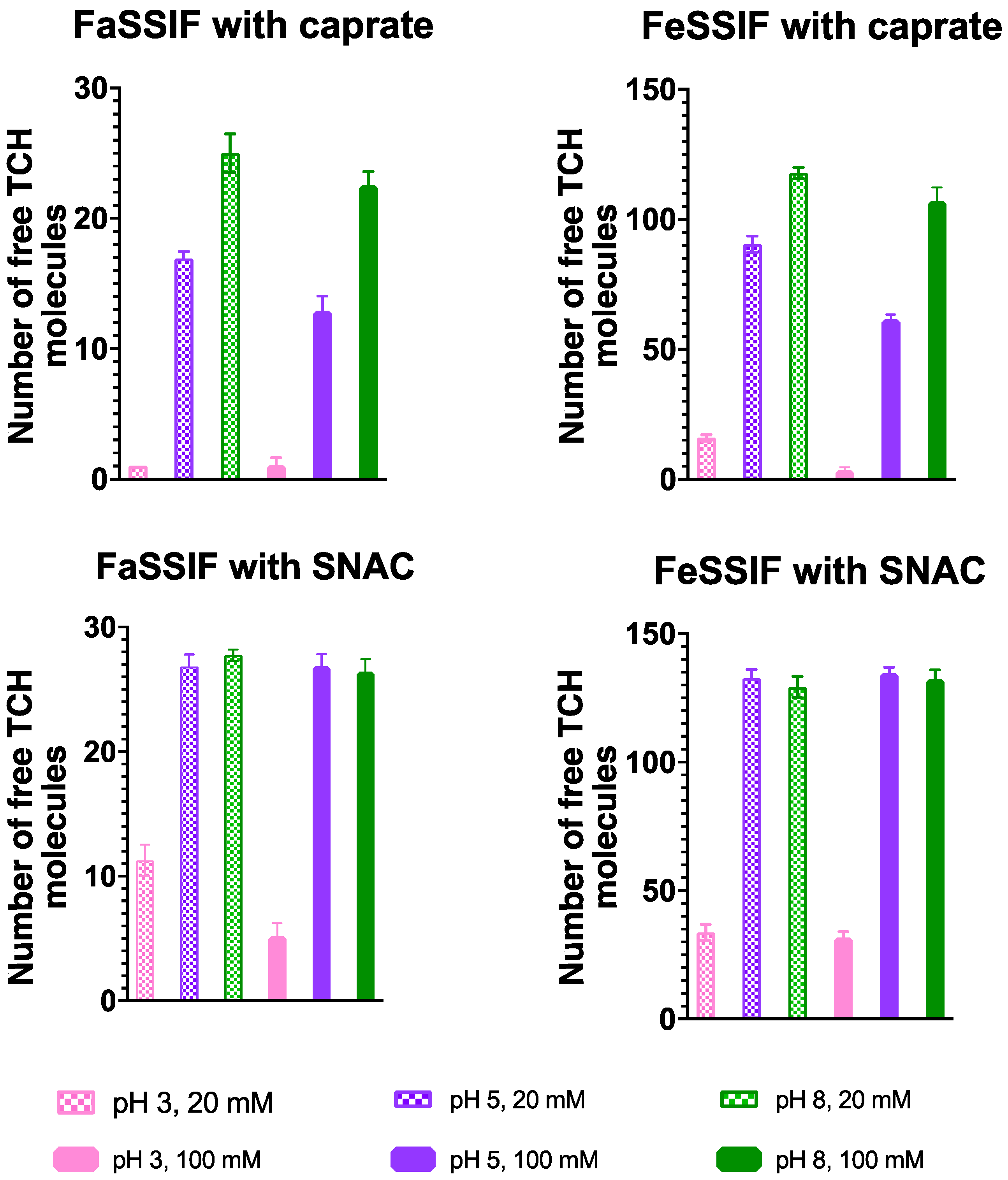
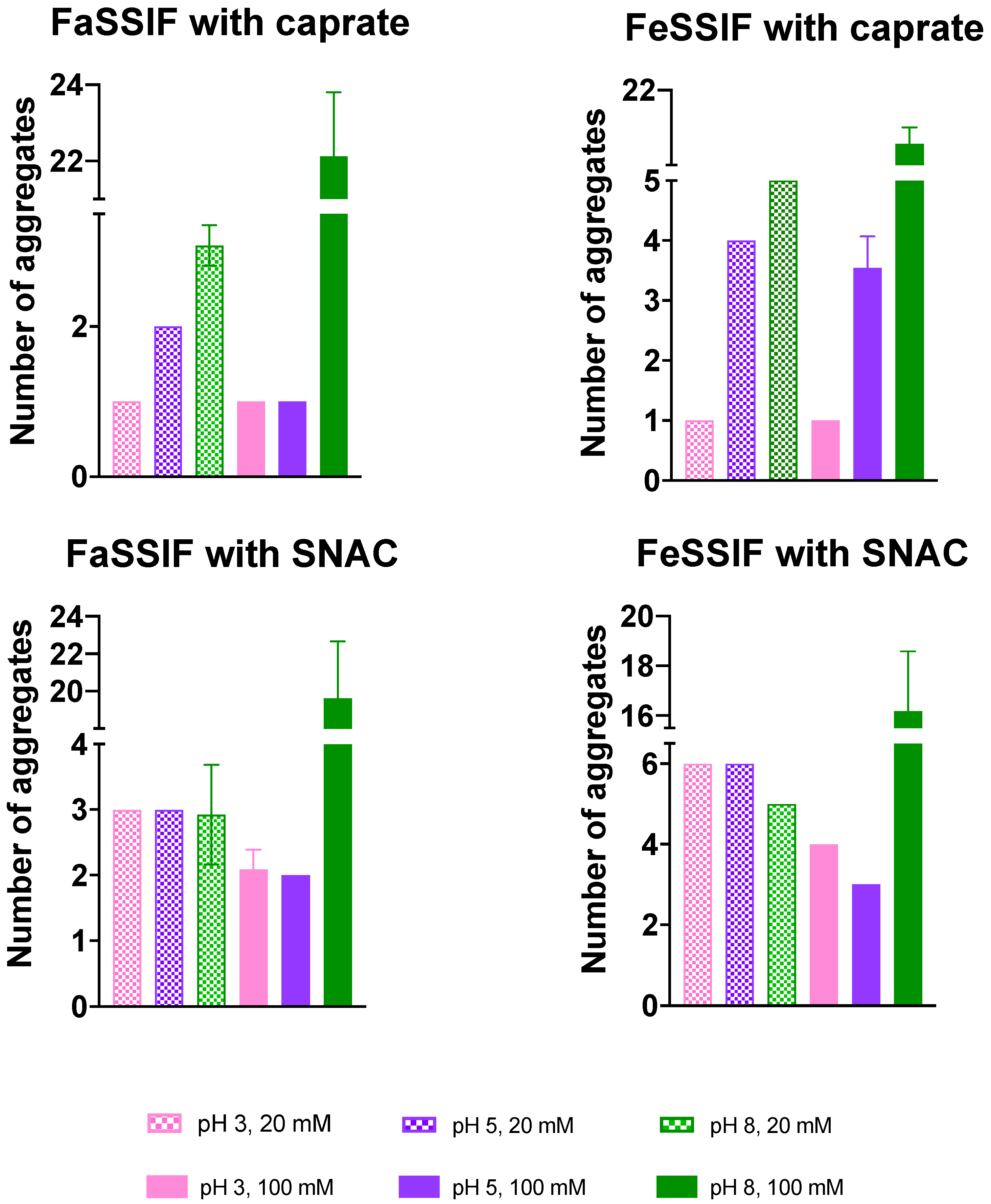
3.1. pH-Dependent Aggregation Dynamics
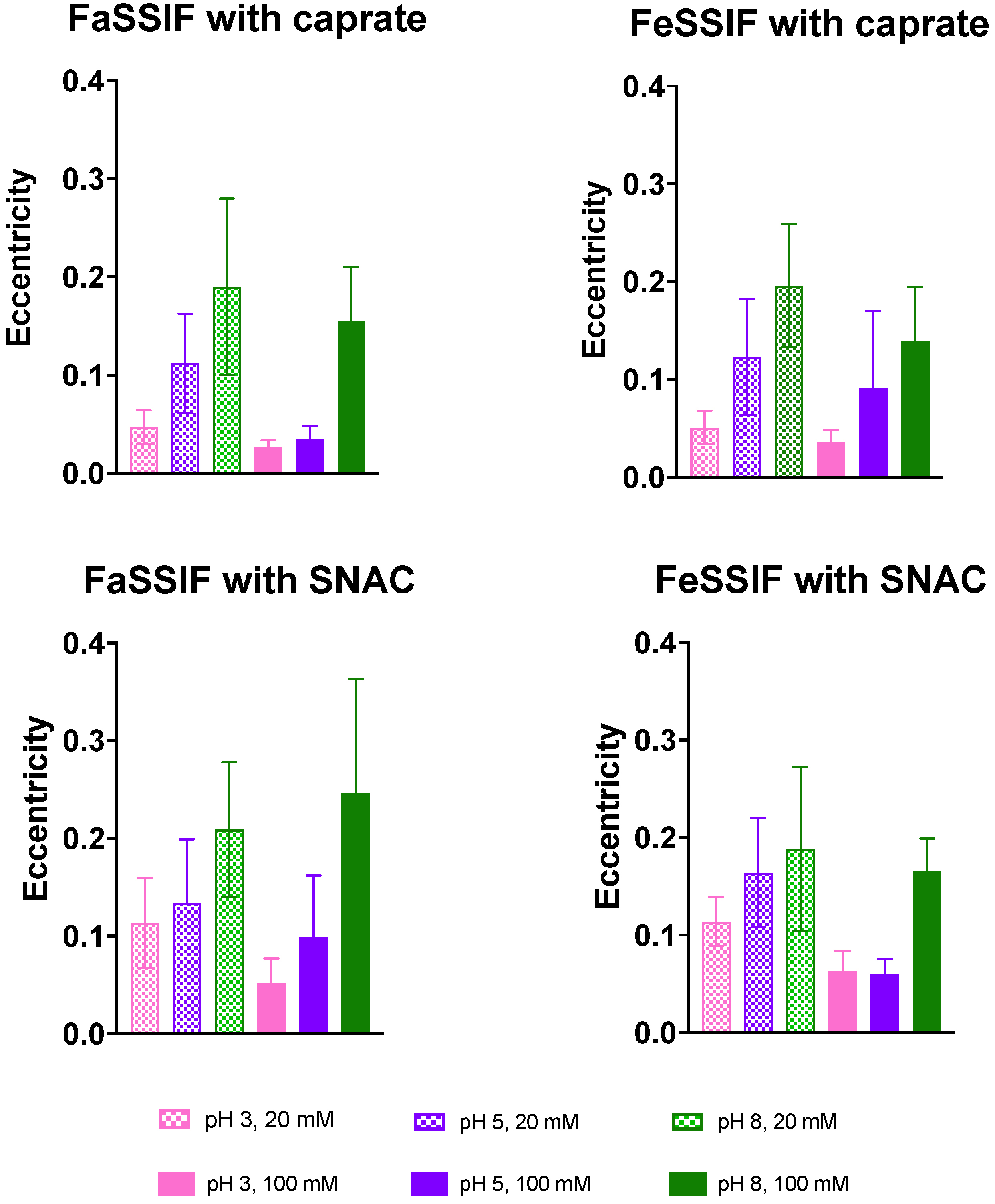
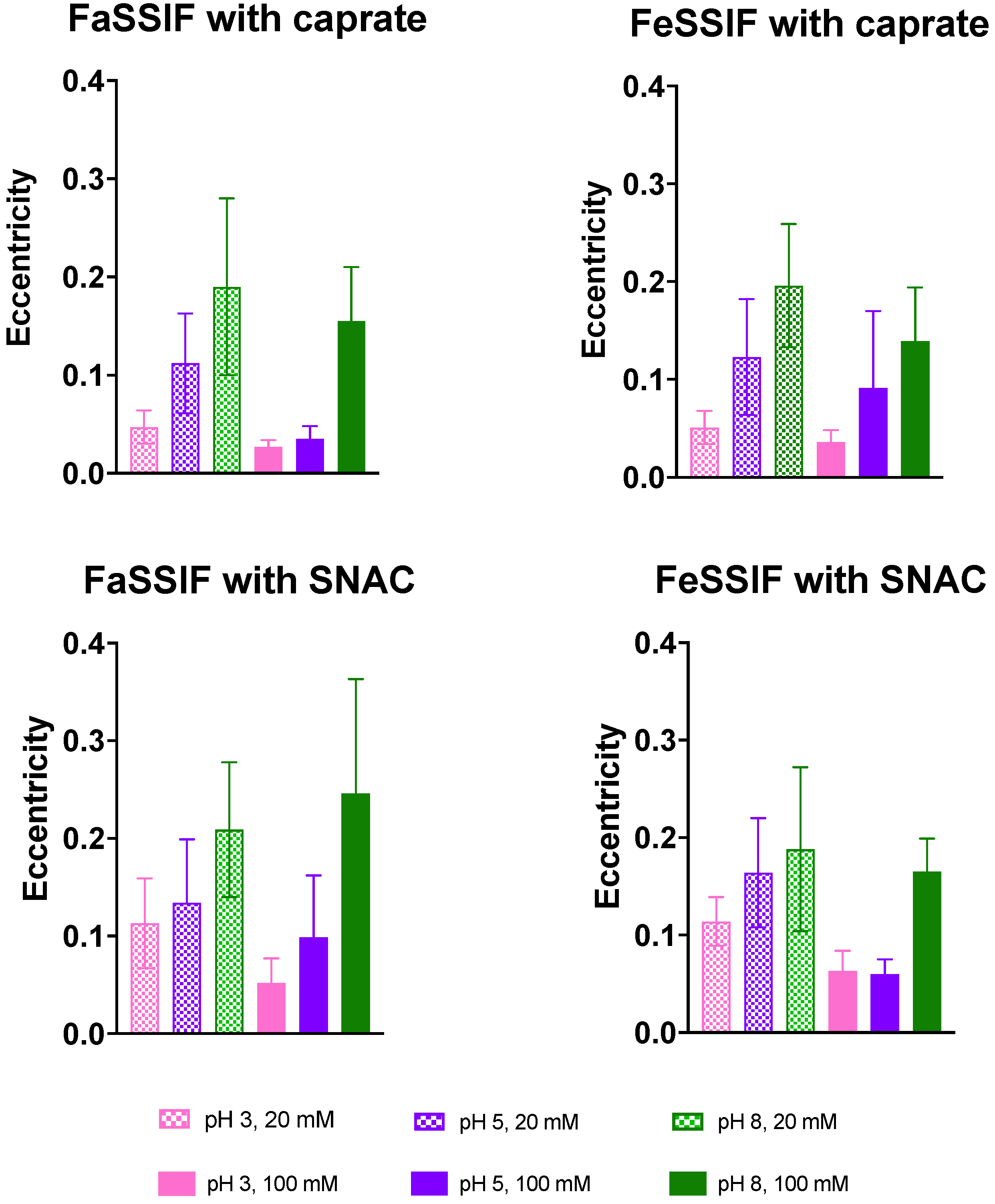
3.2. Impact of pH on Aggregate Shapes
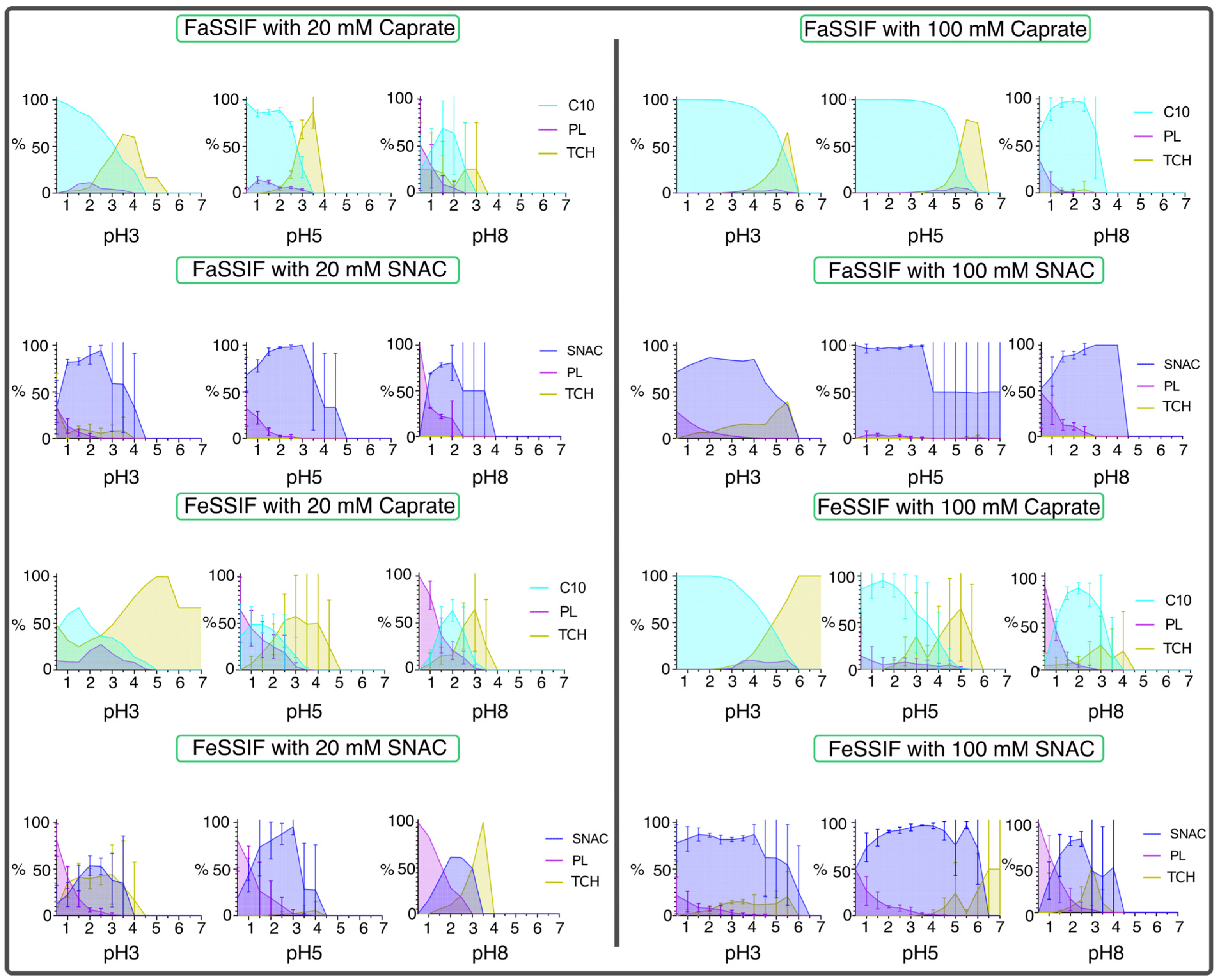
3.3. Impact of pH on Aggregates’ Structural Characteristics
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Morrow, T.; Felcone, L.H. Defining the difference: What makes biologics unique. Biotechnol. Healthc. 2004, 1, 24. [Google Scholar] [PubMed]
- Maher, S.; Mrsny, R.J.; Brayden, D.J. Intestinal permeation enhancers for oral peptide delivery. Adv. Drug Deliv. Rev. 2016, 106, 277–319. [Google Scholar] [CrossRef]
- Danielsen, E.M. Intestinal permeation enhancers: Lessons learned from studies using an organ culture model. Biochim. Biophys. Acta 2021, 1863, 183474. [Google Scholar] [CrossRef] [PubMed]
- Leonard, T.W.; Lynch, J.; McKenna, M.J.; Brayden, D.J. Promoting absorption of drugs in humans using medium-chain fatty acid-based solid dosage forms: GIPETTM. Expert Opin. Drug Deliv. 2006, 3, 685–692. [Google Scholar] [CrossRef]
- Karsdal, M.A.; Henriksen, K.; Bay-Jensen, A.C.; Molloy, B.; Arnold, M.; John, M.R.; Byrjalsen, I.; Azria, M.; Riis, B.J.; Qvist, P.; et al. Lessons learned from the development of oral calcitonin: The first tablet formulation of a protein in phase III clinical trials. J. Clin. Pharmacol. 2011, 51, 460–471. [Google Scholar] [CrossRef]
- McCartney, F.; Gleeson, J.P.; Brayden, D.J. Safety concerns over the use of intestinal permeation enhancers: A mini-review. Tissue Barriers 2016, 4, e1176822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walsh, E.G.; Adamczyk, B.E.; Chalasani, K.B.; Maher, S.; O’Toole, E.B.; Fox, J.S.; Leonard, T.W.; Brayden, D.J. Oral delivery of macromolecules: Rationale underpinning Gastrointestinal Permeation Enhancement Technology (GIPET®). Ther. Deliv. 2011, 2, 1595–1610. [Google Scholar] [CrossRef]
- Aungst, B.J. Absorption Enhancers: Applications and Advances. AAPS J. 2012, 14, 10–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halberg, I.B.; Lyby, K.; Wassermann, K.; Heise, T.; Zijlstra, E.; Plum-Mörschel, L. Efficacy and safety of oral basal insulin versus subcutaneous insulin glargine in type 2 diabetes: A randomised, double-blind, phase 2 trial. Lancet Diabetes Endocrinol. 2019, 7, 179–188. [Google Scholar] [CrossRef]
- Tillman, L.G.; Geary, R.S.; Hardee, G.E. Oral delivery of antisense oligonucleotides in man. J. Pharm. Sci. 2008, 97, 225–236. [Google Scholar] [CrossRef]
- Tuvia, S.; Pelled, D.; Marom, K.; Salama, P.; Levin-Arama, M.; Karmeli, I.; Idelson, G.H.; Landau, I.; Mamluk, R. A novel suspension formulation enhances intestinal absorption of macromolecules via transient and reversible transport mechanisms. Pharm. Res. 2014, 31, 2010–2021. [Google Scholar] [CrossRef] [Green Version]
- Buckley, S.T.; Bækdal, T.A.; Vegge, A.; Maarbjerg, S.J.; Pyke, C.; Ahnfelt-Rønne, J.; Madsen, K.G.; Schéele, S.G.; Alanentalo, T.; Kirk, R.K.; et al. Transcellular stomach absorption of a derivatized glucagon-like peptide-1 receptor agonist. Sci. Transl. Med. 2021, 32, 115942. [Google Scholar] [CrossRef]
- Roos, C.; Dahlgren, D.; Sjögren, E.; Sjöblom, M.; Hedeland, M.; Lennernäs, H. Effects of absorption-modifying excipients on jejunal drug absorption in simulated fasted and fed luminal conditions. Eur. J. Pharm. Biopharm. 2019, 142, 387–395. [Google Scholar] [CrossRef]
- Gradauer, K.; Nishiumi, A.; Unrinin, K.; Higashino, H.; Kataoka, M.; Pedersen, B.L.; Buckley, S.T.; Yamashita, S. Interaction with Mixed Micelles in the Intestine Attenuates the Permeation Enhancing Potential of Alkyl-Maltosides. Mol. Pharm. 2015, 12, 2245–2253. [Google Scholar] [CrossRef] [PubMed]
- Clulow, A.J.; Parrow, A.; Hawley, A.; Khan, J.; Pham, A.C.; Larsson, P.; Bergström, C.A.S.; Boyd, B.J. Characterization of Solubilizing Nanoaggregates Present in Different Versions of Simulated Intestinal Fluid. J. Phys. Chem. B 2017, 121, 10869–10881. [Google Scholar] [CrossRef] [PubMed]
- Hossain, S.; Joyce, P.; Parrow, A.; Jõemetsa, S.; Höök, F.; Larsson, P.; Bergström, C.A.S. Influence of bile composition on membrane incorporation of transient permeability enhancers. Mol. Pharm. 2020, 17, 4226–4240. [Google Scholar] [CrossRef]
- Yoon, B.K.; Jackman, J.A.; Kim, M.C.; Cho, N.J. Spectrum of Membrane Morphological Responses to Antibacterial Fatty Acids and Related Surfactants. Langmuir 2015, 31, 10223–10232. [Google Scholar] [CrossRef]
- Flynn, K.R.; Martin, L.L.; Ackland, M.L.; Torriero, A.A.J. Real-Time Quartz Crystal Microbalance Monitoring of Free Docosahexaenoic Acid Interactions with Supported Lipid Bilayers. Langmuir 2016, 32, 11717–11727. [Google Scholar] [CrossRef] [PubMed]
- Joyce, P.; Jõemetsa, S.; Isaksson, S.; Hossain, S.; Larsson, P.; Bergström, C.; Höök, F. Investigating drug permeation across a lipid membrane supported on mesoporous silica. Angew. Chemie 2020, 133, 2097–2101. [Google Scholar] [CrossRef]
- Voth, G.A. The computer simulation of proton transport in biomolecular systems. Front Biosci. 2003, 8, 1384–1397. [Google Scholar] [CrossRef] [Green Version]
- Tepper, H.L.; Voth, G.A. Protons may leak through pure lipid bilayers via a concerted mechanism. Biophys. J. 2005, 88, 3095–3108. [Google Scholar] [CrossRef] [Green Version]
- Baptista, A.M.; Teixeira, V.H.; Soares, C.M. Constant-p H molecular dynamics using stochastic titration. J. Chem. Phys. 2002, 117, 4184–4200. [Google Scholar] [CrossRef]
- Mongan, J.; Case, D.A.; McCammon, J.A. Constant pH molecular dynamics in generalized Born implicit solvent. J. Comput. Chem. 2004, 25, 2038–2048. [Google Scholar] [CrossRef]
- Lee, M.S.; Salsbury, F.R., Jr.; Brooks, C.L., III. Constant-pH molecular dynamics using continuous titration coordinates. Proteins Struct. Funct. Bioinforma. 2004, 56, 738–752. [Google Scholar] [CrossRef] [PubMed]
- Donnini, S.; Tegeler, F.; Groenhof, G.; Grubmüller, H. Constant pH molecular dynamics in explicit solvent with λ-dynamics. J. Chem. Theory Comput. 2011, 7, 1962–1978. [Google Scholar] [CrossRef]
- Grünewald, F.; Souza, P.C.T.; Abdizadeh, H.; Barnoud, J.; De Vries, A.H.; Marrink, S.J. Titratable Martini model for constant pH simulations. J. Chem. Phys. 2020, 153, 024118. [Google Scholar] [CrossRef]
- Souza, P.C.T.; Alessandri, R.; Barnoud, J.; Thallmair, S.; Faustino, I.; Grünewald, F.; Patmanidis, I.; Abdizadeh, H.; Bruininks, B.M.H.; Wassenaar, T.A.; et al. Martini 3: A general purpose force field for coarse-grained molecular dynamics. Nat. Methods 2021, 18, 382–388. [Google Scholar] [CrossRef] [PubMed]
- Holmboe, M.; Larsson, P.; Anwar, J.; Bergström, C.A.S. Partitioning into Colloidal Structures of Fasted State Intestinal Fluid Studied by Molecular Dynamics Simulations. Langmuir 2016, 32, 12732–12740. [Google Scholar] [CrossRef]
- Carey, M.C. Bile Acids and Bile Salts: Ionization and Solubility Properties. Hepatology 1984, 4, 66S–71S. [Google Scholar] [CrossRef]
- Roda, A.; Grigolo, B.; Minutello, A.; Pellicciari, R.; Natalini, B. Physicochemical and biological properties of natural and synthetic C-22 and C-23 hydroxylated bile acids. J. Lipid Res. 1990, 31, 289–298. [Google Scholar] [CrossRef]
- Martini 3.0. Available online: http://cgmartini.nl/index.php/martini-3-0 (accessed on 23 November 2021).
- Maher, S.; Leonard, T.W.; Jacobsen, J.; Brayden, D.J. Safety and efficacy of sodium caprate in promoting oral drug absorption: From in vitro to the clinic. Adv. Drug Deliv. Rev. 2009, 61, 1427–1449. [Google Scholar] [CrossRef]
- Deng, L.; Dong, H.; Dong, A.; Zhang, J. A strategy for oral chemotherapy via dual pH-sensitive polyelectrolyte complex nanoparticles to achieve gastric survivability, intestinal permeability, hemodynamic stability and intracellular activity. Eur. J. Pharm. Biopharm. 2015, 97, 107–117. [Google Scholar] [CrossRef]
- Riethorst, D.; Mols, R.; Duchateau, G.; Tack, J.; Brouwers, J.; Augustijns, P. Characterization of Human Duodenal Fluids in Fasted and Fed State Conditions. J. Pharm. Sci. 2016, 105, 673–681. [Google Scholar] [CrossRef] [Green Version]
- Kanicky, J.R.; Shah, D.O. Effect of premicellar aggregation on the pKa of fatty acid soap solutions. Langmuir 2003, 19, 2034–2038. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindah, E. Gromacs: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007, 126, 014101. [Google Scholar] [CrossRef] [Green Version]
- Parrinello, M.; Rahman, A. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Hossain, M.S.; Berg, S.; Bergström, C.A.S.; Larsson, P. Aggregation Behavior of Medium Chain Fatty Acids Studied by Coarse-Grained Molecular Dynamics Simulation. AAPS PharmSciTech 2019, 20, 61. [Google Scholar] [CrossRef]
- Sega, M.; Hantal, G.; Fábián, B.; Jedlovszky, P. Pytim: A python package for the interfacial analysis of molecular simulations. J. Comput. Chem. 2018, 39, 2118–2125. [Google Scholar] [CrossRef]
- Twarog, C.; Fattah, S.; Heade, J.; Maher, S.; Fattal, E.; Brayden, D.J. Intestinal Permeation Enhancers for Oral Delivery of Macromolecules: A Comparison between Salcaprozate Sodium (SNAC) and Sodium Caprate (C 10). Pharmaceutics 2019, 11, 78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spivak, W.; Morrison, C.; Devinuto, D.; Yuey, W. Spectrophotometric determination of the critical micellar concentration of bile salts using bilirubin monoglucuronide as a micellar probe. Utility of derivative spectroscopy. Biochem. J. 1988, 252, 275–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lebecque, S.; Crowet, J.M.; Nasir, M.N.; Deleu, M.; Lins, L. Molecular dynamics study of micelles properties according to their size. J. Mol. Graph. Model. 2017, 72, 6–15. [Google Scholar] [CrossRef] [PubMed]
- Yoshii, N.; Okazaki, S. A molecular dynamics study of structural stability of spherical SDS micelle as a function of its size. Chem. Phys. Lett. 2006, 425, 58–61. [Google Scholar] [CrossRef]
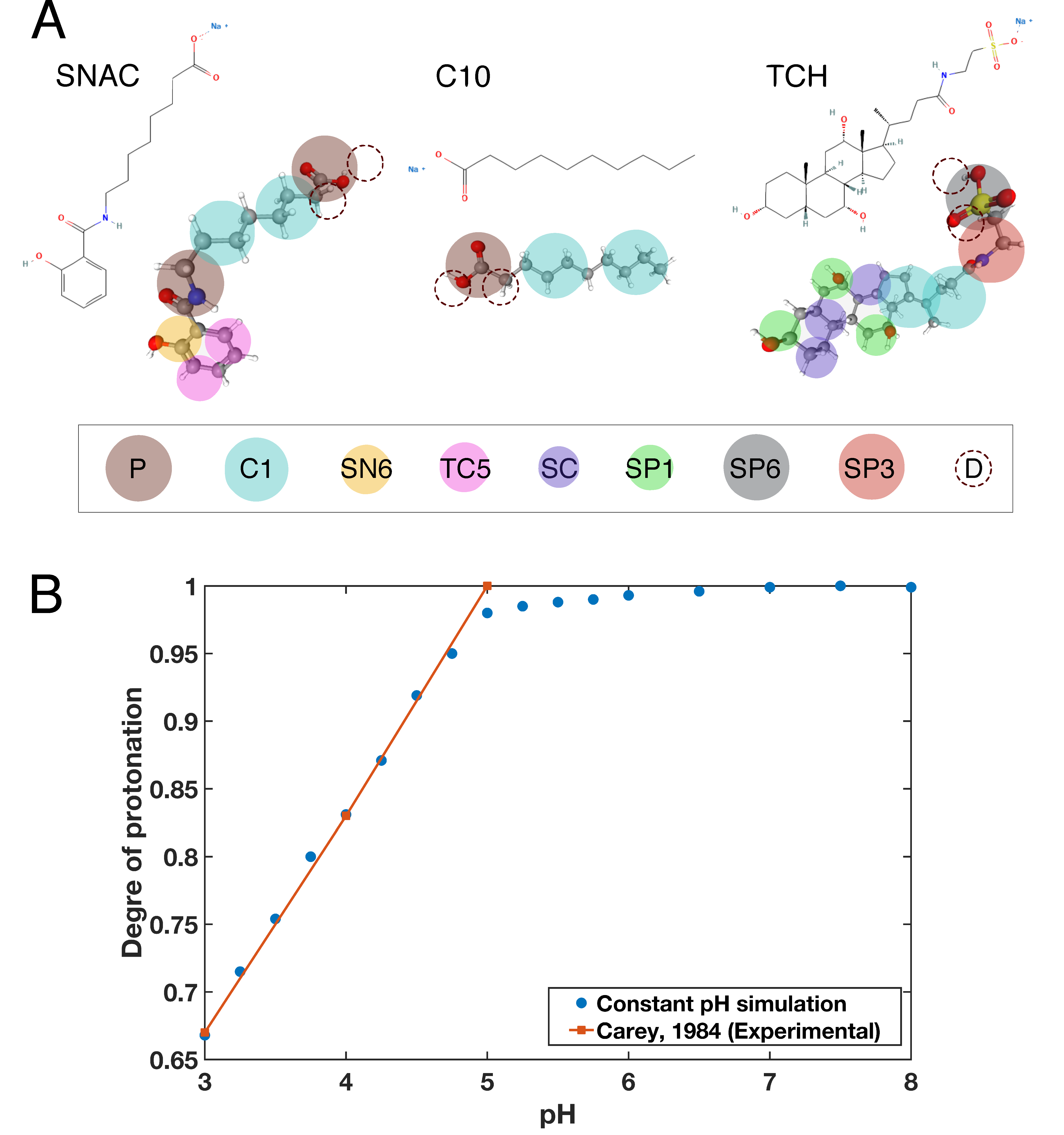
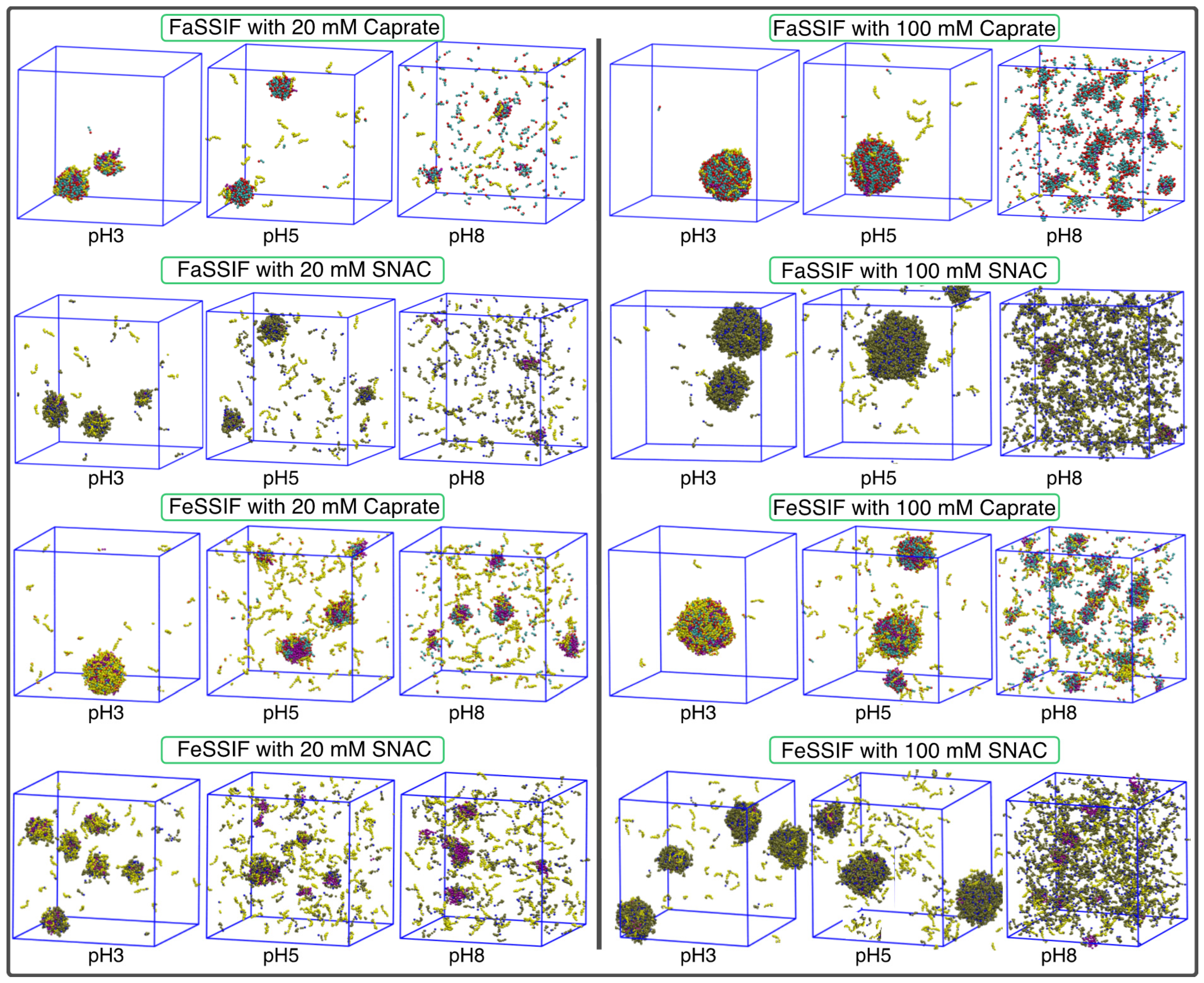
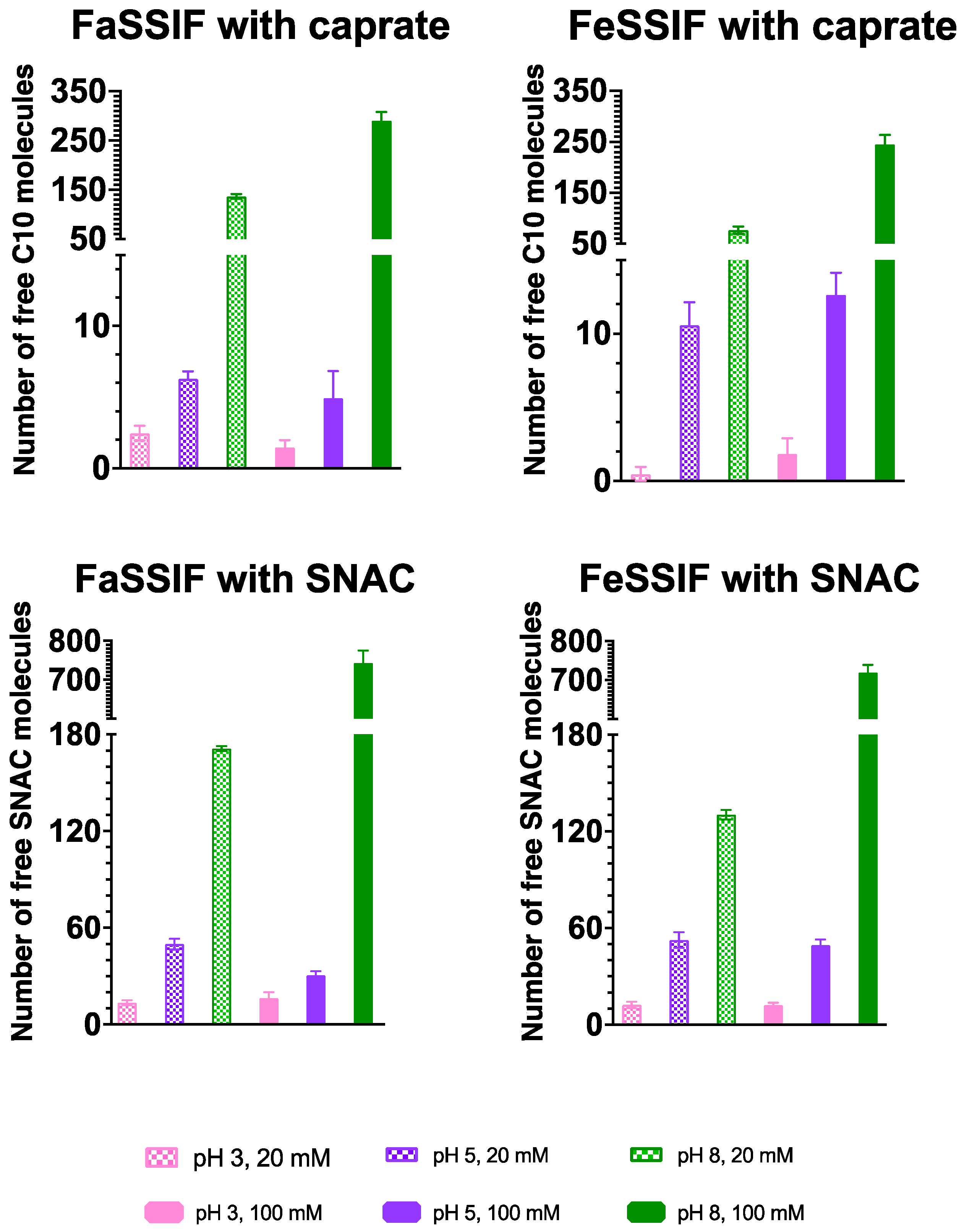
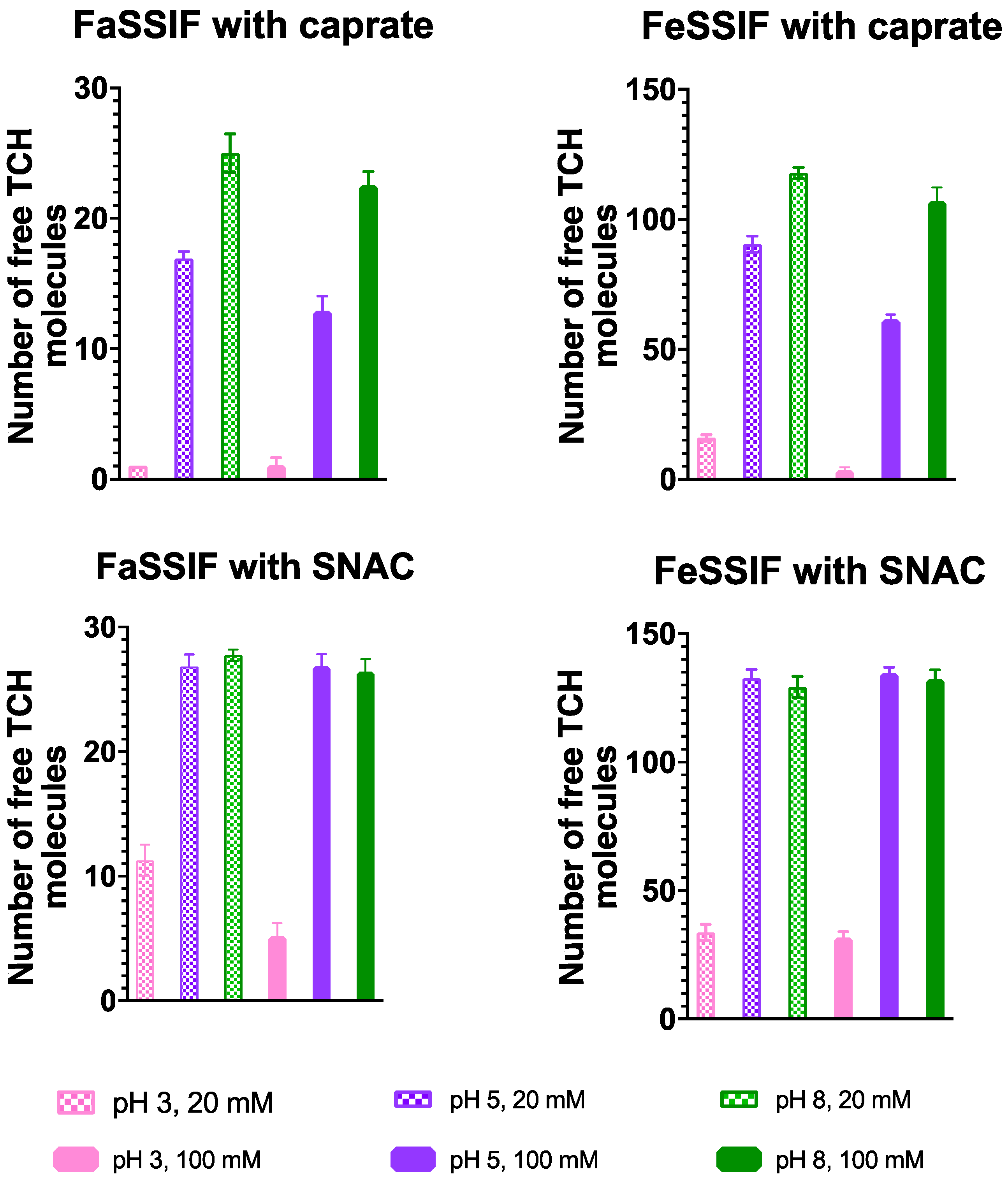

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mediums | Permeation Enhancers | Concentration of Permeation Enhancers | pH |
|---|---|---|---|
| FaSSIF: Sodium taurocholate (3.0 mM) and soy lecithin (0.75 mM) [15]; FeSSIF: Sodium taurocholate (15.0 mM) and soy lecithin (3.75 mM) [15] | Caprate and SNAC | 20 and 100 mM | 3, 5 and 8 |
| Aggregation Number, N (Average ± Std. Error) | Aggregation Number, N (Average ± Std. Error) | ||||||
|---|---|---|---|---|---|---|---|
| Systems | pH 3 | pH 5 | pH 8 | Systems | pH 3 | pH 5 | pH 8 |
| FaSSIF with 20 mM caprate | 220 ± 0 | 100 ± 2 | 21 ± 1 | FaSSIF with 20 mM SNAC | 66 ± 4 | 42 ± 2 | 10 ± 0 |
| FaSSIF with 100 mM caprate | 972 ± 0 | 956 ± 1 | 30 ± 0 | FaSSIF with 100 mM SNAC | 382 ± 36 | 459 ± 52 | 10 ± 0 |
| FeSSIF with 20 mM caprate | 346 ± 0 | 66 ± 5 | 33 ± 2 | FeSSIF with 20 mM SNAC | 53 ± 1 | 29 ± 3 | 21 ± 1 |
| FeSSIF with 100 mM caprate | 1111 ± 0 | 277 ± 17 | 39 ± 1 | FeSSIF with 100 mM SNAC | 269 ± 10 | 311 ± 23 | 18 ± 1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hossain, S.; Parrow, A.; Kabedev, A.; Kneiszl, R.C.; Leng, Y.; Larsson, P. Explicit-pH Coarse-Grained Molecular Dynamics Simulations Enable Insights into Restructuring of Intestinal Colloidal Aggregates with Permeation Enhancers. Processes 2022, 10, 29. https://doi.org/10.3390/pr10010029
Hossain S, Parrow A, Kabedev A, Kneiszl RC, Leng Y, Larsson P. Explicit-pH Coarse-Grained Molecular Dynamics Simulations Enable Insights into Restructuring of Intestinal Colloidal Aggregates with Permeation Enhancers. Processes. 2022; 10(1):29. https://doi.org/10.3390/pr10010029
Chicago/Turabian StyleHossain, Shakhawath, Albin Parrow, Aleksei Kabedev, Rosita Carolina Kneiszl, Yuning Leng, and Per Larsson. 2022. "Explicit-pH Coarse-Grained Molecular Dynamics Simulations Enable Insights into Restructuring of Intestinal Colloidal Aggregates with Permeation Enhancers" Processes 10, no. 1: 29. https://doi.org/10.3390/pr10010029
APA StyleHossain, S., Parrow, A., Kabedev, A., Kneiszl, R. C., Leng, Y., & Larsson, P. (2022). Explicit-pH Coarse-Grained Molecular Dynamics Simulations Enable Insights into Restructuring of Intestinal Colloidal Aggregates with Permeation Enhancers. Processes, 10(1), 29. https://doi.org/10.3390/pr10010029