
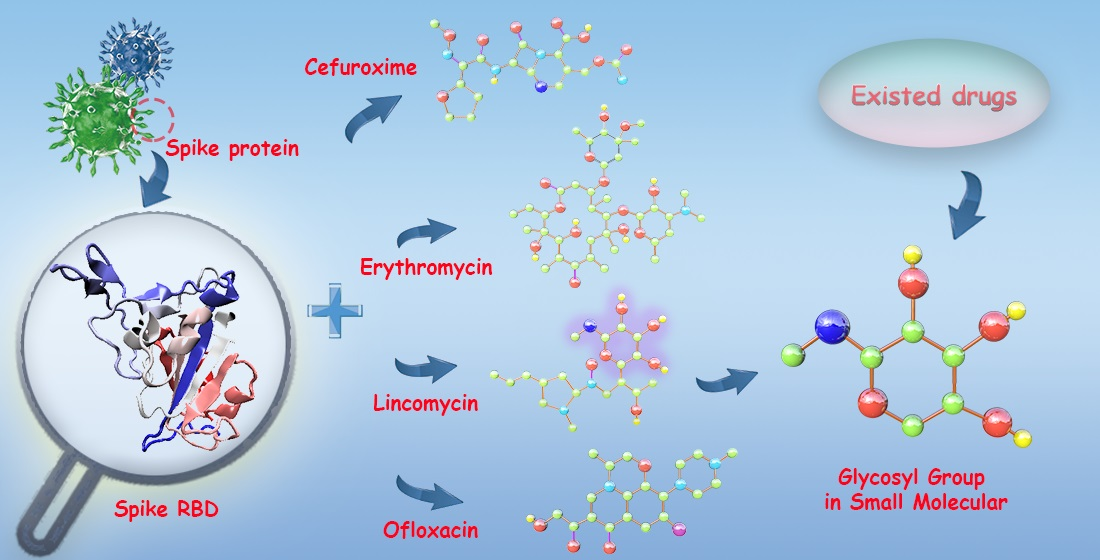
Interactive Mechanism of Potential Inhibitors with Glycosyl for SARS-CoV-2 by Molecular Dynamics Simulation

 and
and
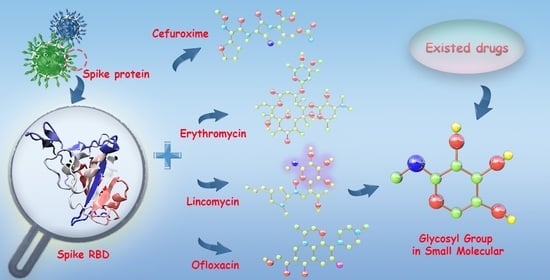
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Research Systems
2.2. Molecular Docking Strategy
2.3. MD Simulation
2.4. Analysis Methods
3. Results and Discussion
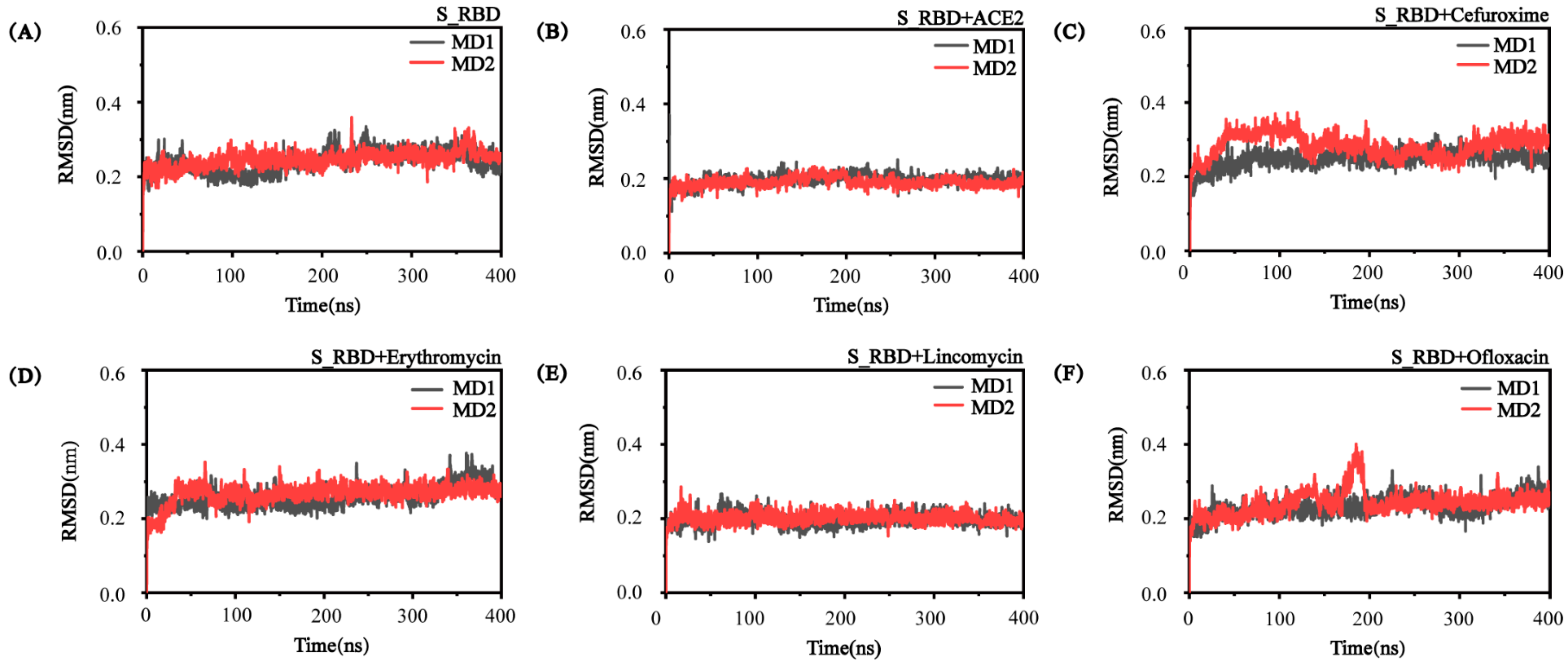
3.1. The Convergence and Stability of S_RBD in the Research Systems
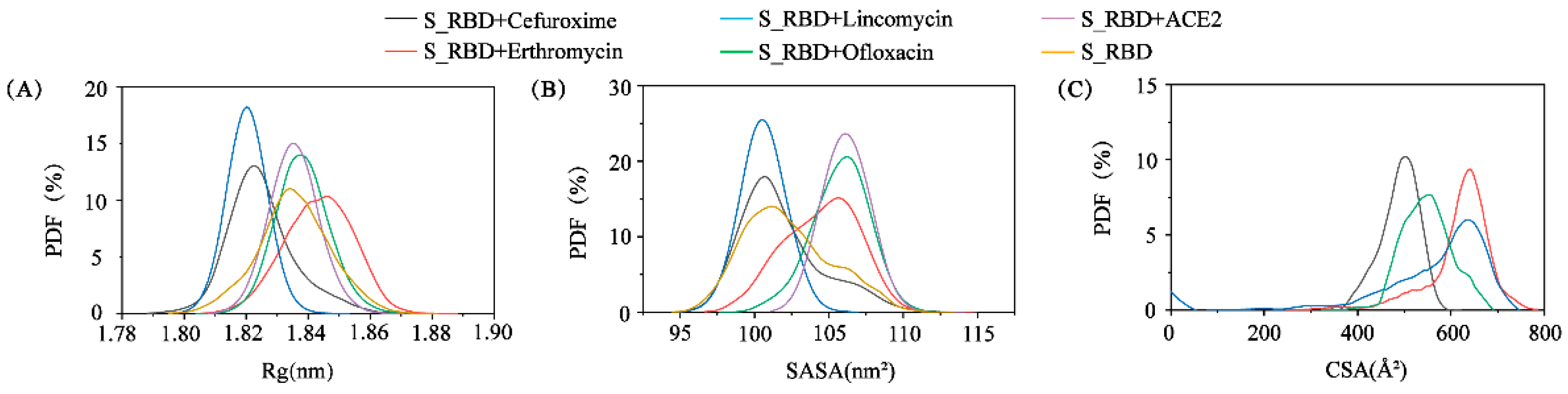
3.2. The Structural Characteristics of S_RBD in the Absence or Presence of the Small Molecules/ACE2
3.3. Analysis of the Interactions among the Residue-Residue of S_RBD
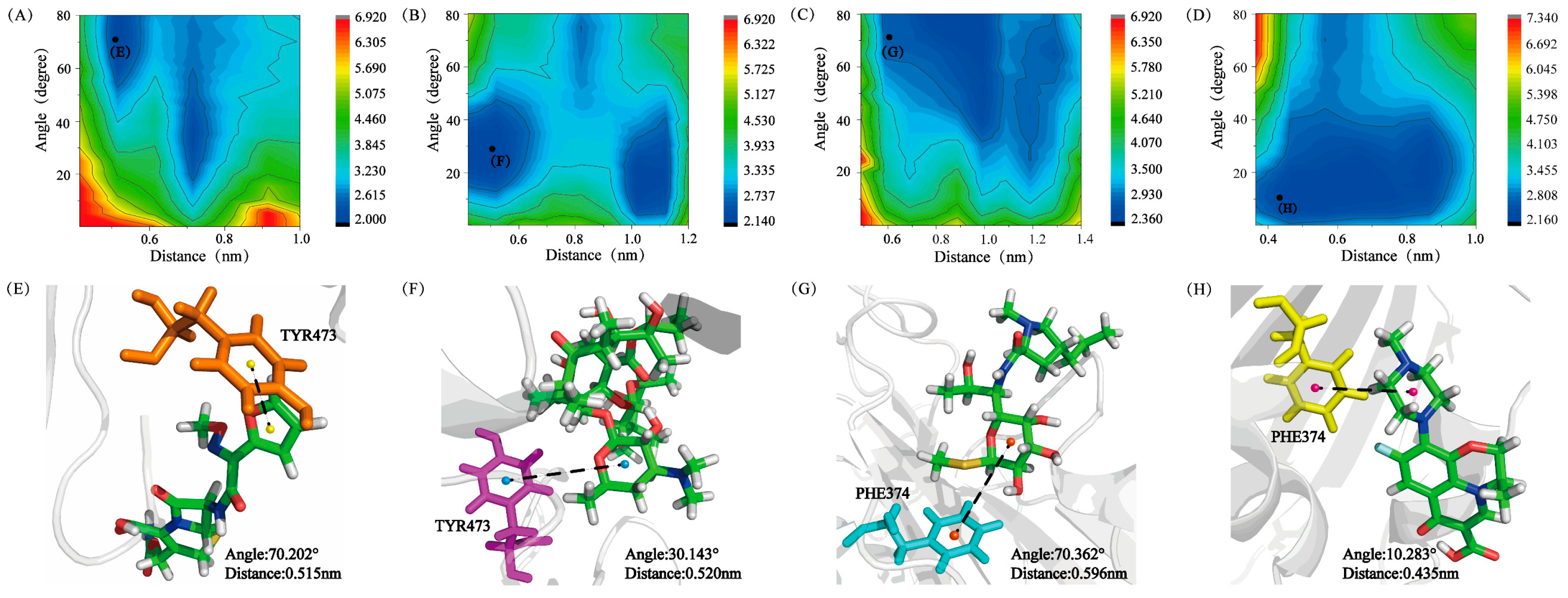
3.4. The Conformational Difference of S_RBD in Different Research Systems
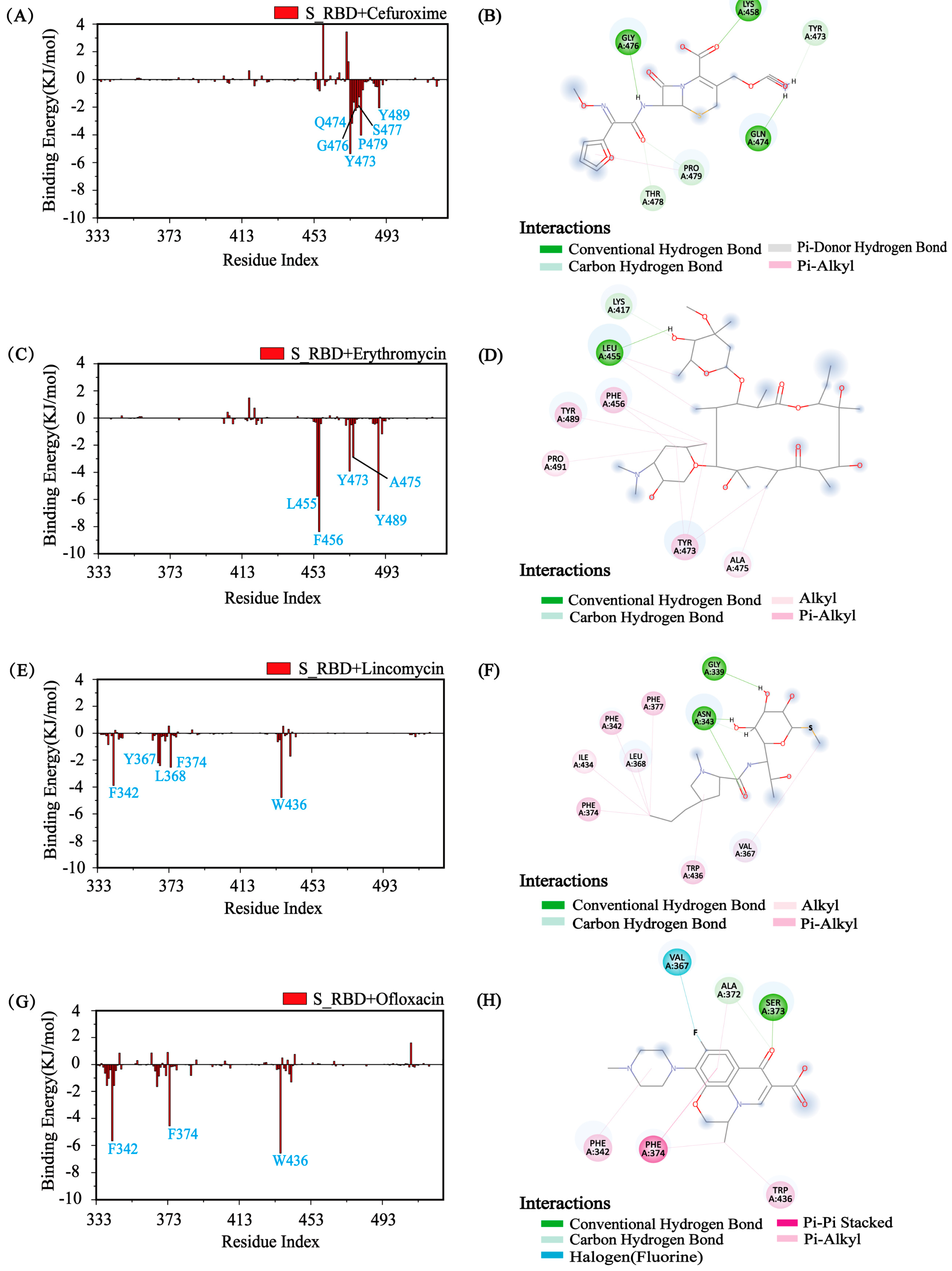
3.5. Identify the Major Interaction Residues of S_RBD by Calculating the Binding Energy
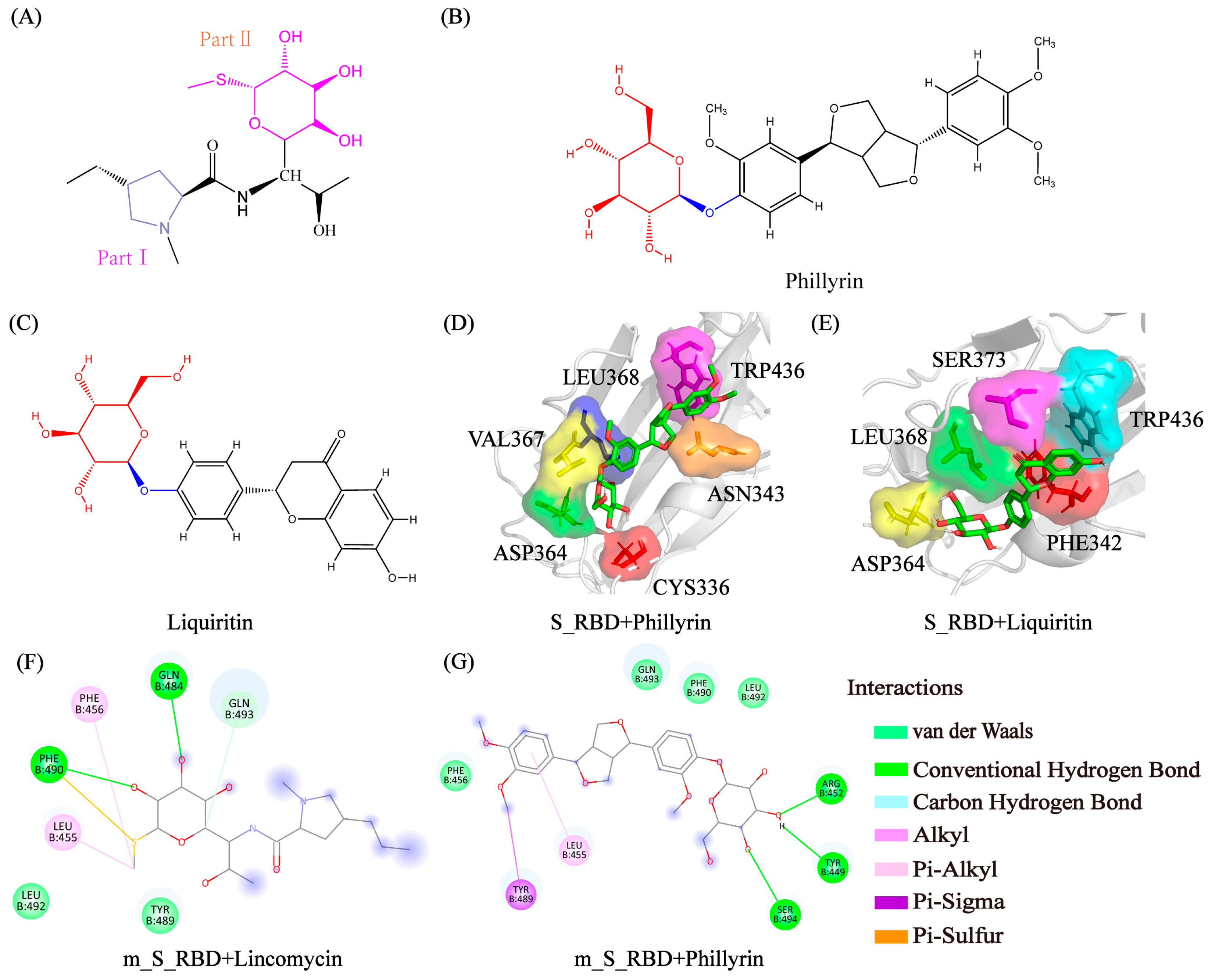
3.6. Analysis of Physical Interactions between the Residues and the Small Molecules
3.7. Prediction Potential Inhibitors with Glycosyl for SARS-CoV-2 and the Genetic Variants of SARS-CoV-2 from Existing Drugs
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef] [Green Version]
- Zhou, P.; Yang, X.; Wang, X.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.; Zhu, Y.; Li, B.; Huang, C.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sinha, S.; Wang, S. Classification of VUS and unclassified variants in BRCA1 BRCT repeats by molecular dynamics simulation. Comput. Struct. Biotechnol. J. 2020, 18, 723–736. [Google Scholar] [CrossRef]
- Han, Y.; Wang, Z.; Ren, J.; Wei, Z.; Li, J. Potential inhibitors for the novel coronavirus (SARS-CoV-2). Brief. Bioinform. 2021, 22, 1225–1231. [Google Scholar] [CrossRef]
- Fischer, A.; Sellner, M.; Neranjan, S.; Smieško, M.; Lill, M.A. Potential Inhibitors for Novel Coronavirus Protease Identified by Virtual Screening of 606 Million Compounds. Int. J. Mol. Sci. 2020, 21, 3626. [Google Scholar] [CrossRef]
- Li, Z.; Li, X.; Huang, Y.-Y.; Wu, Y.; Liu, R.; Zhou, L.; Lin, Y.; Wu, D.; Zhang, L.; Liu, H.; et al. Identify potent SARS-CoV-2 main protease inhibitors via accelerated free energy perturbation-based virtual screening of existing drugs. Proc. Natl. Acad. Sci. USA 2020, 117, 27381–27387. [Google Scholar] [CrossRef]
- Jin, Z.; Du, X.; Xu, Y.; Deng, Y.; Liu, M.; Zhao, Y.; Zhang, B.; Li, X.; Zhang, L.; Peng, C.; et al. Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature 2020, 582, 289–293. [Google Scholar] [CrossRef] [Green Version]
- Adebisi, Y.A.; Jimoh, N.D.; Ogunkola, I.O.; Uwizeyimana, T.; Olayemi, A.H.; Ukor, N.A.; Lucero-Prisno, D.E. The use of antibiotics in COVID-19 management: A rapid review of national treatment guidelines in 10 African countries. Trop. Med. Health 2021, 11, 128–134. [Google Scholar]
- Yacouba, A.; Olowo-Okere, A.; Yunusa, I. Repurposing of antibiotics for clinical management of COVID-19: A narrative review. Ann. Clin. Microbiol. Antimicrob. 2021, 20, 37. [Google Scholar] [CrossRef]
- Martínez-Flores, D.; Zepeda-Cervantes, J.; Cruz-Reséndiz, A.; Aguirre-Sampieri, S.; Sampieri, A.; Vaca, L. SARS-CoV-2 Vaccines Based on the Spike Glycoprotein and Implications of New Viral Variants. Front. Immunol. 2021, 12. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, K.; Massoud, T.F.; Paulmurugan, R. SARS-CoV-2 Vaccine Development: An Overview and Perspectives. ACS Pharm. Transl. Sci 2020, 3, 844–858. [Google Scholar] [CrossRef]
- Creech, C.B.; Walker, S.C.; Samuels, R.J. SARS-CoV-2 Vaccines. JAMA 2021, 325, 1318–1320. [Google Scholar] [CrossRef]
- Shang, J.; Ye, G.; Shi, K.; Wan, Y.; Luo, C.; Aihara, H.; Geng, Q.; Auerbach, A.; Li, F. Structural basis of receptor recognition by SARS-CoV-2. Nature 2020, 581, 221–224. [Google Scholar] [CrossRef] [Green Version]
- Walls, A.C.; Park, Y.-J.; Alejandratortorici, M.; Wall, A.; Mcguire, A.T.; Veesler, D. Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell 2020, 181, 281–292. [Google Scholar] [CrossRef]
- Schütz, D.; Ruiz-Blanco, Y.B.; Münch, J.; Kirchhoff, F.; Sanchez-Garcia, E.; Müller, J.A. Peptide and peptide-based inhibitors of SARS-CoV-2 entry. Adv. Drug Deliv. Rev. 2020, 167, 47–65. [Google Scholar] [CrossRef] [PubMed]
- Pant, S.; Singh, M.; Ravichandiran, V.; Murty, U.S.N.; Srivastava, H.K. Peptide-like and small-molecule inhibitors against Covid-19. J. Biomol. Struct. Dyn. 2020, 39, 2904–2913. [Google Scholar] [CrossRef] [Green Version]
- Elfiky, A.A. SARS-CoV-2 RNA dependent RNA polymerase (RdRp) targeting: An in silico perspective. J. Biomol. Struct. Dyn. 2020, 39, 3204–3212. [Google Scholar]
- Ngo, S.T.; Pham, N.Q.A.; Le, L.T. Computational Determination of Potential Inhibitors of SARS-CoV-2 Main Protease. J. Chem. Inf. Modeling 2020, 60, 5771–5780. [Google Scholar] [CrossRef] [PubMed]
- Durojaiye, A.B.; Clarke, J.-R.D.; Stamatiades, G.A.; Wang, C. Repurposing cefuroxime for treatment of COVID-19: A scoping review of in silico studies. J. Biomol. Struct. Dyn. 2020, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Dayer, M.R. Old Drugs for Newly Emerging Viral Disease, COVID-19: Bioinformatic Prospective. arXiv 2020, arXiv:2003.04524. Available online: https://arxiv.org/abs/2003.04524 (accessed on 22 September 2021).
- Cuesta, S.A.; Mora, J.R.; Márquez, E.A. In Silico Screening of the DrugBank Database to Search for Possible Drugs against SARS-CoV-2. Molecules 2021, 26, 1100. [Google Scholar] [CrossRef]
- Bello, M. Prediction of potential inhibitors of the dimeric SARS-CoV2 main proteinase through the MM/GBSA approach. Elsevier Public Health Emerg. Collect. 2020, 101, 107762. [Google Scholar] [CrossRef]
- Zoufaly, A.; Poglitsch, M.; Aberle, J.H.; Hoepler, W.; Seitz, T.; Traugott, M.; Grieb, A.; Pawelka, E.; Laferl, H.; Wenisch, C.; et al. Human recombinant soluble ACE2 in severe COVID-19. Lancet Respir. Med. 2020, 8, 1154–1158. [Google Scholar] [CrossRef]
- Hu, X.; Zhou, Z.; Li, F.; Xiao, Y.; Wang, Z.; Xu, J.; Dong, F.; Zheng, H.; Yu, R. The study of antiviral drugs targeting SARS-CoV-2 nucleocapsid and spike proteins through large-scale compound repurposing. Heliyon 2021, 7, e06387. [Google Scholar] [CrossRef] [PubMed]
- Batiha, G.E.-S.; Zayed, M.A.; Awad, A.A.; Shaheen, H.M.; Mustapha, S.; Herrera-Calderon, O.; Pagnossa, J.P.; Algammal, A.M.; Zahoor, M.; Adhikari, A.; et al. Management of SARS-CoV-2 Infection: Key Focus in Macrolides Efficacy for COVID-19. Front. Med. 2021, 8, 642313. [Google Scholar] [CrossRef]
- Guvenmez, O.; Keskin, H.; Ay, B.; Birinci, S.; Kanca, M.F. The comparison of the effectiveness of lincocin and azitro in the treatment of covid-19-associated pneumonia: A prospective study. J. Popul. Ther. Clin. Pharmacol. 2020, 27, e5–e10. [Google Scholar] [CrossRef]
- Wang, Q.; Zhang, Y.; Wu, L.; Niu, S.; Song, C.; Zhang, Z.; Lu, G.; Qiao, C.; Hu, Y.; Yuen, K.-Y.; et al. Structural and Functional Basis of SARS-CoV-2 Entry by Using Human ACE2. Cell 2020, 181, 894–904. [Google Scholar] [CrossRef] [PubMed]
- Hornak, V.; Abel, R.; Okur, A.; Strockbine, B.; Roitberg, A.; Simmerling, C. Comparison of multiple Amber force fields and development of improved protein backbone parameters. Proteins Struct. Funct. Bioinform. 2006, 65, 712–725. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, M.W.; Baldridge, K.K.; Boatz, J.A.; Elbert, S.T.; Gordon, M.S.; Jensen, J.H.; Montgomery, J.A., Jr. General atomic and molecular electronic structure system. J. Comput. Chem. 1993, 14, 1347–1363. [Google Scholar] [CrossRef]
- Schüttelkopf, A.W.; Aalten, V.; Mf, D. PRODRG: A tool for high-throughput crystallography of protein–ligand complexes. Acta Crystallogr. Sect. D Biol. Crystallogr. 2004, 60, 1355–1363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mo, Y.; Lu, Y.; Wei, G.; Derreumaux, P. Structural diversity of the soluble trimers of the human amylin (20–29) peptide revealed by molecular dynamics simulations. J. Chem. Phys. 2009, 130, 03B616. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [Green Version]
- Forli, S.; Olson, A.J. A Force Field with Discrete Displaceable Waters and Desolvation Entropy for Hydrated Ligand Docking. J. Med. Chem. 2012, 55, 623–638. [Google Scholar] [CrossRef] [Green Version]
- Lindorff-Larsen, K.; Piana, S.; Palmo, K.; Maragakis, P.; Klepeis, J.L.; Dror, R.O.; Shaw, D.E. Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins: Struct. Funct. Bioinform. 2010, 78, 1950–1958. [Google Scholar] [CrossRef] [Green Version]
- Hess, B.; Kutzner, C.; Spoel, D.V.D.; Lindahl, E. GROMACS 4: Algorithms for Highly Efficient, Load-Balanced, and Scalable Molecular Simulation. J. Chem. Theory Comput. 2008, 4, 435–447. [Google Scholar] [CrossRef] [Green Version]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- DeLano, W.L. PyMOL: An Open-Source Molecular Graphics Tool. CCP4 Newsl. Protein Crystallogr. 2002, 40, 82–92. [Google Scholar]
- Zhan, C.; Chen, Y.; Tang, Y.; Wei, G. Green Tea Extracts EGCG and EGC Display Distinct Mechanisms in Disrupting Aβ42 Protofibril. ACS Chem. Neurosci. 2020, 11, 1841–1851. [Google Scholar] [CrossRef]
- Onufriev, A.; Bashford, D.; Case, D.A. Modification of the Generalized Born Model Suitable for Macromolecules. J. Phys. Chem. B 2000, 104, 3712–3720. [Google Scholar] [CrossRef] [Green Version]
- Onufriev, A.; Bashford, D.; Case, D.A. Exploring protein native states and large-scale conformational changes with a modified generalized born model. Proteins Struct. Funct. Bioinform. 2004, 55, 383–394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salomon-Ferrer, R.; Case, D.A.; Walker, R.C. An overview of the Amber biomolecular simulation package. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2012, 3, 198–210. [Google Scholar] [CrossRef]
- Narang, S.S.; Goyal, D.; Goyal, B. Inhibition of Alzheimer’s amyloid-β42 peptide aggregation by a bi-functional bis-tryptoline triazole: Key insights from molecular dynamics simulations. J. Biomol. Struct. Dyn. 2019, 38, 1598–1611. [Google Scholar] [CrossRef]
- Zhao, S.; Zhu, Y.; Wang, X.; Liu, Y.; Sun, Y.; Zhao, Q.; Li, H. Structural Insight into the Interactions between Structurally Similar Inhibitors and SIRT6. Int. J. Mol. Sci. 2020, 21, 2601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gowder, S.M.; Chatterjee, J.; Chaudhuri, T.; Paul, K. Prediction and Analysis of Surface Hydrophobic Residues in Tertiary Structure of Proteins. Sci. World J. 2014, 14, 971258. [Google Scholar]
- Berhanu, W.M.; Hansmann, U.H. The stability of cylindrin β-barrel amyloid oligomer models—A molecular dynamics study. Proteins: Struct. Funct. Bioinform. 2013, 81, 1542–1555. [Google Scholar] [CrossRef] [Green Version]
- Zhang, T.; Zhang, J. Molecular Mechanism of the Inhibition of EGCG on the Alzheimer A beta(1-42) Dimer. J. Phys. Chem. B 2013, 117, 3993–4002. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Zheng, Q.; Yu, L.; Zhang, H.; Sun, C. A molecular dynamics and computational study of human KAT3 involved in KYN pathway. Sci. China Chem. 2013, 56, 514–523. [Google Scholar] [CrossRef]
- Kitagawa, K.; Shigemura, K.; Nomi, M.; Takami, N.; Yamada, N.; Fujisawa, M. Use of oral third generation cephalosporins and quinolones and occurrence of antibiotic-resistant strains in the neurogenic bladder (NB) outpatient setting: A retrospective chart audit. Spinal Cord 2020, 58, 705–710. [Google Scholar] [CrossRef] [PubMed]
- Leclercq, R. Mechanisms of Resistance to Macrolides and Lincosamides: Nature of the Resistance Elements and Their Clinical Implications. Clin. Infect. Dis. 2002, 34, 482–492. [Google Scholar] [CrossRef] [Green Version]
- Matzov, D.; Eyal, Z.; Benhamou, R.I.; Shalev-Benami, M.; Halfon, Y.; Krupkin, M.; Zimmerman, E.; Rozenberg, H.; Bashan, A.; Fridman, M.; et al. Structural insights of lincosamides targeting the ribosome of Staphylococcus aureus. Nucleic Acids Res. 2017, 45, 10284–10292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andriole, T. The Quinolones: Prospects; Academic Press: Cambridge, MA, USA, 2000; pp. 477–495. [Google Scholar]
- Society and America. Guidelines for the management of adults with hospital-acquired, ventilator-associated, and healthcare-associated pneumonia. Am. J. Respir. Crit. Care Med. 2005, 171, 388–416. [Google Scholar] [CrossRef]
- Bai, C.; Lao, Z.; Chen, Y.; Tang, Y.; Wei, G. Pristine and Hydroxylated Fullerenes Prevent the Aggregation of Human Islet Amyloid Polypeptide and Display Different Inhibitory Mechanisms. Front. Chem. 2020, 8, 51. [Google Scholar] [CrossRef] [PubMed]
- Ali, A.; Vijayan, R. Dynamics of the ACE2–SARS-CoV-2/SARS-CoV spike protein interface reveal unique mechanisms. Sci. Rep. 2020, 10, 14214. [Google Scholar] [CrossRef]
- Sen, D.; Debnath, P.; Debnath, B.; Bhaumik, S.; Debnath, S. Identification of potential inhibitors of SARS-CoV-2 main protease and spike receptor from 10 important spices through structure-based virtual screening and molecular dynamic study. J. Biomol. Struct. Dyn. 2020, 39, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Sixto-López, Y.; Correa-Basurto, J.; Bello, M.; Landeros-Rivera, B.; Garzón-Tiznado, J.A.; Montaño, S. Structural insights into SARS-CoV-2 spike protein and its natural mutants found in Mexican population. Sci. Rep. 2021, 11, 4659. [Google Scholar] [CrossRef]
- Sun, Y.; Qian, Z.; Wei, G. The Inhibitory Mechanism of a Fullerene Derivative against Amyloid-β peptide aggregation: An Atomistic Simulation Study. Phys. Chem. Chem. Phys. 2016, 18, 12582–12591. [Google Scholar] [CrossRef]
- Song, M.; Sun, Y.; Luo, Y.; Zhu, Y.; Liu, Y.; Li, H. Exploring the Mechanism of Inhibition of Au Nanoparticles on the Aggregation of Amyloid-β(16-22) Peptides at the Atom Level by All-Atom Molecular Dynamics. Int. J. Mol. Sci. 2018, 19, 1815. [Google Scholar] [CrossRef] [Green Version]
- Song, M.; Zhu, Y.; Wei, G.; Li, H. Carbon nanotube prevents the secondary structure formation of amyloid-β trimers: An all-atom molecular dynamics study. Mol. Simul. 2017, 43, 1189–1195. [Google Scholar] [CrossRef]
- Jin, Y.; Sun, Y.; Lei, J.; Wei, G. Dihydrochalcone molecules destabilize Alzheimer’s amyloid- β protofibrils through binding to the protofibril cavity. Phys. Chem. Chem. Phys. 2018, 20, 17208–17217. [Google Scholar] [CrossRef] [PubMed]
- Ge, X.; Sun, Y.; Ding, F. Structures and dynamics of β-barrel oligomer intermediates of amyloid-beta16-22 aggregation. Biochim. Et Biophys. Acta (BBA) 2018, 1860, 1687–1697. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Song, M.; Zhao, S.; Li, H.; Zhao, Q.; Shen, J. Molecular dynamics simulations reveal the mechanism of the interactions between the inhibitors and SIRT2 at atom level. Mol. Simul. 2020, 46, 638–649. [Google Scholar] [CrossRef]
- Hu, K.; Guan, W.-J.; Bi, Y.; Zhang, W.; Li, L.; Zhang, B.; Liu, Q.; Song, Y.; Li, X.; Duan, Z.; et al. Efficacy and safety of Lianhuaqingwen capsules, a repurposed Chinese herb, in patients with coronavirus disease 2019: A multicenter, prospective, randomized controlled trial. Phytomedicine 2021, 85, 153242. [Google Scholar] [CrossRef]
- Ni, L.; Wen, Z.; Hu, X.; Tang, W.; Wang, H.; Zhou, L.; Wu, L.; Wang, H.; Xu, C.; Xu, X.; et al. Effects of Shuanghuanglian oral liquids on patients with COVID-19: A randomized, open-label, parallel-controlled, multicenter clinical trial. Front. Med. 2021, 99, 1–4. [Google Scholar]
- Unal, M.A.; Bitirim, C.V.; Summak, G.Y.; Bereketoglu, S.; Cevher Zeytin, I.; Besbinar, O.; Gurcan, C.; Aydos, D.; Goksoy, E.; Kocakaya, E.; et al. Ribavirin shows antiviral activity against SARS-CoV-2 and downregulates the activity of TMPRSS2 and the expression of ACE2 in vitro. Can. J. Physiol. Pharmacol. 2021, 99, 449–460. [Google Scholar] [CrossRef] [PubMed]
- Qu, S.; Wei, X.; Sai, M.; Lan, X.; Chen, M. Chemical constituents from Rhodiola crenulata. Chin. Tradit. Pat. Med. 2020, 42, 3199–3203. [Google Scholar]
- Li, L.; Li, W.; Wang, B.; Sun, Y. Research of Effective Component Content Difference between Honeysuckle and Forsythia Compatibility and Single Herbs. J. Liaoning Univ. TCM 2018, 20, 51–53. [Google Scholar]
- Tomimori, T.; Jin, H.; Miyaichi, Y.; Toyofuku, S.; Namba, T. Studies on the constituents of Scutellaria species. VI. On the flavonoid constituents of the root of Scutellaria baicalensis Georgi (5). Quantitative analysis of flavonoids in Scutellaria roots by high-performance liquid chromatography. J. Stage 1985, 105, 148–155. [Google Scholar]
- Li, K.; Shi, Q.; Zhu, H.; Tang, D. Chemical Compositions in Bitter Almond. J. Northwest. For. Univ. 2004, 19, 124–126. [Google Scholar]
- Tian, Q.; Zhang, B.; Liu, H. Research Advances on Pharmacological Activities of Components in Licorice. Nat. Prod. Res. Dev. 2006, 18, 343–347. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| English Name | Cefuroxime | Erythromycin | Lincomycin | Ofloxacin |
|---|---|---|---|---|
| Molecular formula | C16H16N4O8S | C37H67NO13 | C18H34N2O6S | C18H20FN3O4 |
| Binding energy (KJ/mol) | −15.104 | −9.330 | −13.656 | −18.995 |
| Chemical structure | 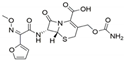 |  | 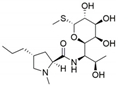 | 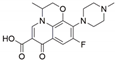 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Y.; Chen, L.; Wang, X.; Zhu, Y.; Liu, Y.; Li, H.; Zhao, Q. Interactive Mechanism of Potential Inhibitors with Glycosyl for SARS-CoV-2 by Molecular Dynamics Simulation. Processes 2021, 9, 1749. https://doi.org/10.3390/pr9101749
Zhang Y, Chen L, Wang X, Zhu Y, Liu Y, Li H, Zhao Q. Interactive Mechanism of Potential Inhibitors with Glycosyl for SARS-CoV-2 by Molecular Dynamics Simulation. Processes. 2021; 9(10):1749. https://doi.org/10.3390/pr9101749
Chicago/Turabian StyleZhang, Yuqi, Li Chen, Xiaoyu Wang, Yanyan Zhu, Yongsheng Liu, Huiyu Li, and Qingjie Zhao. 2021. "Interactive Mechanism of Potential Inhibitors with Glycosyl for SARS-CoV-2 by Molecular Dynamics Simulation" Processes 9, no. 10: 1749. https://doi.org/10.3390/pr9101749
APA StyleZhang, Y., Chen, L., Wang, X., Zhu, Y., Liu, Y., Li, H., & Zhao, Q. (2021). Interactive Mechanism of Potential Inhibitors with Glycosyl for SARS-CoV-2 by Molecular Dynamics Simulation. Processes, 9(10), 1749. https://doi.org/10.3390/pr9101749