
Scalable Interactive Visualization for Connectomics

 ,
,  ,
,
Abstract
1. Introduction
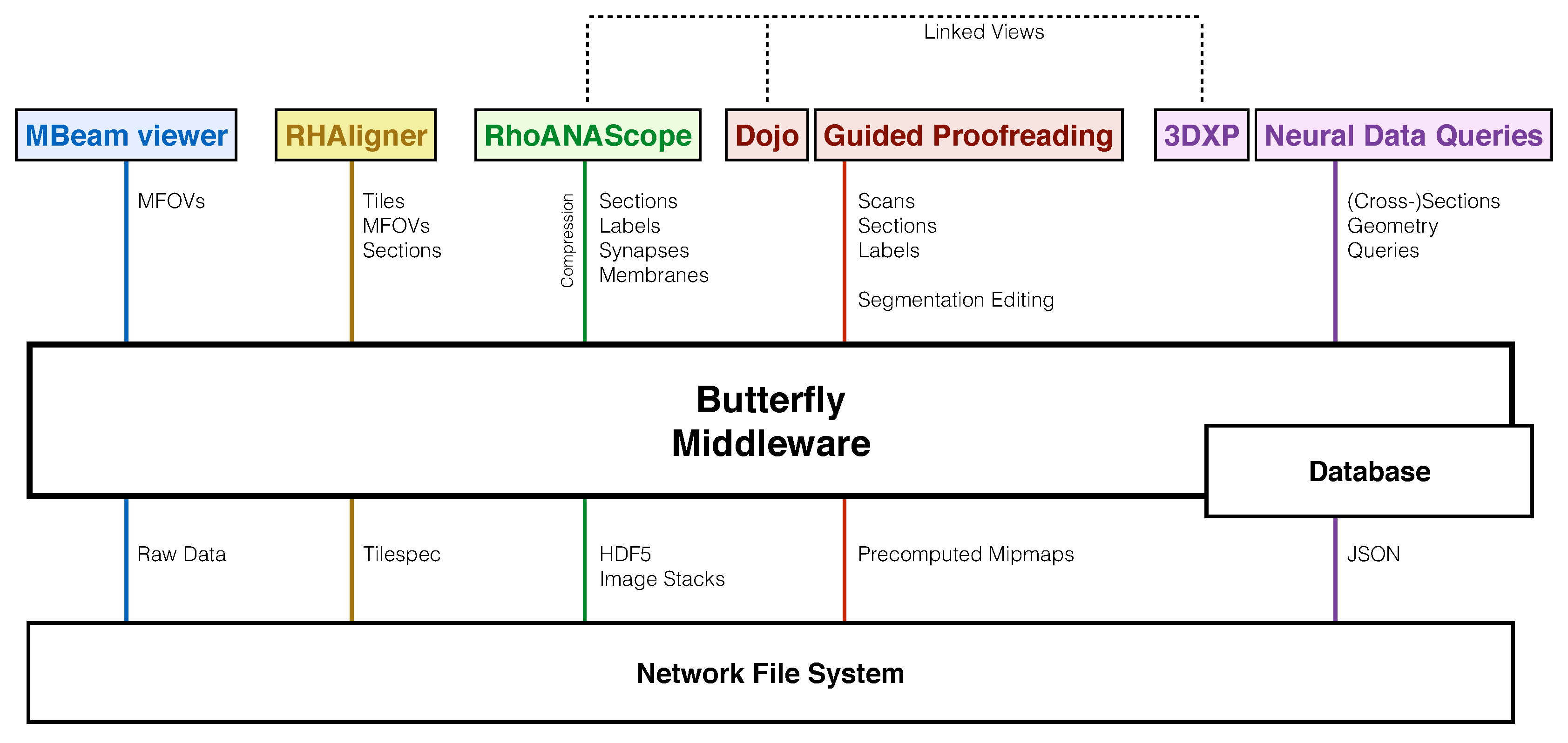
- MBeam viewer, a tool to quickly assess image quality and contrast during acquisition.
- RHAligner, visualization scripts for debugging the registration process.
- RhoANAScope, a visualizer for image data and label overlays during segmentation.
- Dojo, an interactive proofreading tool with multi-user support [11].
- Guided Proofreading, a machine learning proofreading tool to correct errors quickly [12].
- 3DXP, a 3D visualization of neuron geometries.
- Neural Data Queries, a system for semantic queries of neurons and their connections.
2. Related Work
3. Overview
3.1. Volumetric EM Image Data
3.2. Data Management Concepts
3.3. Scalability Through Demand-Driven and Display-Aware Web Applications
4. Visualization during Acquisition
4.1. Motivation
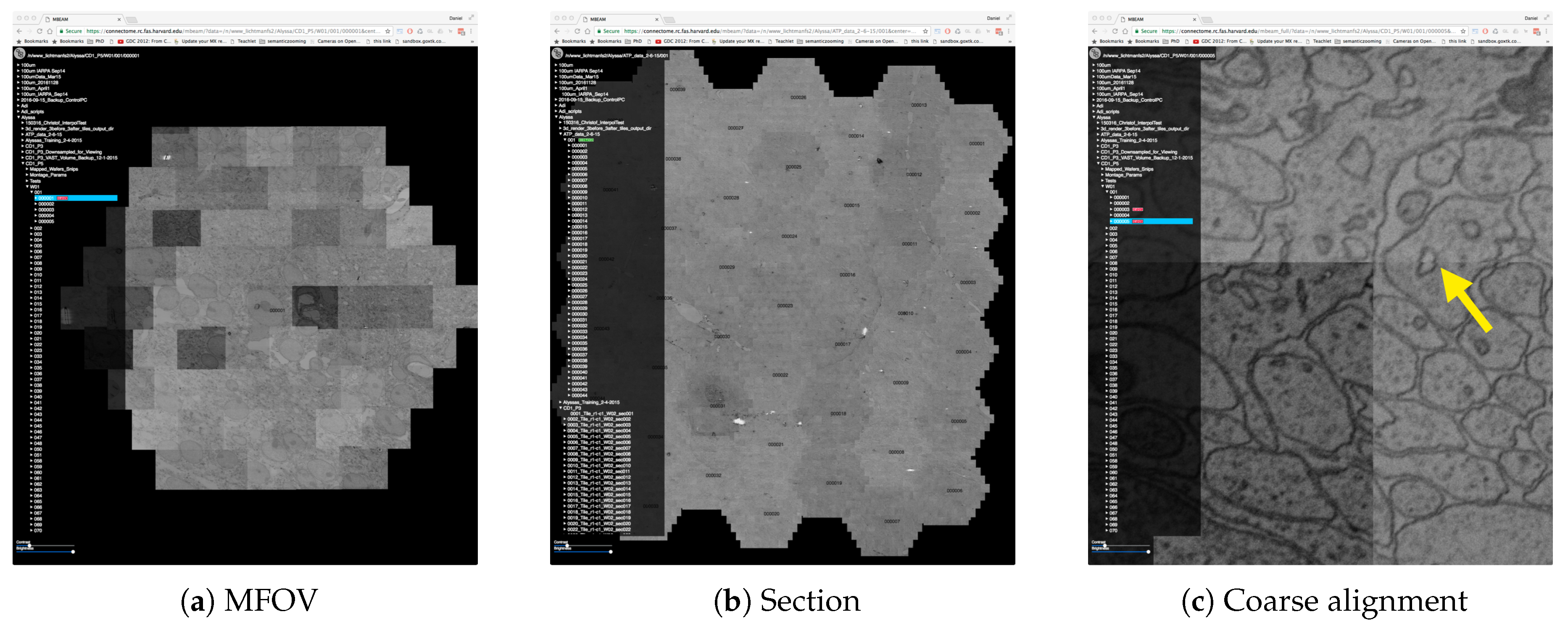
4.2. Data
4.3. The MBeam Viewer
5. Visualization of Registration
5.1. Motivation
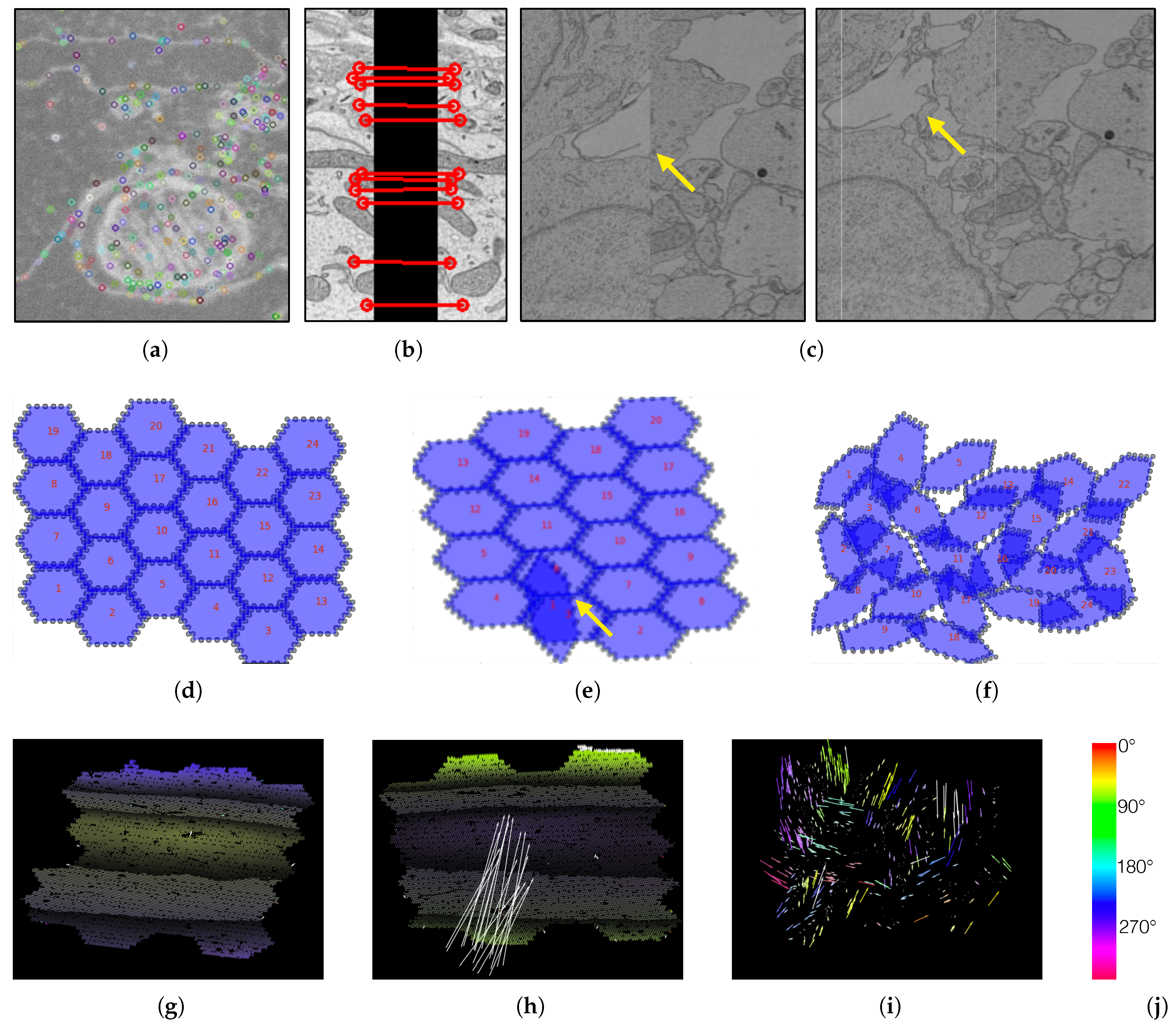
5.2. Data
5.3. The RHAligner Plugin
6. Visualization of Segmentation
6.1. Motivation
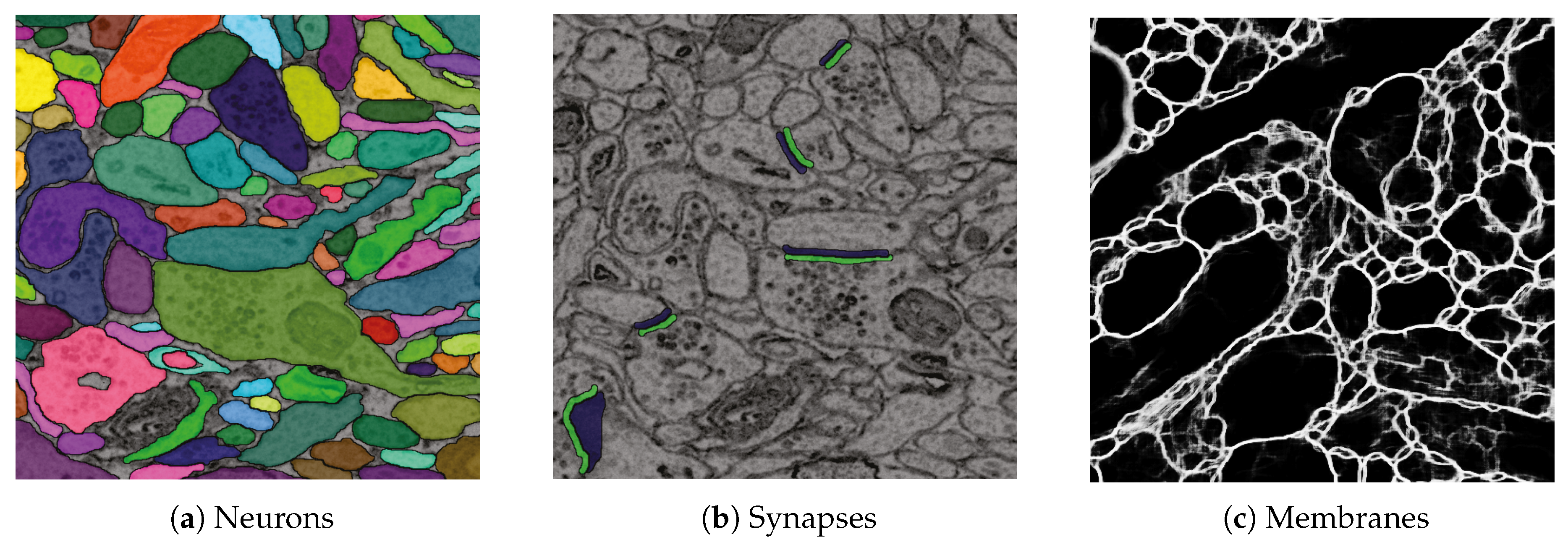
6.2. Data
6.3. RhoANAScope
7. Interactive Visualization for Proofreading
7.1. Motivation
7.2. Data
7.3. Proofreading Applications
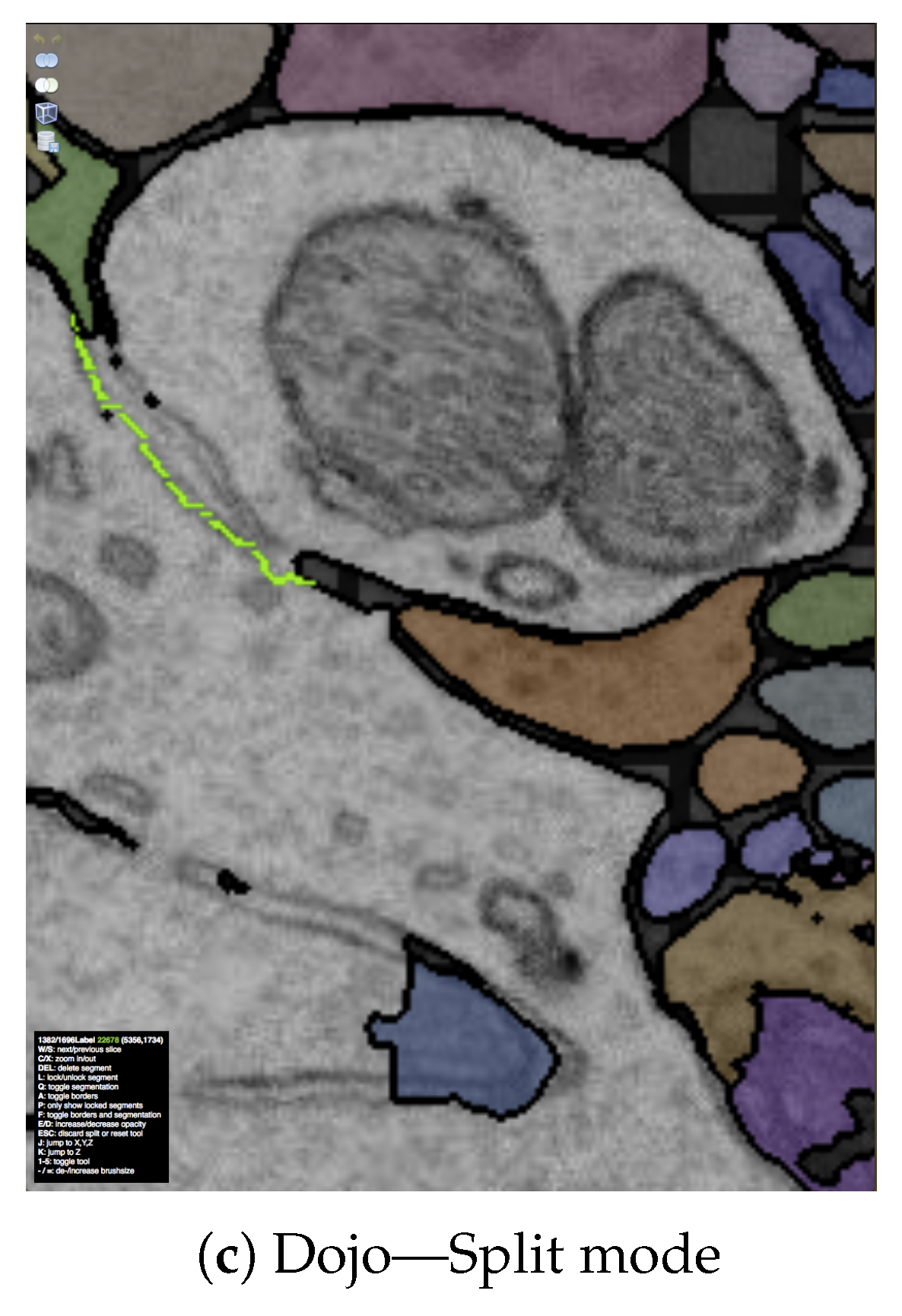
7.3.1. Interactive Proofreading Using Dojo
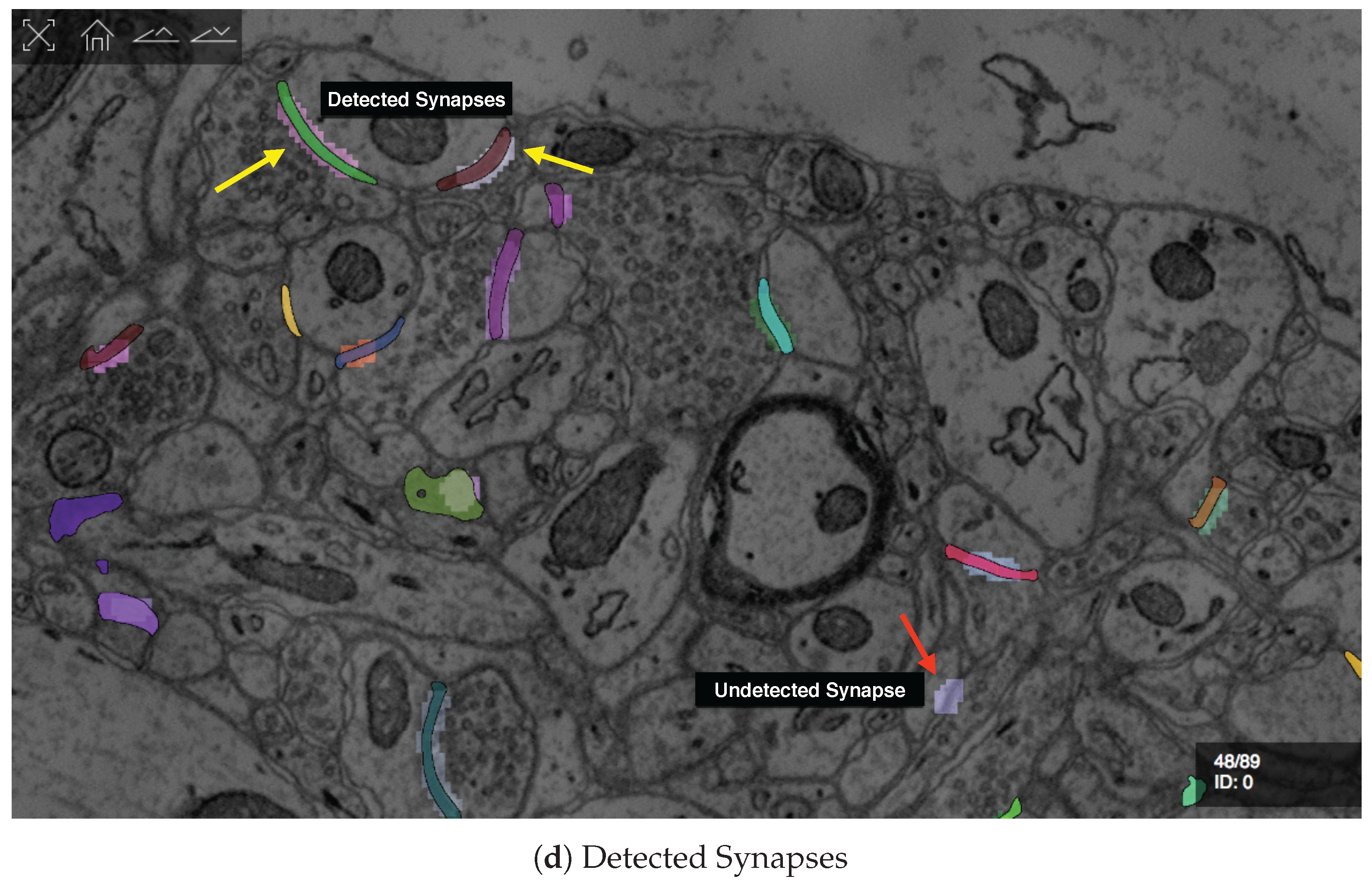
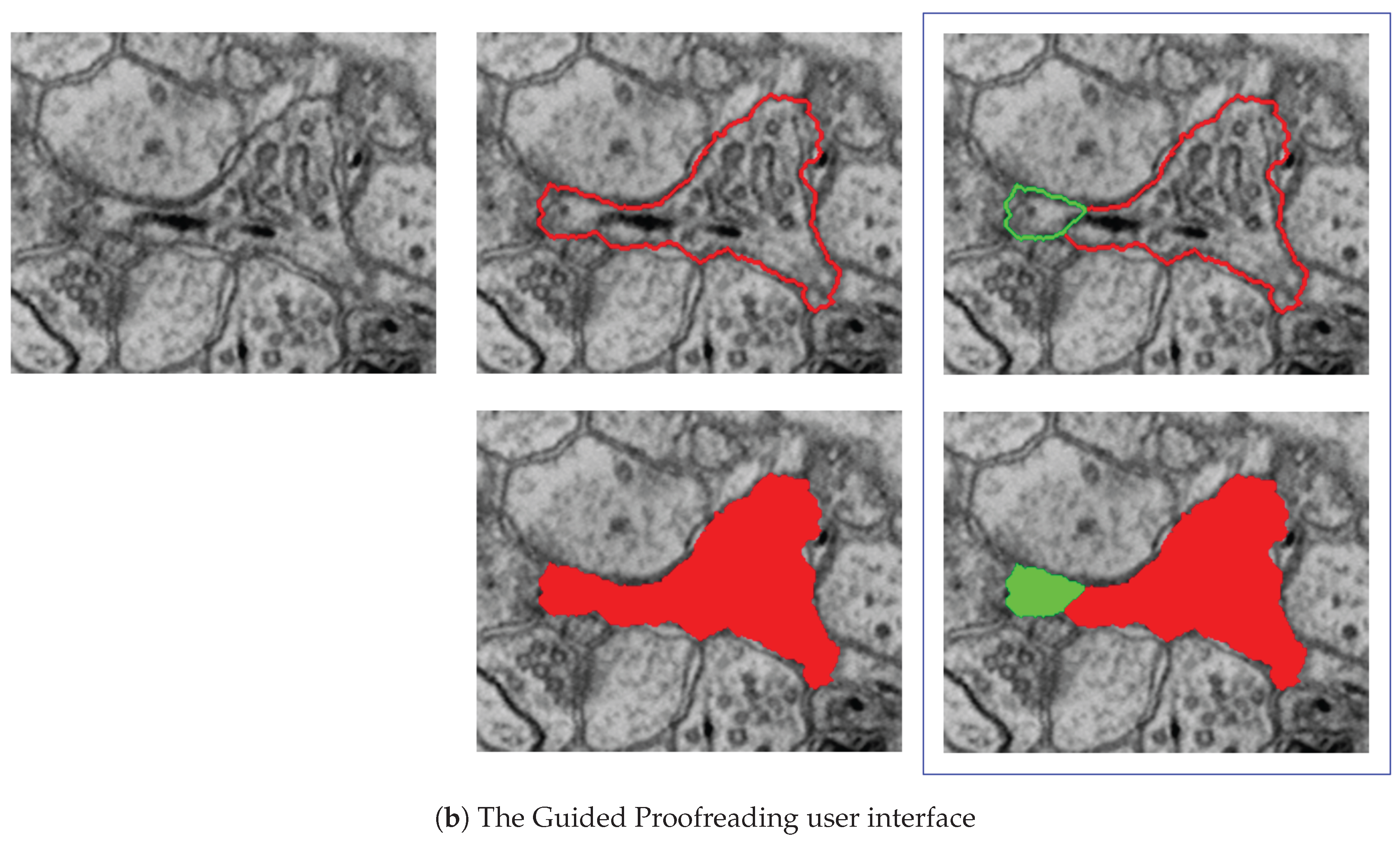
7.3.2. Guided Proofreading
8. Network Analysis
8.1. Motivation
8.2. Data
8.3. Tools for Network Analysis
8.3.1. 3DXP
8.3.2. Neural Data Queries
9. Performance and Scalability Experiments
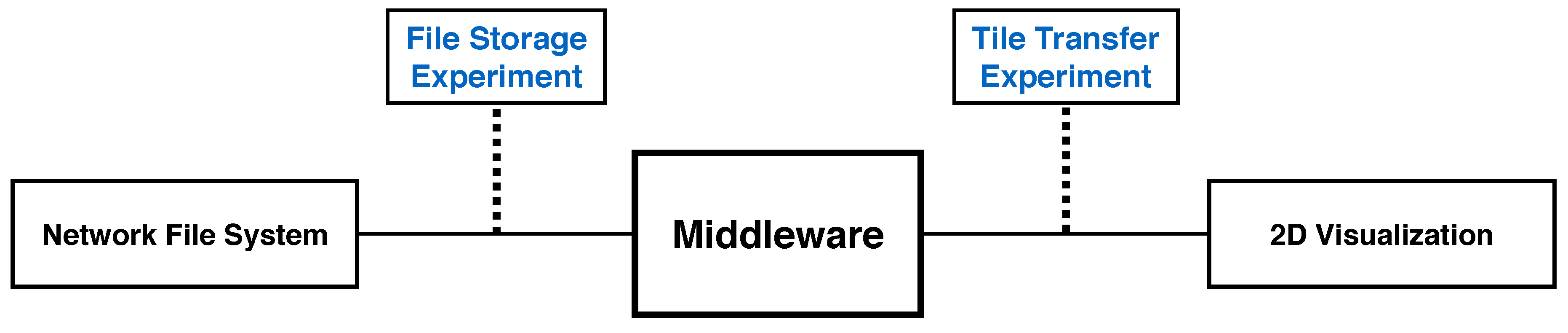
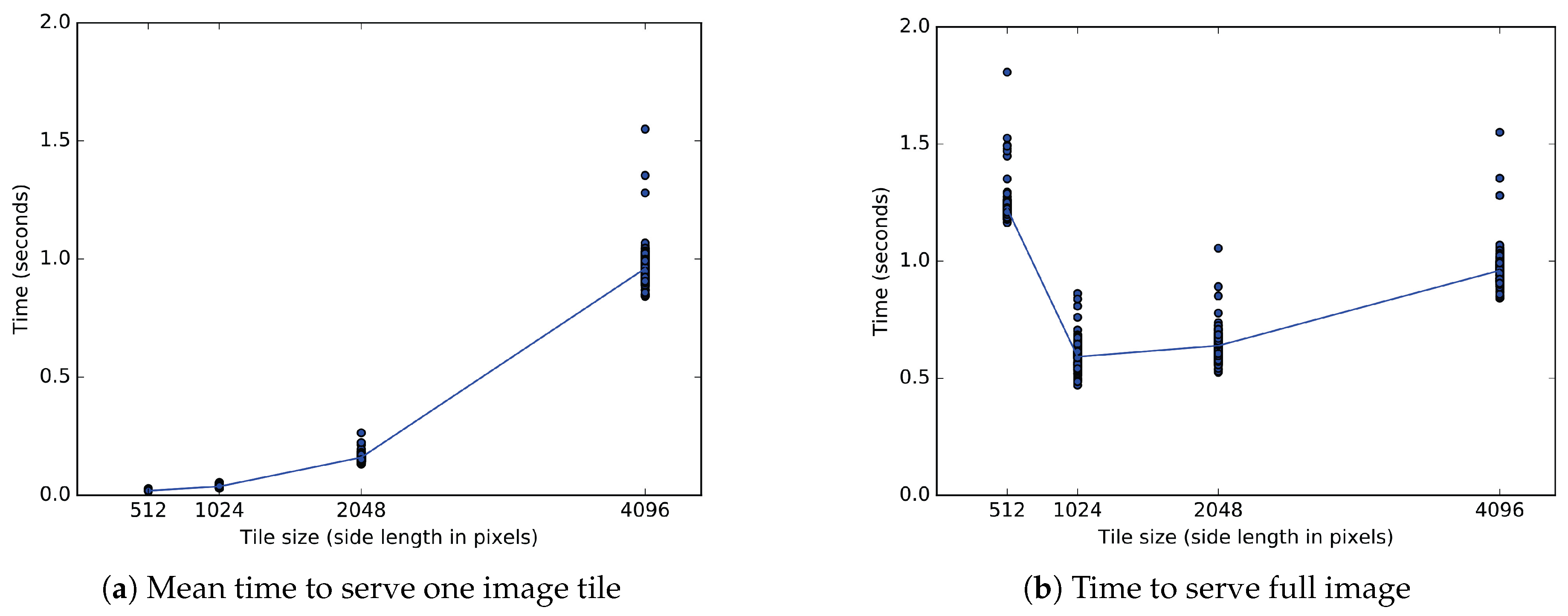
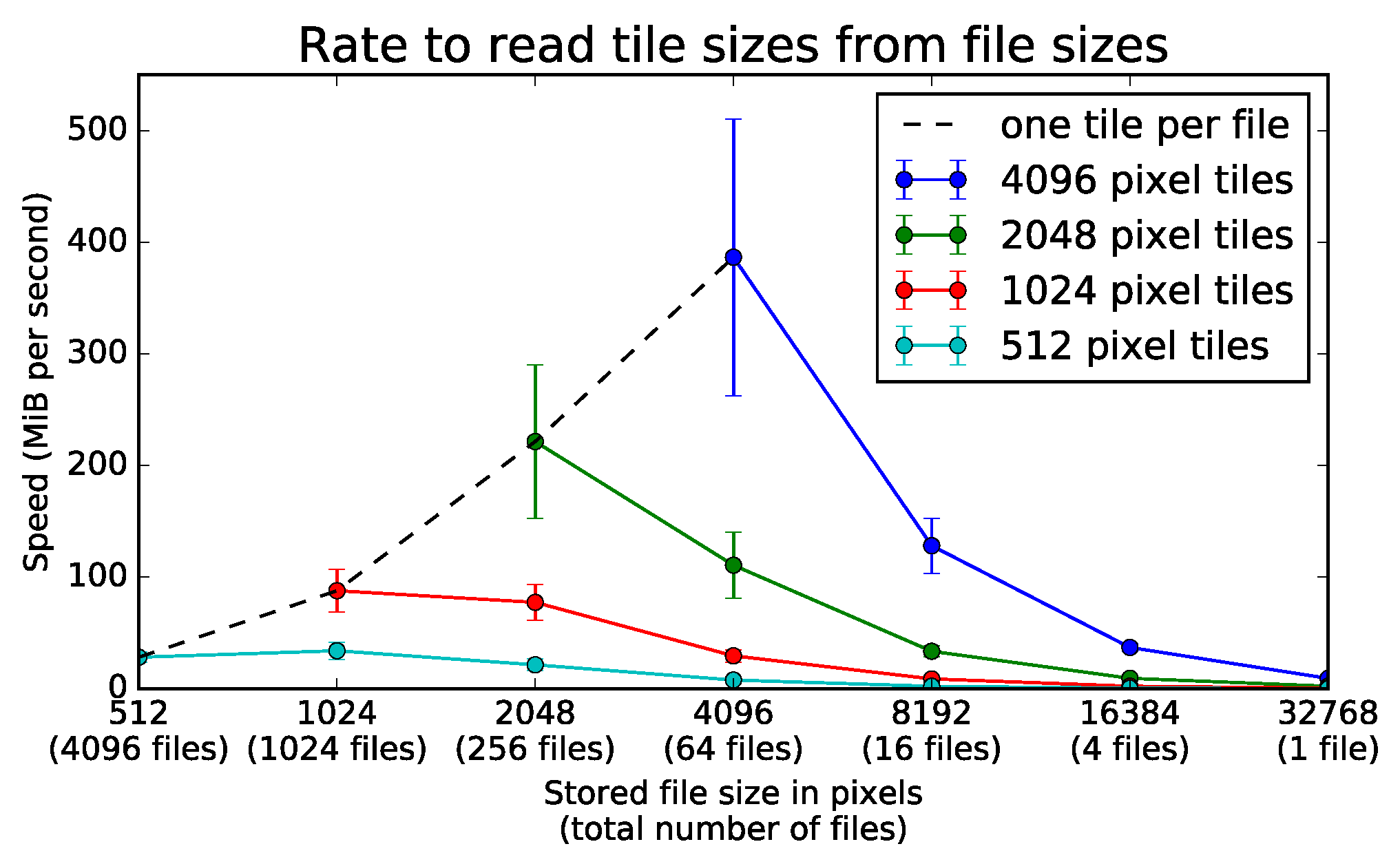
9.1. Client Tile Transfer Experiment
9.1.1. Experimental Setup
9.1.2. Experimental Results
9.2. File Storage Experiment
9.2.1. Experimental Setup
9.2.2. Experimental Results
10. Implementations and Distribution
10.1. Data Access API
10.2. Distribution
11. Use Case: Splitting Merged Somas
12. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Lichtman, J.W. The Big and the Small: Challenges of Imaging the Brain’s Circuits. Science 2011, 334, 618–623. [Google Scholar] [CrossRef]
- Seung, S. Connectome: How the Brain’s Wiring Makes Us Who We Are; Houghton Mifflin Harcourt: Boston, MA, USA, 2012. [Google Scholar]
- Hagmann, P. From Diffusion MRI to Brain Connectomics. Ph.D. Thesis, Université de Lausanne de Nationalité Suisse et Originaire de Däniken, Lausanne, Switzerland, 2005. [Google Scholar]
- Sporns, O.; Tononi, G.; Kötter, R. The Human Connectome: A Structural Description of the Human Brain. PLoS Comput. Biol. 2005, 1. [Google Scholar] [CrossRef] [PubMed]
- Kasthuri, N.; Hayworth, K.J.; Berger, D.R.; Schalek, R.L.; Conchello, J.A.; Knowles-Barley, S.; Lee, D.; Vázquez-Reina, A.; Kaynig, V.; Jones, T.R.; et al. Saturated reconstruction of a volume of neocortex. Cell 2015, 162, 648–661. [Google Scholar] [CrossRef] [PubMed]
- Suissa-Peleg, A.; Haehn, D.; Knowles-Barley, S.; Kaynig, V.; Jones, T.R.; Wilson, A.; Schalek, R.; Lichtman, J.W.; Pfister, H. Automatic Neural Reconstruction from Petavoxel of Electron Microscopy Data. Microsc. Microanal. 2016, 22, 536–537. [Google Scholar] [CrossRef]
- Schalek, R.; Lee, D.; Kasthuri, N.; Peleg, A.; Jones, T.; Kaynig, V.; Haehn, D.; Pfister, H.; Cox, D.; Lichtman, J. Imaging a 1 mm3 Volume of Rat Cortex Using a MultiBeam SEM. Microsc. Microanal. 2016, 22, 582–583. [Google Scholar] [CrossRef]
- Kaynig, V.; Vazquez-Reina, A.; Knowles-Barley, S.; Roberts, M.; Jones, T.R.; Kasthuri, N.; Miller, E.; Lichtman, J.; Pfister, H. Large-scale automatic reconstruction of neuronal processes from electron microscopy images. Med. Image Anal. 2015, 22, 77–88. [Google Scholar] [CrossRef] [PubMed]
- Knowles-Barley, S.; Kaynig, V.; Jones, T.R.; Wilson, A.; Morgan, J.; Lee, D.; Berger, D.; Kasthuri, N.; Lichtman, J.W.; Pfister, H. RhoanaNet Pipeline: Dense Automatic Neural Annotation. arXiv, 2016; arXiv:1611.06973. [Google Scholar]
- IEEE ISBI Challenge: SNEMI3D—3D Segmentation of Neurites in EM Images. 2013. Available online: http://brainiac2.mit.edu/SNEMI3D (accessed on 21 August 2017).
- Haehn, D.; Knowles-Barley, S.; Roberts, M.; Beyer, J.; Kasthuri, N.; Lichtman, J.; Pfister, H. Design and Evaluation of Interactive Proofreading Tools for Connectomics. IEEE Trans. Vis. Comput. Graph. 2014, 20, 2466–2475. [Google Scholar] [CrossRef] [PubMed]
- Haehn, D.; Kaynig, V.; Tompkin, J.; Lichtman, J.W.; Pfister, H. Guided Proofreading of Automatic Segmentations for Connectomics. arXiv, 2017; arXiv:1704.00848. [Google Scholar]
- Al-Awami, A.K.; Beyer, J.; Haehn, D.; Kasthuri, N.; Lichtman, J.W.; Pfister, H.; Hadwiger, M. NeuroBlocks—Visual Tracking of Segmentation and Proofreading for Large Connectomics Projects. IEEE Trans. Vis. Comput. Graph. 2016, 22, 738–746. [Google Scholar] [CrossRef] [PubMed]
- Al-Awami, A.; Beyer, J.; Strobelt, H.; Kasthuri, N.; Lichtman, J.; Pfister, H.; Hadwiger, M. NeuroLines: A Subway Map Metaphor for Visualizing Nanoscale Neuronal Connectivity. IEEE Trans. Vis. Comput. Graph. 2014, 20, 2369–2378. [Google Scholar] [CrossRef] [PubMed]
- Beyer, J.; Al-Awami, A.; Kasthuri, N.; Lichtman, J.W.; Pfister, H.; Hadwiger, M. ConnectomeExplorer: Query-Guided Visual Analysis of Large Volumetric Neuroscience Data. IEEE Trans. Vis. Comput. Graph. 2013, 19, 2868–2877. [Google Scholar] [CrossRef] [PubMed]
- Lichtman, J.W.; Pfister, H.; Shavit, N. The big data challenges of connectomics. Nat. Neurosci. 2014, 17, 1448–1454. [Google Scholar] [CrossRef]
- Pfister, H.; Kaynig, V.; Botha, C.P.; Bruckner, S.; Dercksen, V.J.; Hege, H.C.; Roerdink, J.B. Visualization in Connectomics. arXiv, 2012; arXiv:1206.1428v2. [Google Scholar]
- Margulies, D.S.; Böttger, J.; Watanabe, A.; Gorgolewski, K.J. Visualizing the human connectome. NeuroImage 2013, 80, 445–461. [Google Scholar] [CrossRef] [PubMed]
- Hayworth, K.J.; Morgan, J.L.; Schalek, R.; Berger, D.R.; Hildebrand, D.G.C.; Lichtman, J.W. Imaging ATUM ultrathin section libraries with WaferMapper: A multi-scale approach to EM reconstruction of neural circuits. Front. Neural Circuits 2014, 8. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, H.E. Nanoscience: The Science of the Small in Physics, Engineering, Chemistry, Biology and Medicine; Springer: Berlin/Heidelberg, Germany, 2010; Charpter 2. [Google Scholar]
- Janelia Farm. Raveler. 2014. Available online: https://openwiki.janelia.org/wiki/display/flyem/Raveler (accessed on 27 August 2017).
- Knowles-Barley, S.; Roberts, M.; Kasthuri, N.; Lee, D.; Pfister, H.; Lichtman, J.W. Mojo 2.0: Connectome Annotation Tool. Front. Neuroinform. 2013. [Google Scholar] [CrossRef]
- NeuTu: Software Package for Neuron Reconstruction and Visualization. 2013. Available online: https://github.com/janelia-flyem/NeuTu (accessed on 20 May 2017).
- Hadwiger, M.; Beyer, J.; Jeong, W.K.; Pfister, H. Interactive Volume Exploration of Petascale Microscopy Data Streams Using a Visualization-Driven Virtual Memory Approach. IEEE Trans. Vis. Comput. Graph. 2012, 18, 2285–2294. [Google Scholar] [CrossRef] [PubMed]
- Beyer, J.; Hadwiger, M.; Al-Awami, A.; Jeong, W.K.; Kasthuri, N.; Lichtman, J.W.; Pfister, H. Exploring the Connectome: Petascale Volume Visualization of Microscopy Data Streams. IEEE Comput. Graph. Appl. 2013, 33, 50–61. [Google Scholar] [CrossRef] [PubMed]
- Sicat, R.; Hadwiger, M.; Mitra, N.J. Graph Abstraction for Simplified Proofreading of Slice-based Volume Segmentation. In Proceedings of the 34th Annual Conference of the European Association for Computer Graphics, Girona, Spain, 6–10 May 2013. [Google Scholar]
- Kim, J.S.; Greene, M.J.; Zlateski, A.; Lee, K.; Richardson, M.; Turaga, S.C.; Purcaro, M.; Balkam, M.; Robinson, A.; Behabadi, B.F.; et al. Space-time wiring specificity supports direction selectivity in the retina. Nature 2014, 509, 331–336. [Google Scholar] [CrossRef] [PubMed]
- Giuly, R.J.; Kim, K.Y.; Ellisman, M.H. DP2: Distributed 3D image segmentation using micro-labor workforce. Bioinformatics 2013, 29, 1359–1360. [Google Scholar] [CrossRef] [PubMed]
- Saalfeld, S.; Cardona, A.; Hartenstein, V.; Tomančák, P. CATMAID: Collaborative annotation toolkit for massive amounts of image data. Bioinformatics 2009, 25, 1984–1986. [Google Scholar] [CrossRef] [PubMed]
- Anderson, J.; Mohammed, S.; Grimm, B.; Jones, B.; Koshevoy, P.; Tasdizen, T.; Whitaker, R.; Marc, R. The Viking Viewer for connectomics: Scalable multi-user annotation and summarization of large volume data sets. J. Micros. 2011, 241, 13–28. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.Y.; Tsai, K.L.; Wang, S.C.; Hsieh, C.H.; Chang, H.M.; Chiang, A.S. The Neuron Navigator: Exploring the information pathway through the neural maze. In Proceedings of the 2011 IEEE Pacific Visualization Symposium, Hong Kong, China, 1–4 March 2011; pp. 35–42. [Google Scholar]
- Ginsburg, D.; Gerhard, S.; Calle, J.E.C.; Pienaar, R. Realtime Visualization of the Connectome in the Browser using WebGL. Front. Neuroinform. 2011. [Google Scholar] [CrossRef]
- Neuroglancer: WebGL-Based Viewer for Volumetric Data. 2017. Available online: https://github.com/google/neuroglancer (accessed on 29 May 2017).
- Khronos Group. WebGL Specification. 2014. Available online: http://www.khronos.org/registry/webgl/specs (accessed on 31 March 2014).
- Haehn, D.; Rannou, N.; Ahtam, B.; Grant, E.; Pienaar, R. Neuroimaging in the Browser using the X Toolkit. Front. Neuroinform. 2012. [Google Scholar] [CrossRef]
- Haehn, D. Slice:Drop: Collaborative medical imaging in the browser. In Proceedings of the ACM SIGGRAPH 2013 Computer Animation Festival, Anaheim, CA, USA, 21–25 July 2013; p. 1. [Google Scholar]
- Bakker, R.; Tiesinga, P.; Kötter, R. The Scalable Brain Atlas: Instant Web-Based Access to Public Brain Atlases and Related Content. Neuroinformatics 2015, 13, 353–366. [Google Scholar] [CrossRef] [PubMed]
- Stephan, K.E.; Kamper, L.; Bozkurt, A.; Burns, G.A.P.C.; Young, M.P.; Kötter, R. Advanced database methodology for the Collation of Connectivity data on the Macaque brain (CoCoMac). Philos. Trans. R. Soc. Lond. B Biol. Sci. 2001, 356, 1159–1186. [Google Scholar] [CrossRef] [PubMed]
- Bota, M.; Dong, H.W.; Swanson, L.W. Brain architecture management system. Neuroinformatics 2005, 3, 15–47. [Google Scholar] [CrossRef]
- Schmitt, O.; Eipert, P. neuroVIISAS: Approaching Multiscale Simulation of the Rat Connectome. Neuroinformatics 2012, 10, 243–267. [Google Scholar] [CrossRef] [PubMed]
- Gerhard, S.; Daducci, A.; Lemkaddem, A.; Meuli, R.; Thiran, J.; Hagmann, P. The connectome viewer toolkit: An open source framework to manage, analyze, and visualize connectomes. Front. Neuroinform. 2011, 5. [Google Scholar] [CrossRef] [PubMed]
- Sorger, J.; Buhler, K.; Schulze, F.; Liu, T.; Dickson, B. neuroMap—Interactive graph-visualization of the fruit fly’s neural circuit. In Proceedings of the 2013 IEEE Symposium on Biological Data Visualization (BioVis), Atlanta, GA, USA, 13–14 October 2013; pp. 73–80. [Google Scholar]
- DVID. Distributed, Versioned, Image-Oriented Dataservice. 2016. Available online: https://github.com/janelia-flyem/dvid/wiki (accessed on 14 January 2016).
- The Boss: A Cloud Based Storage Service Developed for the IARPA MICrONS Program. 2017. Available online: https://docs.theboss.io/ (accessed on 29 May 2017).
- Matejek, B.; Haehn, D.; Lekschas, F.; Mitzenmacher, M.; Pfister, H. Compresso: Efficient Compression of Segmentation Data For Connectomics. In Proceedings of the International Conference on Medical Image Computing and Computer-Assisted Intervention, Quebec City, QC, Canada, 10–14 September 2017. [Google Scholar]
- Williams, L. Pyramidal parametrics. In Proceedings of the 10th Annual Conference on Computer Graphics and Interactive Techniques, Detroit, MI, USA, 25–29 July 1983; ACM: New York, NY, USA, 1983; Volume 17, pp. 1–11. [Google Scholar]
- Kaiser, G.E. Cooperative Transactions for Multiuser Environments. In Modern Database Systems; ACM Press/Addison-Wesley Publishing Co.: New York, NY, USA, 1995; pp. 409–433. [Google Scholar]
- Jeong, W.K.; Johnson, M.K.; Yu, I.; Kautz, J.; Pfister, H.; Paris, S. Display-aware image editing. In Proceedings of the 2011 IEEE International Conference on Computational Photography (ICCP), Pittsburgh, PA, USA, 8–10 April 2011; pp. 1–8. [Google Scholar]
- Beyer, J.; Hadwiger, M.; Jeong, W.K.; Pfister, H.; Lichtman, J. Demand-driven volume rendering of terascale EM data. In Proceedings of the International Conference on Computer Graphics and Interactive Techniques, SIGGRAPH 2011, Vancouver, BC, Canada, 7–11 August 2011; p. 57. [Google Scholar]
- Saalfeld, S.; Fetter, R.; Cardona, A.; Tomancak, P. Elastic volume reconstruction from series of ultra-thin microscopy sections. Nat. Methods 2012, 9, 717–720. [Google Scholar] [CrossRef] [PubMed]
- Lowe, D.G. Object Recognition from Local Scale-Invariant Features. In Proceedings of the Seventh IEEE International Conference on Computer Vision, Kerkyra, Greece, 20–25 September 1999; IEEE Computer Society: Washington, DC, USA, 1999; p. 1150. [Google Scholar]
- Janelia Farm. The Tilespec JSON Data Model. 2015. Available online: https://github.com/saalfeldlab/render/blob/master/docs/src/site/markdown/data- model.md (accessed on 27 August 2017).
- Nunez-Iglesias, J.; Kennedy, R.; Parag, T.; Shi, J.; Chklovskii, D.B. Machine Learning of Hierarchical Clustering to Segment 2D and 3D Images. PLoS ONE 2013, 8. [Google Scholar] [CrossRef]
- Ronneberger, O.; Fischer, P.; Brox, T. U-Net: Convolutional Networks for Biomedical Image Segmentation. In Proceedings of the International Conference on Medical Image Computing and Computer-Assisted Intervention (MICCAI), Munich, Germany, 5–9 October 2015; Springer: Berlin, Germany, 2015; Volume 9351, pp. 234–241. [Google Scholar]
- Nguyen, Q. Parallel and Scalable Neural Image Segmentation for Connectome Graph Extraction; Massachusetts Institute of Technology: Cambridge, MA, USA, 2015. [Google Scholar]
- Nunez-Iglesias, J.; Kennedy, R.; Plaza, S.M.; Chakraborty, A.; Katz, W.T. Graph-based active learning of agglomeration (GALA): A Python library to segment 2D and 3D neuroimages. Front. Neuroinform. 2014, 8. [Google Scholar] [CrossRef] [PubMed]
- Parag, T.; Chakraborty, A.; Plaza, S.; Scheffer, L. A Context-Aware Delayed Agglomeration Framework for Electron Microscopy Segmentation. PLoS ONE 2015, 10. [Google Scholar] [CrossRef] [PubMed]
- Santurkar, S.; Budden, D.M.; Matveev, A.; Berlin, H.; Saribekyan, H.; Meirovitch, Y.; Shavit, N. Toward Streaming Synapse Detection with Compositional ConvNets. arXiv, 2017; arXiv:1702.07386. [Google Scholar]
- Lorensen, W.E.; Cline, H.E. Marching Cubes: A High Resolution 3D Surface Construction Algorithm. In Proceedings of the 14th Annual Conference on Computer Graphics and Interactive Techniques, Anaheim, CA, USA, 27–31 July 1987. [Google Scholar]
- Limper, M.; Jung, Y.; Behr, J.; Alexa, M. The POP Buffer: Rapid Progressive Clustering by Geometry Quantization. Comput. Graph. Forum 2013, 32, 197–206. [Google Scholar] [CrossRef]
- Dory, M.; Parrish, A.; Berg, B. Introduction to Tornado; O’Reilly Media, Inc.: Sebastopol, CA, USA, 2012. [Google Scholar]
- OpenSeaDragon. 2016. Available online: http://openseadragon.github.io/ (accessed on 27 August 2017).
- Nouri, D. Nolearn: Scikit-Learn Compatible Neural Network Library. 2016. Available online: https://github.com/dnouri/nolearn (accessed on 27 August 2017).
- Behr, J.; Eschler, P.; Jung, Y.; Zöllner, M. X3DOM: A DOM-based HTML5/X3D Integration Model. In Proceedings of the 14th International Conference on 3D Web Technology, Darmstadt, Germany, 16–17 June 2009; ACM: New York, NY, USA, 2009; pp. 127–135. [Google Scholar]
- Chodorow, K.; Dirolf, M. MongoDB: The Definitive Guide, 1st ed.; O’Reilly Media, Inc.: Sebastopol, CA, USA, 2010. [Google Scholar]

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type | Digital Dimensions | Physical Dimensions | Image Size | Segmentation Size (Compressed) |
|---|---|---|---|---|
| Tile | pixels | MB | MB ( KB) | |
| MFOV | pixels | 520 MB | GB ( MB) | |
| Section | pixels | GB | GB ( MB) | |
| Scan A (m) | voxels | TB | TB ( GB) | |
| Scan B (1 mm) | 33,940 voxels | PB | PB ( TB) |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Haehn, D.; Hoffer, J.; Matejek, B.; Suissa-Peleg, A.; Al-Awami, A.K.; Kamentsky, L.; Gonda, F.; Meng, E.; Zhang, W.; Schalek, R.; et al. Scalable Interactive Visualization for Connectomics. Informatics 2017, 4, 29. https://doi.org/10.3390/informatics4030029
Haehn D, Hoffer J, Matejek B, Suissa-Peleg A, Al-Awami AK, Kamentsky L, Gonda F, Meng E, Zhang W, Schalek R, et al. Scalable Interactive Visualization for Connectomics. Informatics. 2017; 4(3):29. https://doi.org/10.3390/informatics4030029
Chicago/Turabian StyleHaehn, Daniel, John Hoffer, Brian Matejek, Adi Suissa-Peleg, Ali K. Al-Awami, Lee Kamentsky, Felix Gonda, Eagon Meng, William Zhang, Richard Schalek, and et al. 2017. "Scalable Interactive Visualization for Connectomics" Informatics 4, no. 3: 29. https://doi.org/10.3390/informatics4030029
APA StyleHaehn, D., Hoffer, J., Matejek, B., Suissa-Peleg, A., Al-Awami, A. K., Kamentsky, L., Gonda, F., Meng, E., Zhang, W., Schalek, R., Wilson, A., Parag, T., Beyer, J., Kaynig, V., Jones, T. R., Tompkin, J., Hadwiger, M., Lichtman, J. W., & Pfister, H. (2017). Scalable Interactive Visualization for Connectomics. Informatics, 4(3), 29. https://doi.org/10.3390/informatics4030029