
Discovering Entities Similarities in Biological Networks Using a Hybrid Immune Algorithm



Abstract
1. Introduction
2. Community Detection
Modularity Optimization in Networks
- for each vertex will denote the degree of i, i.e., the total number of edges in E that have i as one of the endpoints;
- for each community will denote the sum of the degrees of the vertices belonging to community , i.e.,
- for each community will denote the number of edges or links inside the community
- for each vertex and each community will denote the number of edges or links from i to vertices in
3. Hybrid-IA: The Hybrid Immune Algorithm
| Algorithm 1 Pseudo-code of Hybrid-IA. |
|
3.1. The Cloning Operator
3.2. The Hypermutation Operator
3.3. The Static Aging Operator
3.4. The Selection Operator
3.5. Local Search
4. Biological Networks Data Set
4.1. Protein–Protein Interaction (PPI) Networks
4.2. Metabolic Networks
4.3. Transcriptional Regulatory Networks
4.4. Synthetic Networks
5. Experimental Results
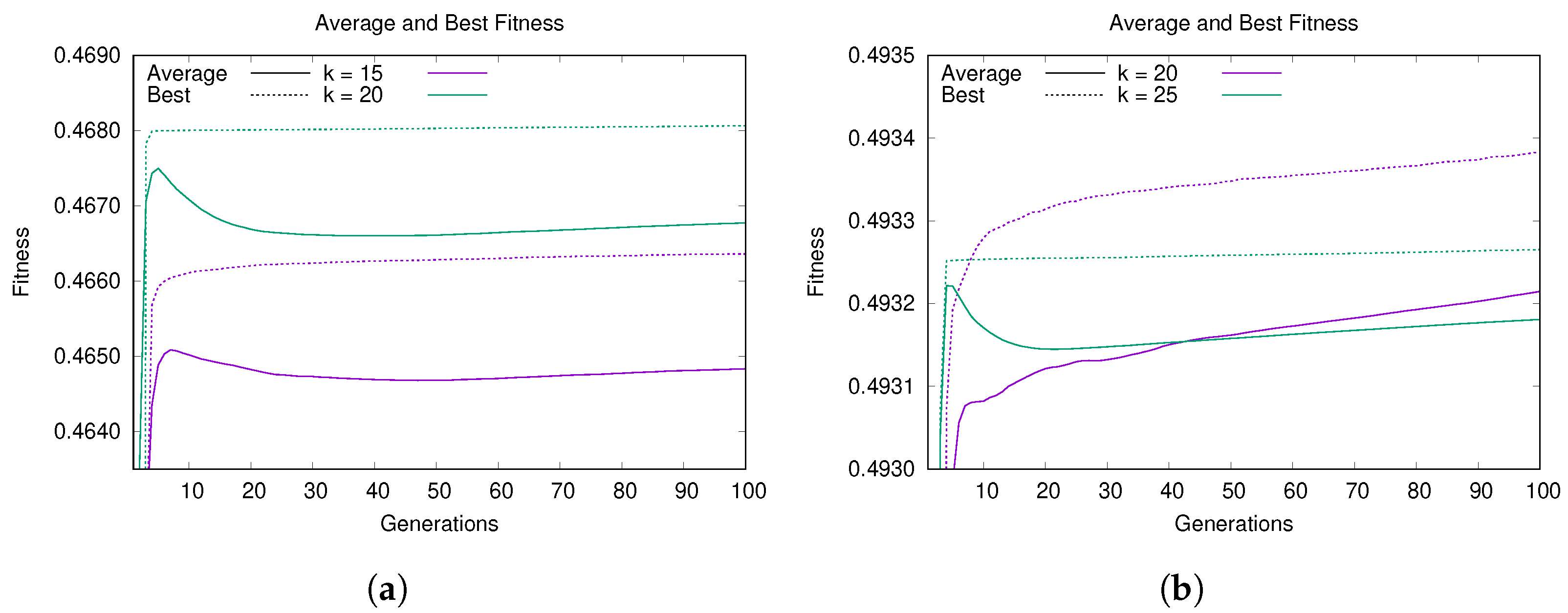
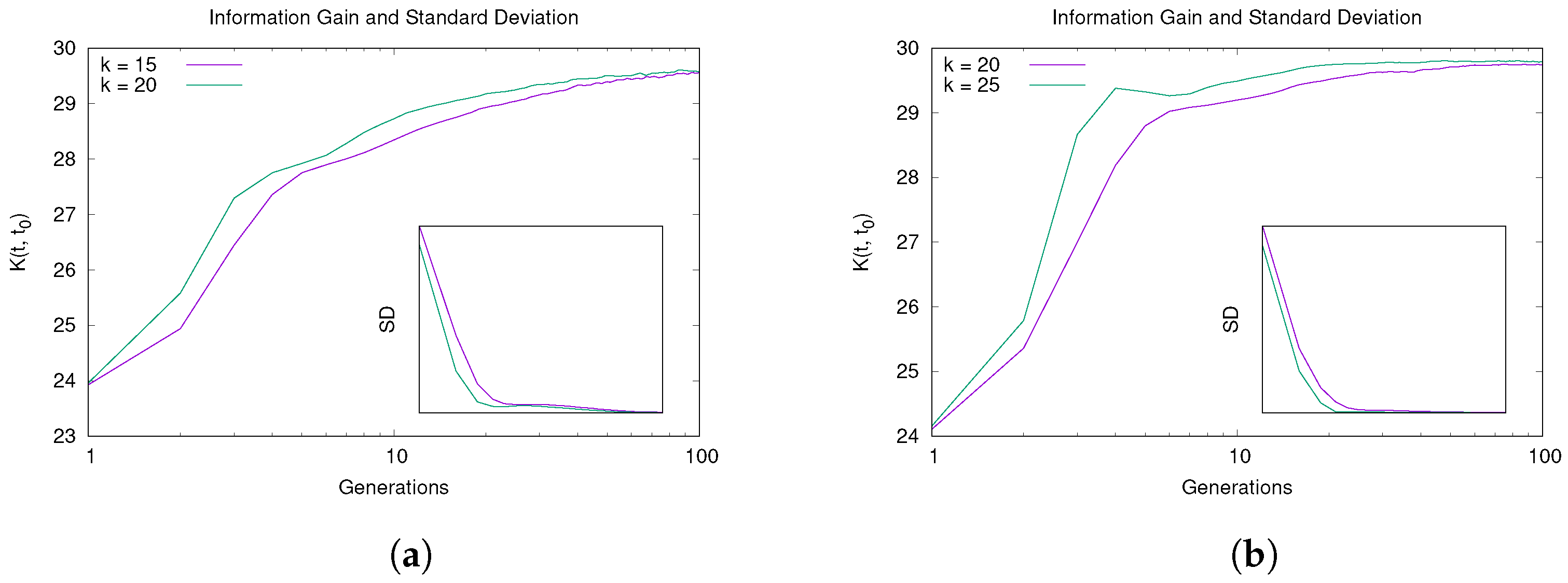
5.1. Convergence and Learning Analysis
5.2. The Biological Networks
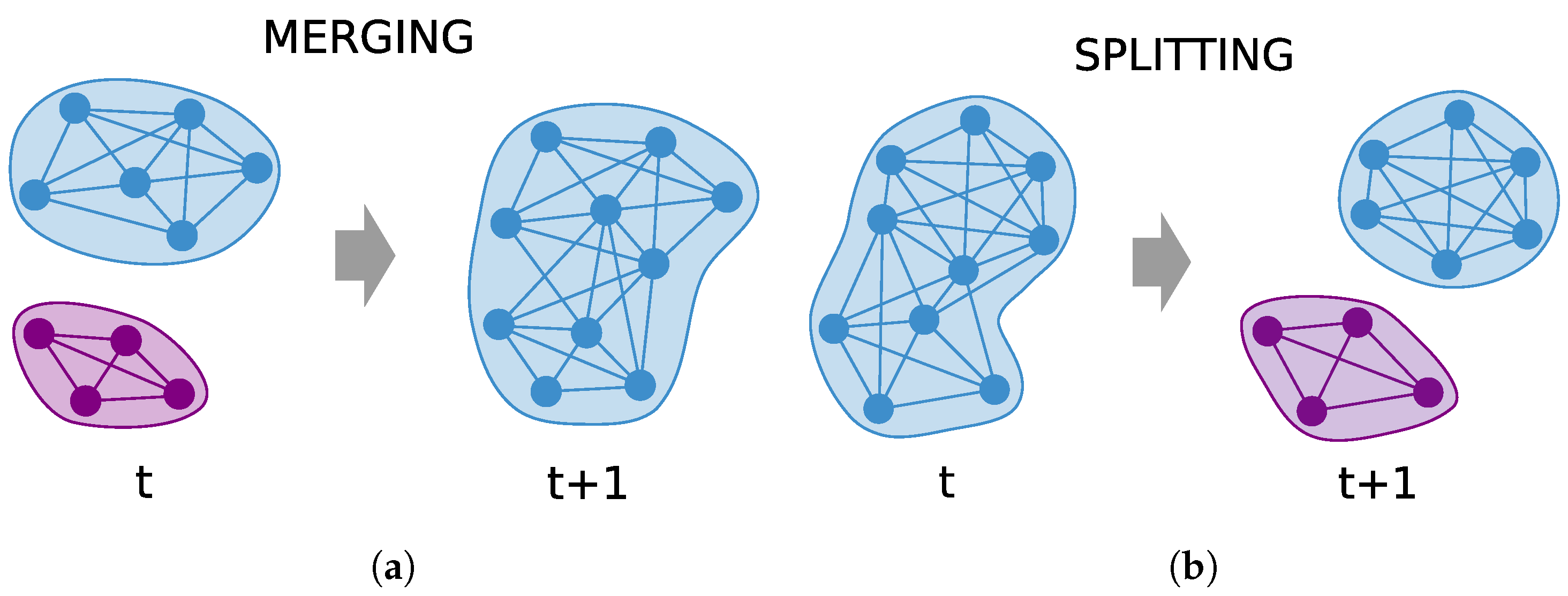
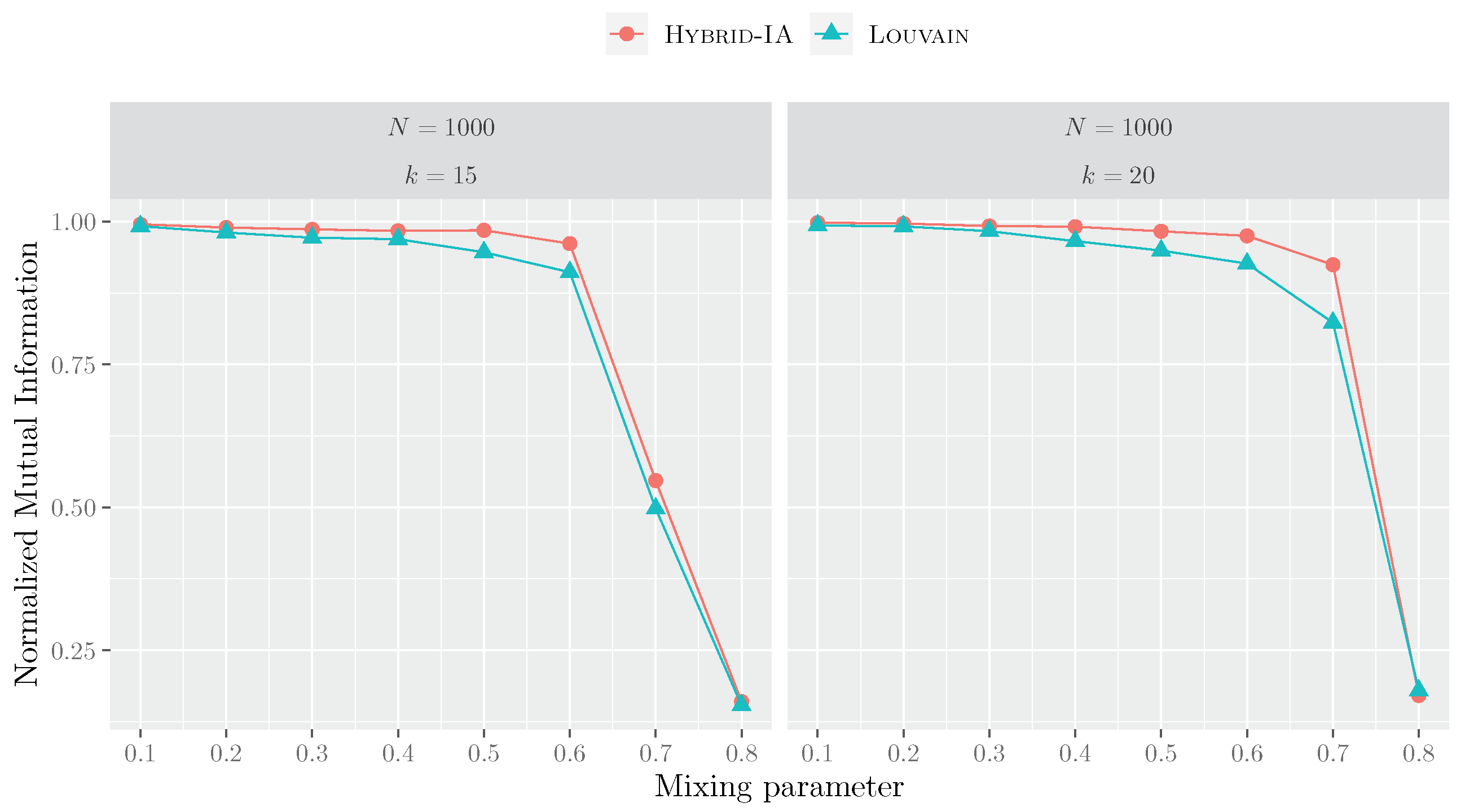
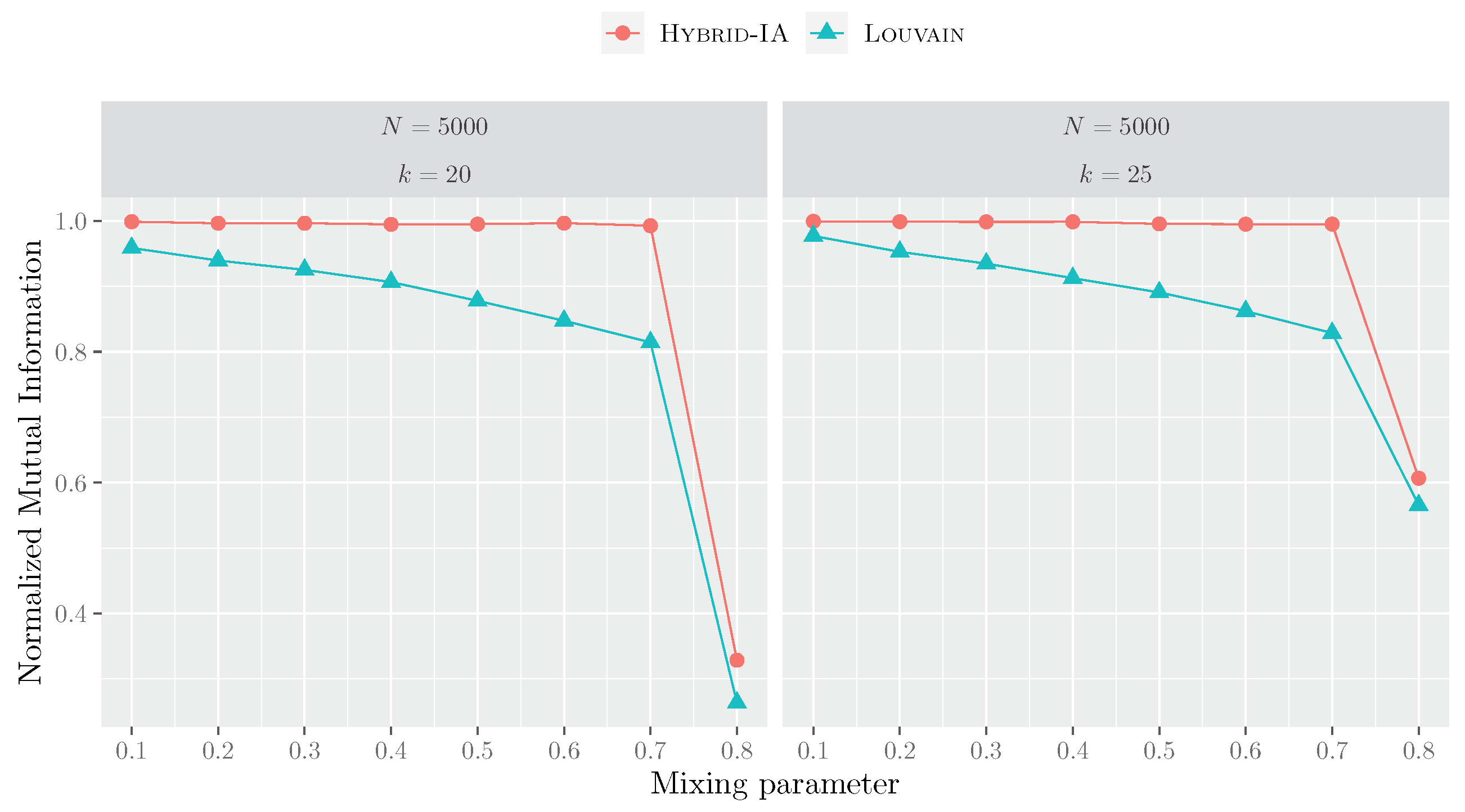
5.3. Functional Sensitivity of Community Detection
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Acknowledgments
Conflicts of Interest
References
- Von Mering, C.; Krause, R.; Snel, B.; Cornell, M.; Oliver, S.G.; Fields, S.; Bork, P. Comparative assessment of large-scale data sets of protein–protein interactions. Nature 2002, 417, 399–403. [Google Scholar] [CrossRef] [PubMed]
- Barabási, A.L.; Oltvai, Z.N. Network biology: Understanding the cell’s functional organization. Nat. Rev. Genet. 2004, 5, 101–113. [Google Scholar] [CrossRef] [PubMed]
- Barabási, A.L.; Gulbahce, N.; Loscalzo, J. Network medicine: A network-based approach to human disease. Nat. Rev. Genet. 2011, 12, 56–68. [Google Scholar] [CrossRef] [PubMed]
- Goh, K.I.; Cusick, M.E.; Valle, D.; Childs, B.; Vidal, M.; Barabási, A.L. The human disease network. Proc. Natl. Acad. Sci. USA 2007, 104, 8685–8690. [Google Scholar] [CrossRef] [PubMed]
- Aureli, M.; Masilamani, A.P.; Illuzzi, G.; Loberto, N.; Scandroglio, F.; Prinetti, A.; Chigorno, V.; Sonnino, S. Activity of plasma membrane β-galactosidase and β-glucosidase. FEBS Lett. 2009, 583, 2469–2473. [Google Scholar] [CrossRef]
- Spampinato, A.G.; Scollo, R.A.; Cavallaro, S.; Pavone, M.; Cutello, V. An Immunological Algorithm for Graph Modularity Optimization. In Advances in Intelligent Systems and Computing, Proceedings of the Advances in Computational Intelligence Systems (UKCI 2019), Portsmouth, UK, 4–6 September 2019; Ju, Z., Yang, L., Yang, C., Gegov, A., Zhou, D., Eds.; Springer: Cham, Switzerland, 2020; Volume 1043, pp. 235–247. [Google Scholar] [CrossRef]
- Cutello, V.; Fargetta, G.; Pavone, M.; Scollo, R.A. Optimization Algorithms for Detection of Social Interactions. Algorithms 2020, 13, 139. [Google Scholar] [CrossRef]
- Girvan, M.; Newman, M.E.J. Community structure in social and biological networks. Proc. Natl. Acad. Sci. USA 2002, 99, 7821–7826. [Google Scholar] [CrossRef]
- Gulbahce, N.; Lehmann, S. The art of community detection. BioEssays 2008, 30, 934–938. [Google Scholar] [CrossRef]
- Didimo, W.; Montecchiani, F. Fast layout computation of clustered networks: Algorithmic advances and experimental analysis. Inf. Sci. 2014, 260, 185–199. [Google Scholar] [CrossRef]
- Buchin, K.; Buchin, M.; Gudmundsson, J.; Löffler, M.; Luo, J. Detecting commuting patterns by clustering subtrajectories. Int. J. Comput. Geom. Appl. 2011, 21, 253–282. [Google Scholar] [CrossRef]
- Cavallaro, C.; Vizzari, G. A Novel Spatial–Temporal Analysis Approach to Pedestrian Groups Detection. Procedia Comput. Sci. 2022, 207, 2364–2373. [Google Scholar] [CrossRef]
- Cavallaro, C.; Vitrià, J. Corridor Detection from Large GPS Trajectories Datasets. Appl. Sci. 2020, 10, 5003. [Google Scholar] [CrossRef]
- Cavallaro, C.; Verga, G.; Tramontana, E.; Muscato, O. Multi-agent architecture for point of interest detection and recommendation. In Proceedings of the CEUR Workshop, Parma, Italy, 26–28 June 2019; Volume 2404, pp. 98–104. [Google Scholar]
- Cavallaro, C.; Verga, G.; Tramontana, E.; Muscato, O. Eliciting cities points of interest from people movements and suggesting effective itineraries. Intell. Artif. 2020, 14, 49–61. [Google Scholar] [CrossRef]
- Newman, M.E.J.; Girvan, M. Finding and evaluating community structure in networks. Phys. Rev. E 2004, 69, 026113. [Google Scholar] [CrossRef] [PubMed]
- Brandes, U.; Delling, D.; Gaertler, M.; Görke, R.; Hoefer, M.; Nikoloski, Z.; Wagner, D. On Modularity Clustering. IEEE Trans. Knowl. Data Eng. 2008, 20, 172–188. [Google Scholar] [CrossRef]
- Newman, M.E.J. Fast algorithm for detecting community structure in networks. Phys. Rev. E 2004, 69, 066133. [Google Scholar] [CrossRef]
- Newman, M.E.J. Finding community structure in networks using the eigenvectors of matrices. Phys. Rev. E 2006, 74, 036104. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.; Pan, X.; Tang, Z.; Luo, X. A Fast Fuzzy Clustering Algorithm for Complex Networks via a Generalized Momentum Method. IEEE Trans. Fuzzy Syst. 2022, 30, 3473–3485. [Google Scholar] [CrossRef]
- Hu, L.; Chan, K.C.C. Fuzzy Clustering in a Complex Network Based on Content Relevance and Link Structures. IEEE Trans. Fuzzy Syst. 2016, 24, 456–470. [Google Scholar] [CrossRef]
- Xu, Z.; Ke, Y.; Wang, Y.; Cheng, H.; Cheng, J. GBAGC: A General Bayesian Framework for Attributed Graph Clustering. ACM Trans. Knowl. Discov. Data 2014, 9, 1–43. [Google Scholar] [CrossRef]
- Hu, L.; Zhang, J.; Pan, X.; Yan, H.; You, Z.H. HiSCF: Leveraging higher-order structures for clustering analysis in biological networks. Bioinformatics 2020, 37, 542–550. [Google Scholar] [CrossRef]
- Hu, L.; Chan, K.C.C.; Yuan, X.; Xiong, S. A Variational Bayesian Framework for Cluster Analysis in a Complex Network. IEEE Trans. Knowl. Data Eng. 2020, 32, 2115–2128. [Google Scholar] [CrossRef]
- Pan, X.; Hu, L.; Hu, P.; You, Z.H. Identifying Protein Complexes From Protein–Protein Interaction Networks Based on Fuzzy Clustering and GO Semantic Information. IEEE/ACM Trans. Comput. Biol. Bioinform. 2022, 19, 2882–2893. [Google Scholar] [CrossRef] [PubMed]
- Talbi, E.G. Metaheuristics: From Design to Implementation; Wiley Publishing: Hoboken, NJ, USA, 2009. [Google Scholar]
- Scollo, R.A.; Cutello, V.; Pavone, M. Where the Local Search Affects Best in an Immune Algorithm. In Lecture Notes in Artificial Intelligence, Proceedings of the AIxIA 2020—Advances in Artificial Intelligence (AIxIA 2020), Virtual, 25–27 November 2020; Baldoni, M., Bandini, S., Eds.; Springer: Cham, Swizterland, 2021; Volume 12414, pp. 99–114. [Google Scholar] [CrossRef]
- Fortunato, S.; Barthélemy, M. Resolution limit in community detection. Proc. Natl. Acad. Sci. USA 2007, 104, 36–41. [Google Scholar] [CrossRef] [PubMed]
- Costanza, J.; Cutello, V.; Pavone, M. A Memetic Immunological Algorithm for Resource Allocation Problem. In Lecture Notes in Computer Science, Proceedings of the 10th International Conference on Artificial Immune Systems (ICARIS 2011), Cambridge, UK, 18–21 July 2011; Liò, P., Nicosia, G., Stibor, T., Eds.; Springer: Berlin/Heidelberg, Germany, 2011; Volume 6825, pp. 308–320. [Google Scholar] [CrossRef]
- Stracquadanio, G.; Greco, O.; Conca, P.; Cutello, V.; Pavone, M.; Nicosia, G. Packing equal disks in a unit square: An immunological optimization approach. In Proceedings of the International Workshop on Artificial Immune Systems (AIS), Taormina-Sicily, Italy, 17–18 July 2015; pp. 1–5. [Google Scholar] [CrossRef]
- Fouladvand, S.; Osareh, A.; Shadgar, B.; Pavone, M.; Sharafi, S. DENSA: An effective negative selection algorithm with flexible boundaries for self-space and dynamic number of detectors. Eng. Appl. Artif. Intell. 2017, 62, 359–372. [Google Scholar] [CrossRef]
- Pavone, M.; Narzisi, G.; Nicosia, G. Clonal selection: An immunological algorithm for global optimization over continuous spaces. J. Glob. Optim. 2012, 53, 769–808. [Google Scholar] [CrossRef]
- Cutello, V.; Oliva, M.; Pavone, M.; Scollo, R.A. An Immune Metaheuristics for Large Instances of the Weighted Feedback Vertex Set Problem. In Proceedings of the IEEE Symposium Series on Computational Intelligence (SSCI 2019), Xiamen, China, 6–9 December 2019; pp. 1928–1936. [Google Scholar] [CrossRef]
- Di Stefano, A.; Vitale, A.; Cutello, V.; Pavone, M. How long should offspring lifespan be in order to obtain a proper exploration? In Proceedings of the IEEE Symposium Series on Computational Intelligence (SSCI 2016), Athens, Greece, 6–9 December 2016; pp. 1–8. [Google Scholar] [CrossRef]
- Vitale, A.; Di Stefano, A.; Cutello, V.; Pavone, M. The Influence of Age Assignments on the Performance of Immune Algorithms. In Advances in Computational Intelligence Systems, Proceedings of the 18th UK Workshop on Computational Intelligence, Nottingham, UK, 5–7 September 2018; Lotfi, A., Bouchachia, H., Gegov, A., Langensiepen, C., McGinnity, M., Eds.; Springer International Publishing: Cham, Switzerland, 2019; pp. 16–28. [Google Scholar]
- Cutello, V.; Nicosia, G.; Pavone, M.; Stracquadanio, G. An Information-Theoretic Approach for Clonal Selection Algorithms. In Lecture Notes in Computer Science, Proceedings of the 9th International Conference on Artificial Immune Systems (ICARIS 2010), Edinburgh, UK, 26–29 July 2010; Hart, E., McEwan, C., Timmis, J., Hone, A., Eds.; Springer: Berlin/Heidelberg, Germany, 2010; Volume 6209, pp. 144–157. [Google Scholar] [CrossRef]
- Kernighan, B.W.; Lin, S. An efficient heuristic procedure for partitioning graphs. Bell Syst. Tech. J. 1970, 49, 291–307. [Google Scholar] [CrossRef]
- Zhang, Y.; Gao, P.; Yuan, J.S. Plant Protein–Protein Interaction Network and Interactome. Curr. Genom. 2010, 11, 40–46. [Google Scholar] [CrossRef]
- Gu, H.; Zhu, P.; Jiao, Y.; Meng, Y.; Chen, M. PRIN: A predicted rice interactome network. BMC Bioinform. 2011, 12, 161. [Google Scholar] [CrossRef]
- Lim, Y.H.; Charette, J.M.; Baserga, S.J. Assembling a Protein–Protein Interaction Map of the SSU Processome from Existing Datasets. PLoS ONE 2011, 6, e17701. [Google Scholar] [CrossRef]
- Mullard, A. Protein–protein interaction inhibitors get into the groove. Nat. Rev. Drug Discov. 2012, 11, 173–175. [Google Scholar] [CrossRef] [PubMed]
- Taylor, I.W.; Linding, R.; Warde-Farley, D.; Liu, Y.; Pesquita, C.; Faria, D.; Bull, S.; Pawson, T.; Morris, Q.; Wrana, J.L. Dynamic modularity in protein interaction networks predicts breast cancer outcome. Nat. Biotechnol. 2009, 27, 199–204. [Google Scholar] [CrossRef] [PubMed]
- Diss, G.; Filteau, M.; Freschi, L.; Leducq, J.B.; Rochette, S.; Torres-Quiroz, F.; Landry, C.R. Integrative avenues for exploring the dynamics and evolution of protein interaction networks. Curr. Opin. Biotechnol. 2013, 24, 775–783. [Google Scholar] [CrossRef] [PubMed]
- Cattle Protein–Protein Interactions. 2009. Available online: https://biit.cs.ut.ee/graphweb/welcome.cgi?t=examples (accessed on 30 October 2022).
- Xenarios, I.; Rice, D.W.; Salwinski, L.; Baron, M.K.; Marcotte, E.M.; Eisenberg, D. DIP: The Database of Interacting Proteins. Nucleic Acids Res. 2000, 28, 289–291. [Google Scholar] [CrossRef]
- Rain, J.C.; Selig, L.; De Reuse, H.; Battaglia, V.; Reverdy, C.; Simon, S.; Lenzen, G.; Petel, F.; Wojcik, J.; Schächter, V.; et al. The protein—Protein interaction map of Helicobacter pylori. Nature 2001, 409, 211–215. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Braun, P.; Yildirim, M.A.; Lemmens, I.; Venkatesan, K.; Sahalie, J.; Hirozane-Kishikawa, T.; Gebreab, F.; Li, N.; Simonis, N.; et al. High-Quality Binary Protein Interaction Map of the Yeast Interactome Network. Science 2008, 322, 104–110. [Google Scholar] [CrossRef] [PubMed]
- Bu, D.; Zhao, Y.; Cai, L.; Xue, H.; Zhu, X.; Lu, H.; Zhang, J.; Sun, S.; Ling, L.; Zhang, N.; et al. Topological structure analysis of the protein–protein interaction network in budding yeast. Nucleic Acids Res. 2003, 31, 2443–2450. [Google Scholar] [CrossRef]
- Lee, D.S.; Park, J.; Kay, K.A.; Christakis, N.A.; Oltvai, Z.N.; Barabási, A.L. The implications of human metabolic network topology for disease comorbidity. Proc. Natl. Acad. Sci. USA 2008, 105, 9880–9885. [Google Scholar] [CrossRef]
- Ross, R.; Dagnone, D.; Jones, P.J.H.; Smith, H.; Paddags, A.; Hudson, R.; Janssen, I. Reduction in Obesity and Related Comorbid Conditions after Diet-Induced Weight Loss or Exercise-Induced Weight Loss in Men. Ann. Intern. Med. 2000, 133, 92–103. [Google Scholar] [CrossRef]
- Yanrui, D.; Zhen, Z.; Wenchao, W.; Yujie, C. Identifying the Communities in the Metabolic Network Using ’Component’ Definition and Girvan-Newman Algorithm. In Proceedings of the 14th International Symposium on Distributed Computing and Applications for Business Engineering and Science (DCABES 2015), Guiyang, China, 18–24 August 2015; pp. 42–45. [Google Scholar] [CrossRef]
- Fortunato, S. Community detection in graphs. Phys. Rep. 2010, 486, 75–174. [Google Scholar] [CrossRef]
- Duch, J.; Arenas, A. Community detection in complex networks using extremal optimization. Phys. Rev. E 2005, 72, 027104. [Google Scholar] [CrossRef] [PubMed]
- Schellenberger, J.; Park, J.O.; Conrad, T.M.; Palsson, B.Ø. BiGG: A Biochemical Genetic and Genomic knowledgebase of large scale metabolic reconstructions. BMC Bioinform. 2010, 11, 213. [Google Scholar] [CrossRef] [PubMed]
- Barabási, A.L.; Albert, R.; Jeong, H. Scale-free characteristics of random networks: The topology of the world-wide web. Phys. A Stat. Mech. Appl. 2000, 281, 69–77. [Google Scholar] [CrossRef]
- Ravasi, T.; Suzuki, H.; Cannistraci, C.V.; Katayama, S.; Bajic, V.B.; Tan, K.; Akalin, A.; Schmeier, S.; Kanamori-Katayama, M.; Bertin, N.; et al. An Atlas of Combinatorial Transcriptional Regulation in Mouse and Man. Cell 2010, 140, 744–752. [Google Scholar] [CrossRef]
- Cantini, L.; Medico, E.; Fortunato, S.; Caselle, M. Detection of gene communities in multi-networks reveals cancer drivers. Sci. Rep. 2015, 5, 17386. [Google Scholar] [CrossRef] [PubMed]
- Marbach, D.; Costello, J.C.; Küffner, R.; Vega, N.M.; Prill, R.J.; Camacho, D.M.; Allison, K.R.; Kellis, M.; Collins, J.J.; Stolovitzky, G. Wisdom of crowds for robust gene network inference. Nat. Methods 2012, 9, 796–804. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, D.M.; Huberman, B.A. A method for finding communities of related genes. Proc. Natl. Acad. Sci. USA 2004, 101, 5241–5248. [Google Scholar] [CrossRef]
- Zhu, J.; Zhang, B.; Smith, E.N.; Drees, B.; Brem, R.B.; Kruglyak, L.; Bumgarner, R.E.; Schadt, E.E. Integrating large-scale functional genomic data to dissect the complexity of yeast regulatory networks. Nat. Genet. 2008, 40, 854–861. [Google Scholar] [CrossRef]
- Tang, B.; Hsu, H.K.; Hsu, P.Y.; Bonneville, R.; Chen, S.S.; Huang, T.H.M.; Jin, V.X. Hierarchical Modularity in ERα Transcriptional Network Is Associated with Distinct Functions and Implicates Clinical Outcomes. Sci. Rep. 2012, 2, 875. [Google Scholar] [CrossRef]
- Shen-Orr, S.S.; Milo, R.; Mangan, S.; Alon, U. Network motifs in the transcriptional regulation network of Escherichia coli. Nat. Genet. 2002, 31, 64–68. [Google Scholar] [CrossRef]
- Milo, R.; Shen-Orr, S.; Itzkovitz, S.; Kashtan, N.; Chklovskii, D.; Alon, U. Network Motifs: Simple Building Blocks of Complex Networks. Science 2002, 298, 824–827. [Google Scholar] [CrossRef] [PubMed]
- Lancichinetti, A.; Fortunato, S.; Radicchi, F. Benchmark graphs for testing community detection algorithms. Phys. Rev. E 2008, 78, 046110. [Google Scholar] [CrossRef] [PubMed]
- Lancichinetti, A.; Fortunato, S. Benchmarks for testing community detection algorithms on directed and weighted graphs with overlapping communities. Phys. Rev. E 2009, 80, 016118. [Google Scholar] [CrossRef] [PubMed]
- Good, B.H.; De Montjoye, Y.A.; Clauset, A. Performance of modularity maximization in practical contexts. Phys. Rev. E 2010, 81, 046106. [Google Scholar] [CrossRef]
- Kullback, S. Information Theory and Statistics; Wiley: Hoboken, NJ, USA, 1959. [Google Scholar]
- Cutello, V.; Nicosia, G.; Pavone, M.; Stracquadanio, G. Entropic divergence for population based optimization algorithms. In Proceedings of the IEEE Congress on Evolutionary Computation, Barcelona, Spain, 18–23 July 2010; pp. 1–8. [Google Scholar] [CrossRef]
- Zito, F.; Cutello, V.; Pavone, M. Optimizing Multi-Variable Time Series Forecasting using Metaheuristics. In Lecture Notes in Computer Science, Proceedings of the 14th Metaheuristics International Conference (MIC 2022), Ortigia-Syracuse, Italy, 11–14 July 2022; Di Gaspero, L., Festa, P., Nakib, A., Pavone, M., Eds.; Springer: Cham, Swizterland, 2023; Volume 13838. [Google Scholar]
- Altuntas, V.; Gök, M.; Kahveci, T. Stability Analysis of Biological Networks’ Diffusion State. IEEE/ACM Trans. Comput. Biol. Bioinform. 2020, 17, 1406–1418. [Google Scholar] [CrossRef]
- Penas, D.R.; González, P.; Egea, J.A.; Doallo, R.; Banga, J.R. Parameter estimation in large-scale systems biology models: A parallel and self-adaptive cooperative strategy. BMC Bioinform. 2017, 18, 52. [Google Scholar] [CrossRef]
- Barros, R.C.; Winck, A.T.; Machado, K.S.; Basgalupp, M.P.; de Carvalho, A.C.; Ruiz, D.D.; de Souza, O.N. Automatic design of decision-tree induction algorithms tailored to flexible-receptor docking data. BMC Bioinform. 2012, 13, 310. [Google Scholar] [CrossRef]
- Cutillas-Lozano, J.M.; Giménez, D.; Almeida, F. Hyperheuristics Based on Parametrized Metaheuristic Schemes. In Proceedings of the Annual Conference on Genetic and Evolutionary Computation (GECCO ’15), Madrid, Spain, 11–15 July 2015; Association for Computing Machinery: New York, NY, USA, 2015; pp. 361–368. [Google Scholar] [CrossRef]
- Bonidia, R.P.; Avila Santos, A.P.; de Almeida, B.L.S.; Stadler, P.F.; da Rocha, U.N.; Sanches, D.S.; de Carvalho, A.C.P.L.F. BioAutoML: Automated feature engineering and metalearning to predict noncoding RNAs in bacteria. Briefings Bioinform. 2022, 23, 1–13. [Google Scholar] [CrossRef]
- Atay, Y.; Koc, I.; Babaoglu, I.; Kodaz, H. Community detection from biological and social networks: A comparative analysis of metaheuristic algorithms. Appl. Soft Comput. 2017, 50, 194–211. [Google Scholar] [CrossRef]
- Civicioglu, P. Transforming geocentric cartesian coordinates to geodetic coordinates by using differential search algorithm. Comput. Geosci. 2012, 46, 229–247. [Google Scholar] [CrossRef]
- Storn, R.; Price, K. Differential Evolution – A Simple and Efficient Heuristic for global Optimization over Continuous Spaces. J. Glob. Optim. 1997, 11, 341–359. [Google Scholar] [CrossRef]
- Glover, F. Heuristics for integer programming using surrogate constraints. Decis. Sci. 1977, 8, 156–166. [Google Scholar] [CrossRef]
- Martí, R.; Laguna, M.; Glover, F. Principles of scatter search. Eur. J. Oper. Res. 2006, 169, 359–372. [Google Scholar] [CrossRef]
- Erol, O.K.; Eksin, I. A new optimization method: Big Bang–Big Crunch. Adv. Eng. Softw. 2006, 37, 106–111. [Google Scholar] [CrossRef]
- Yang, X.S. A New Metaheuristic Bat-Inspired Algorithm. In Studies in Computational Intelligence, Proceedings of the Nature Inspired Cooperative Strategies for Optimization (NICSO 2010), Granada, Spain, 12–14 May 2010; González, J.R., Pelta, D.A., Cruz, C., Terrazas, G., Krasnogor, N., Eds.; Springer: Berlin/Heidelberg, Germany, 2010; pp. 65–74. [Google Scholar] [CrossRef]
- Rashedi, E.; Nezamabadi-pour, H.; Saryazdi, S. GSA: A Gravitational Search Algorithm. Inf. Sci. 2009, 179, 2232–2248. [Google Scholar] [CrossRef]
- Blondel, V.D.; Guillaume, J.L.; Lambiotte, R.; Lefebvre, E. Fast unfolding of communities in large networks. J. Stat. Mech. Theory Exp. 2008, 2008, P10008. [Google Scholar] [CrossRef]
- Danon, L.; Díaz-Guilera, A.; Duch, J.; Arenas, A. Comparing community structure identification. J. Stat. Mech. Theory Exp. 2005, 2005, P09008. [Google Scholar] [CrossRef]
- Hubert, L.; Arabic, P. Comparing Partitions. J. Classif. 1985, 2, 193–218. [Google Scholar] [CrossRef]
- Meilă, M. Comparing clusterings–an information based distance. J. Multivar. Anal. 2007, 98, 873–895. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Reference | |||
|---|---|---|---|---|
| Cattle PPI | [44] | 268 | 303 | 0.85% |
| E. coli TRN | [62] | 418 | 519 | 0.60% |
| C. elegans MRN | [53] | 453 | 2025 | 1.98% |
| Yeast TRN | [63] | 688 | 1078 | 0.46% |
| Helicobacter pylori PPI | [45,46] | 724 | 1403 | 0.54% |
| E. coli MRN | [54] | 1039 | 4741 | 0.88% |
| Yeast PPI (1) | [47] | 2018 | 2705 | 0.13% |
| Yeast PPI (2) | [48] | 2284 | 6646 | 0.25% |
| Name | Hybrid-IA | Louvain | HDSA | BADE | SSGA | BB-BC | BA | GSA | |
|---|---|---|---|---|---|---|---|---|---|
| Cattle PPI | k | 40 | 40 | 40 | 41 | 40 | 48 | 42 | 43 |
| Best | 0.7195 | 0.7195 | 0.7195 | 0.7183 | 0.7118 | 0.7095 | 0.7143 | 0.7053 | |
| Worst | 0.7011 | 0.7181 | 0.7194 | 0.7059 | 0.7052 | 0.7079 | 0.7063 | 0.6949 | |
| Mean | 0.7154 | 0.7193 | 0.7195 | 0.7138 | 0.7079 | 0.7084 | 0.7100 | 0.6983 | |
| StD | 0.0037 | 0.0005 | 0.0001 | 0.0051 | 0.0025 | 0.0007 | 0.0035 | 0.0041 | |
| E. coli TRN | k | 43 | 41 | 47 | 58 | 61 | 71 | 56 | 61 |
| Best | 0.7785 | 0.7793 | 0.7822 | 0.7680 | 0.7507 | 0.7520 | 0.7629 | 0.7416 | |
| Worst | 0.7563 | 0.7747 | 0.7808 | 0.7560 | 0.7412 | 0.7452 | 0.7542 | 0.7328 | |
| Mean | 0.7701 | 0.7779 | 0.7815 | 0.7621 | 0.7457 | 0.7485 | 0.7599 | 0.7375 | |
| StD | 0.0049 | 0.0011 | 0.0006 | 0.0043 | 0.0035 | 0.0026 | 0.0034 | 0.0034 | |
| C. elegans MRN | k | 10 | 10 | 13 | 25 | 22 | 21 | 22 | 24 |
| Best | 0.4506 | 0.4490 | 0.4185 | 0.3473 | 0.3336 | 0.3374 | 0.3514 | 0.3063 | |
| Worst | 0.4321 | 0.4216 | 0.3962 | 0.3335 | 0.3124 | 0.3194 | 0.3356 | 0.2974 | |
| Mean | 0.4437 | 0.4365 | 0.4074 | 0.3385 | 0.3220 | 0.3266 | 0.3438 | 0.3039 | |
| StD | 0.0040 | 0.0049 | 0.0010 | 0.0054 | 0.0077 | 0.0074 | 0.0073 | 0.0037 | |
| Yeast TRN | k | 33 | 26 | - | - | - | - | - | - |
| Best | 0.7668 | 0.7683 | - | - | - | - | - | - | |
| Worst | 0.7363 | 0.7489 | - | - | - | - | - | - | |
| Mean | 0.7569 | 0.7607 | - | - | - | - | - | - | |
| StD | 0.0050 | 0.0033 | - | - | - | - | - | - | |
| Helicobacter pylori PPI | k | 51 | 24 | 52 | 69 | 70 | 75 | 62 | 77 |
| Best | 0.5359 | 0.5462 | 0.5086 | 0.4926 | 0.4726 | 0.4681 | 0.4900 | 0.4600 | |
| Worst | 0.5104 | 0.5356 | 0.5048 | 0.4809 | 0.4659 | 0.4642 | 0.4738 | 0.4549 | |
| Mean | 0.5240 | 0.5410 | 0.5078 | 0.4854 | 0.4695 | 0.4660 | 0.4814 | 0.4567 | |
| StD | 0.0056 | 0.0025 | 0.0017 | 0.0047 | 0.0021 | 0.0018 | 0.0073 | 0.0020 | |
| E. coli MRN | k | 13 | 8 | - | - | - | - | - | - |
| Best | 0.3817 | 0.3734 | - | - | - | - | - | - | |
| Worst | 0.3598 | 0.3450 | - | - | - | - | - | - | |
| Mean | 0.3695 | 0.3583 | - | - | - | - | - | - | |
| StD | 0.0042 | 0.0058 | - | - | - | - | - | - | |
| Yeast PPI (2) | k | 159 | 46 | - | - | - | - | - | - |
| Best | 0.5796 | 0.5961 | - | - | - | - | - | - | |
| Worst | 0.5524 | 0.5870 | - | - | - | - | - | - | |
| Mean | 0.5652 | 0.5925 | - | - | - | - | - | - | |
| StD | 0.0052 | 0.0019 | - | - | - | - | - | - | |
| Yeast PPI (1) | k | 353 | 213 | - | - | - | - | - | - |
| Best | 0.7002 | 0.7648 | - | - | - | - | - | - | |
| Worst | 0.6602 | 0.7519 | - | - | - | - | - | - | |
| Mean | 0.6798 | 0.7609 | - | - | - | - | - | - | |
| StD | 0.0078 | 0.0022 | - | - | - | - | - | - |
| Hybrid-IA | Louvain | Hybrid-IA | Louvain | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Q | NMI | Q | NMI | Q | NMI | Q | NMI | ||
| 0.8608 | 0.9951 | 0.8608 | 0.9918 | 0.8923 | 0.9988 | 0.8934 | 0.9586 | ||
| 0.7621 | 0.9894 | 0.7623 | 0.9807 | 0.7927 | 0.9965 | 0.7949 | 0.9394 | ||
| 0.6646 | 0.9862 | 0.6651 | 0.9716 | 0.6929 | 0.9965 | 0.6960 | 0.9252 | ||
| 0.5654 | 0.9836 | 0.5660 | 0.9691 | 0.5931 | 0.9948 | 0.5976 | 0.9065 | ||
| 0.4670 | 0.9847 | 0.4688 | 0.9462 | 0.4936 | 0.9951 | 0.5003 | 0.8779 | ||
| 0.3688 | 0.9612 | 0.3718 | 0.9113 | 0.3939 | 0.9966 | 0.4030 | 0.8474 | ||
| 0.2712 | 0.5467 | 0.2675 | 0.4977 | 0.2932 | 0.9927 | 0.3056 | 0.8145 | ||
| 0.2415 | 0.1600 | 0.2354 | 0.1536 | 0.2084 | 0.3285 | 0.2102 | 0.2634 | ||
| 0.8606 | 0.9980 | 0.8607 | 0.9931 | 0.8922 | 0.9993 | 0.8925 | 0.9770 | ||
| 0.7622 | 0.9964 | 0.7622 | 0.9914 | 0.7925 | 0.9988 | 0.7936 | 0.9527 | ||
| 0.6656 | 0.9921 | 0.6658 | 0.9830 | 0.6929 | 0.9986 | 0.6948 | 0.9348 | ||
| 0.5668 | 0.9910 | 0.5676 | 0.9656 | 0.5931 | 0.9987 | 0.5966 | 0.9125 | ||
| 0.4685 | 0.9829 | 0.4700 | 0.9491 | 0.4934 | 0.9955 | 0.4983 | 0.8907 | ||
| 0.3688 | 0.9748 | 0.3712 | 0.9263 | 0.3939 | 0.9950 | 0.4008 | 0.8621 | ||
| 0.2714 | 0.9244 | 0.2737 | 0.8230 | 0.2940 | 0.9951 | 0.3037 | 0.8285 | ||
| 0.2169 | 0.1708 | 0.2069 | 0.1793 | 0.1872 | 0.6067 | 0.1942 | 0.5654 | ||
| Hybrid-IA | Louvain | |||||||
|---|---|---|---|---|---|---|---|---|
| Q | NMI | ARI | NVI | Q | NMI | ARI | NVI | |
| 0.8608 | 0.9951 | 0.9873 | 0.0098 | 0.8608 | 0.9918 | 0.9785 | 0.0161 | |
| 0.7621 | 0.9894 | 0.9716 | 0.0209 | 0.7623 | 0.9807 | 0.9483 | 0.0377 | |
| 0.6646 | 0.9862 | 0.9567 | 0.0271 | 0.6651 | 0.9716 | 0.9175 | 0.0550 | |
| 0.5654 | 0.9836 | 0.9490 | 0.0322 | 0.5660 | 0.9691 | 0.9104 | 0.0598 | |
| 0.4670 | 0.9847 | 0.9467 | 0.0301 | 0.4688 | 0.9462 | 0.8274 | 0.1019 | |
| 0.3688 | 0.9612 | 0.8664 | 0.0742 | 0.3718 | 0.9113 | 0.7368 | 0.1627 | |
| 0.2712 | 0.5467 | 0.2379 | 0.6220 | 0.2675 | 0.4977 | 0.2168 | 0.6664 | |
| 0.2415 | 0.1600 | 0.0219 | 0.9130 | 0.2354 | 0.1536 | 0.0218 | 0.9167 | |
| 0.8606 | 0.9980 | 0.9948 | 0.0040 | 0.8607 | 0.9931 | 0.9842 | 0.0135 | |
| 0.7622 | 0.9964 | 0.9918 | 0.0071 | 0.7622 | 0.9914 | 0.9782 | 0.0170 | |
| 0.6656 | 0.9921 | 0.9771 | 0.0156 | 0.6658 | 0.9830 | 0.9525 | 0.0332 | |
| 0.5668 | 0.9910 | 0.9707 | 0.0179 | 0.5676 | 0.9656 | 0.8961 | 0.0664 | |
| 0.4685 | 0.9829 | 0.9426 | 0.0337 | 0.4700 | 0.9491 | 0.8363 | 0.0968 | |
| 0.3688 | 0.9748 | 0.9038 | 0.0491 | 0.3712 | 0.9263 | 0.7641 | 0.1370 | |
| 0.2714 | 0.9244 | 0.7561 | 0.1399 | 0.2737 | 0.8230 | 0.5403 | 0.3002 | |
| 0.2169 | 0.1708 | 0.0288 | 0.9064 | 0.2069 | 0.1793 | 0.0300 | 0.9012 | |
| 0.8923 | 0.9988 | 0.9940 | 0.0024 | 0.8934 | 0.9586 | 0.8194 | 0.0794 | |
| 0.7927 | 0.9965 | 0.9816 | 0.0070 | 0.7949 | 0.9394 | 0.7302 | 0.1142 | |
| 0.6929 | 0.9965 | 0.9808 | 0.0070 | 0.6960 | 0.9252 | 0.6678 | 0.1392 | |
| 0.5931 | 0.9948 | 0.9711 | 0.0104 | 0.5976 | 0.9065 | 0.5941 | 0.1709 | |
| 0.4936 | 0.9951 | 0.9728 | 0.0098 | 0.5003 | 0.8779 | 0.4959 | 0.2177 | |
| 0.3939 | 0.9966 | 0.9797 | 0.0068 | 0.4030 | 0.8474 | 0.4048 | 0.2648 | |
| 0.2932 | 0.9927 | 0.9539 | 0.0145 | 0.3056 | 0.8145 | 0.3327 | 0.3130 | |
| 0.2084 | 0.3285 | 0.0176 | 0.8027 | 0.2102 | 0.2634 | 0.0195 | 0.8481 | |
| 0.8922 | 0.9993 | 0.9966 | 0.0013 | 0.8925 | 0.9770 | 0.8928 | 0.0449 | |
| 0.7925 | 0.9988 | 0.9935 | 0.0024 | 0.7936 | 0.9527 | 0.7821 | 0.0902 | |
| 0.6929 | 0.9986 | 0.9921 | 0.0029 | 0.6948 | 0.9348 | 0.7026 | 0.1225 | |
| 0.5931 | 0.9987 | 0.9915 | 0.0027 | 0.5966 | 0.9125 | 0.6158 | 0.1609 | |
| 0.4934 | 0.9955 | 0.9747 | 0.0089 | 0.4983 | 0.8907 | 0.5366 | 0.1970 | |
| 0.3939 | 0.9950 | 0.9666 | 0.0100 | 0.4008 | 0.8621 | 0.4473 | 0.2424 | |
| 0.2940 | 0.9951 | 0.9653 | 0.0097 | 0.3037 | 0.8285 | 0.3604 | 0.2927 | |
| 0.1872 | 0.6067 | 0.0667 | 0.5607 | 0.1942 | 0.5654 | 0.1065 | 0.6058 | |
| 0.8938 | 0.9995 | 0.9982 | 0.0010 | 0.8945 | 0.9686 | 0.8874 | 0.0609 | |
| 0.7938 | 0.9981 | 0.9925 | 0.0037 | 0.7951 | 0.9538 | 0.8198 | 0.0883 | |
| 0.6940 | 0.9980 | 0.9925 | 0.0040 | 0.6960 | 0.9407 | 0.7563 | 0.1119 | |
| 0.5941 | 0.9977 | 0.9900 | 0.0045 | 0.5972 | 0.9202 | 0.6682 | 0.1478 | |
| 0.4942 | 0.9988 | 0.9945 | 0.0024 | 0.4986 | 0.8982 | 0.5793 | 0.1847 | |
| 0.3943 | 0.9996 | 0.9974 | 0.0008 | 0.4004 | 0.8720 | 0.4872 | 0.2269 | |
| 0.2909 | 0.9847 | 0.8556 | 0.0301 | 0.3013 | 0.8287 | 0.3737 | 0.2925 | |
| 0.2064 | 0.2621 | 0.0085 | 0.8491 | 0.2094 | 0.1704 | 0.0079 | 0.9068 | |
| 0.8937 | 0.9995 | 0.9984 | 0.0010 | 0.8940 | 0.9792 | 0.9253 | 0.0407 | |
| 0.7940 | 0.9993 | 0.9968 | 0.0014 | 0.7947 | 0.9627 | 0.8524 | 0.0719 | |
| 0.6941 | 0.9990 | 0.9955 | 0.0020 | 0.6955 | 0.9497 | 0.7946 | 0.0958 | |
| 0.5942 | 0.9992 | 0.9969 | 0.0015 | 0.5964 | 0.9320 | 0.7179 | 0.1273 | |
| 0.4944 | 0.9982 | 0.9917 | 0.0036 | 0.4978 | 0.9083 | 0.6199 | 0.1679 | |
| 0.3943 | 0.9984 | 0.9897 | 0.0032 | 0.3989 | 0.8841 | 0.5313 | 0.2077 | |
| 0.2941 | 0.9981 | 0.9860 | 0.0038 | 0.3008 | 0.8516 | 0.4299 | 0.2584 | |
| 0.1849 | 0.4316 | 0.0227 | 0.7245 | 0.1867 | 0.3352 | 0.0247 | 0.7985 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Scollo, R.A.; Spampinato, A.G.; Fargetta, G.; Cutello, V.; Pavone, M. Discovering Entities Similarities in Biological Networks Using a Hybrid Immune Algorithm. Informatics 2023, 10, 18. https://doi.org/10.3390/informatics10010018
Scollo RA, Spampinato AG, Fargetta G, Cutello V, Pavone M. Discovering Entities Similarities in Biological Networks Using a Hybrid Immune Algorithm. Informatics. 2023; 10(1):18. https://doi.org/10.3390/informatics10010018
Chicago/Turabian StyleScollo, Rocco A., Antonio G. Spampinato, Georgia Fargetta, Vincenzo Cutello, and Mario Pavone. 2023. "Discovering Entities Similarities in Biological Networks Using a Hybrid Immune Algorithm" Informatics 10, no. 1: 18. https://doi.org/10.3390/informatics10010018
APA StyleScollo, R. A., Spampinato, A. G., Fargetta, G., Cutello, V., & Pavone, M. (2023). Discovering Entities Similarities in Biological Networks Using a Hybrid Immune Algorithm. Informatics, 10(1), 18. https://doi.org/10.3390/informatics10010018