
Cleft Palate and Aortic Dilatation as Clues for Loeys–Dietz Syndrome


 ,
,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
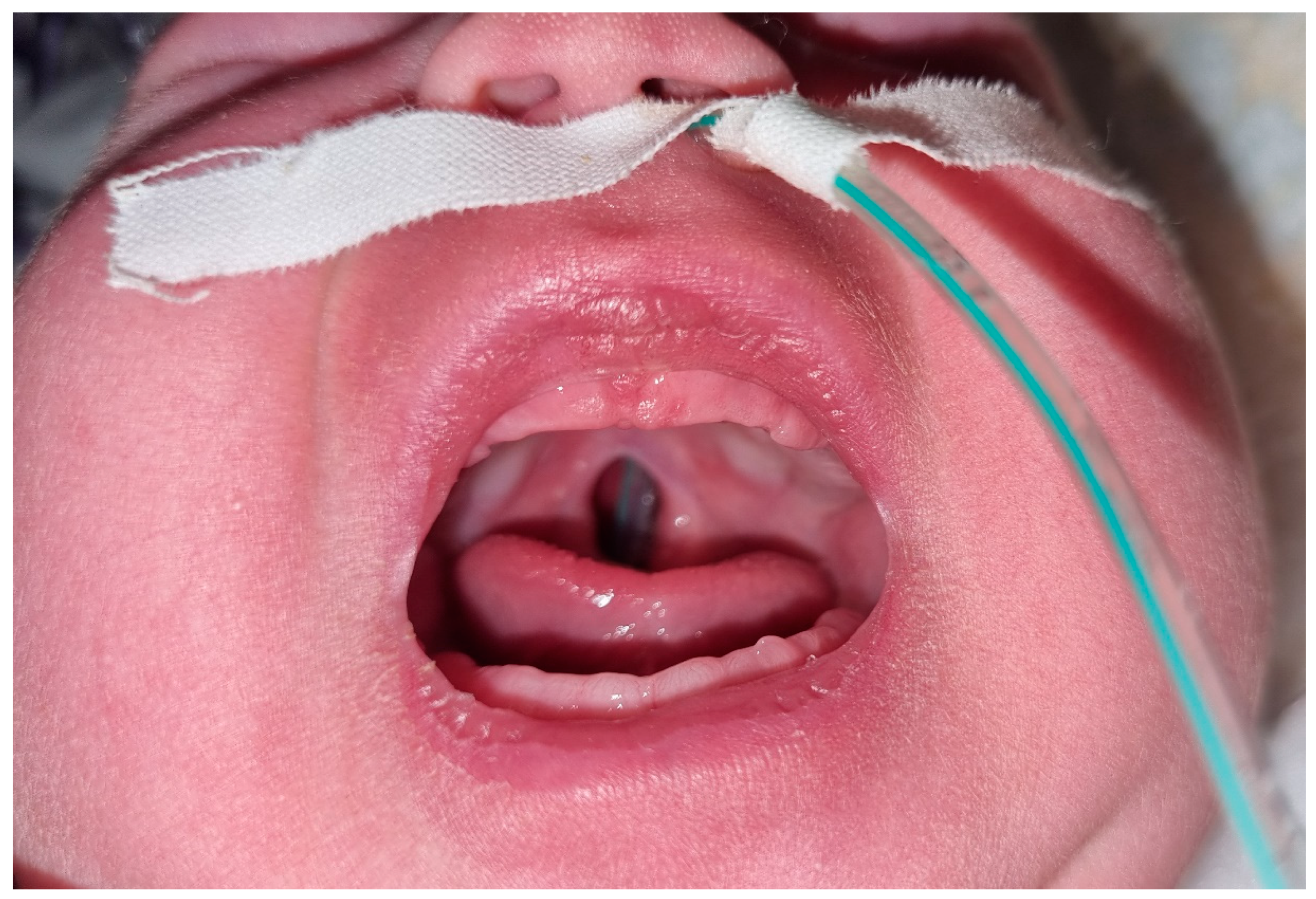
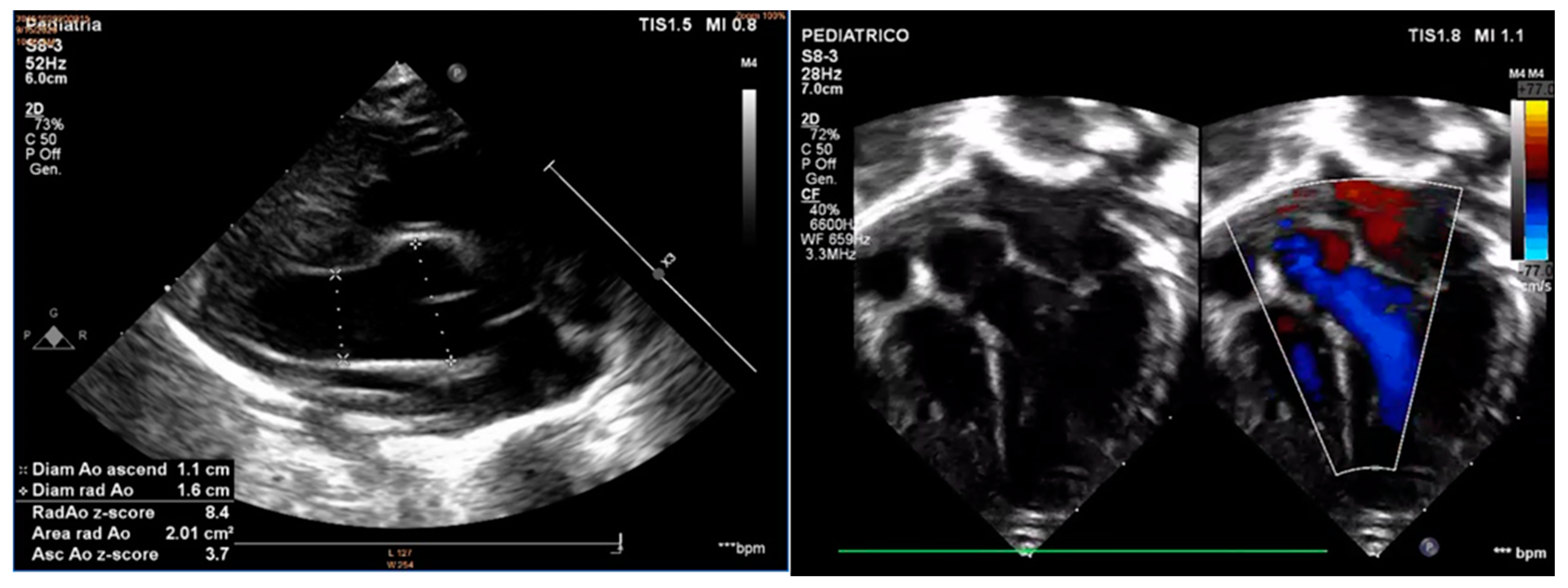
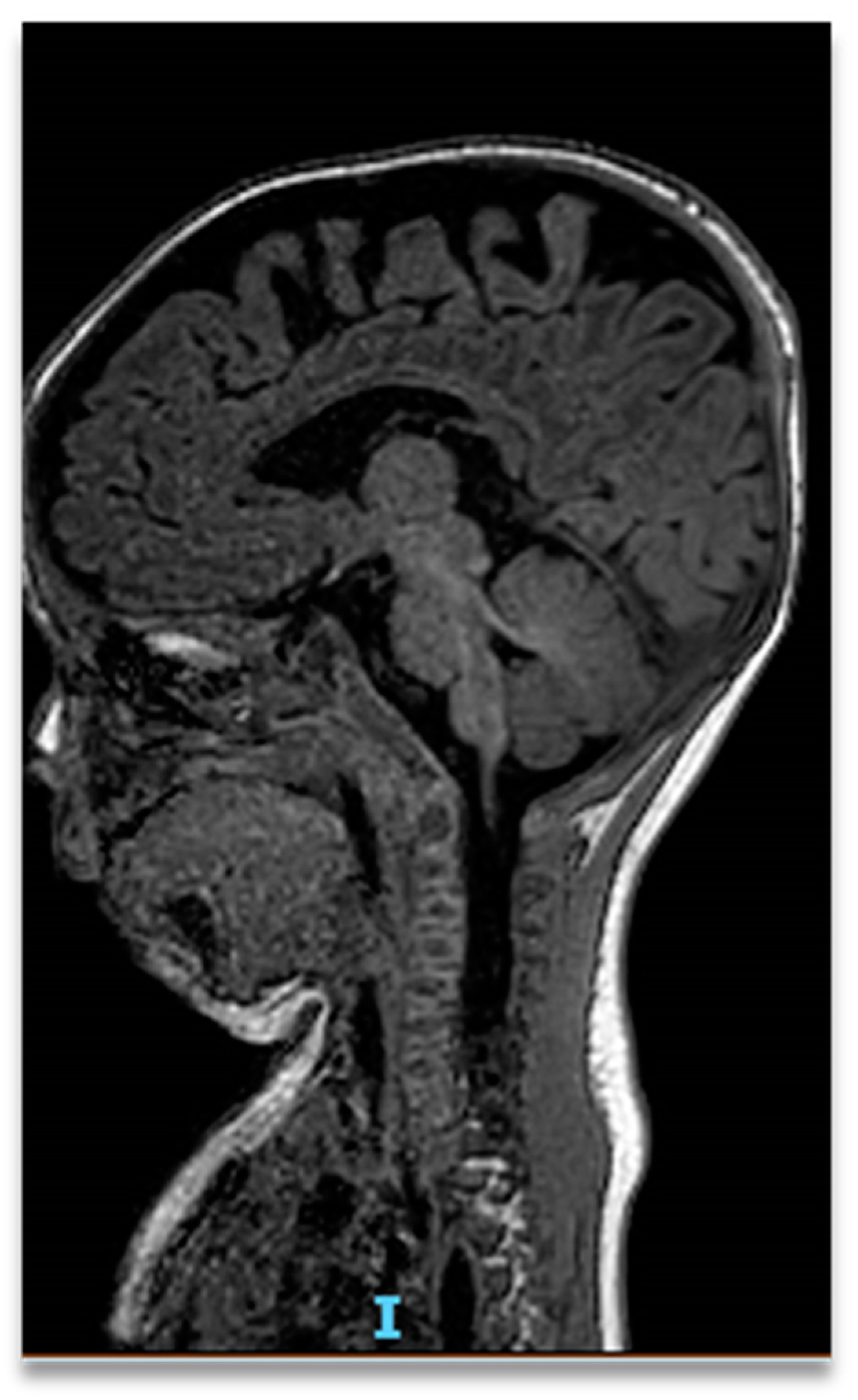
2. Case Report
3. Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Van Laer, L.; Dietz, H.; Loeys, B. Loeys-Dietz Syndrome. In Progress in Heritable Soft Connective Tissue Diseases; Halper, J., Ed.; Advances in Experimental Medicine and Biology; Springer: Dordrecht, The Netherlands, 2014; Volume 802, pp. 95–105. [Google Scholar]
- Loeys, B.L.; Dietz, H.C. Loeys-Dietz Syndrome. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Mirzaa, G.M., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 2008; pp. 1993–2022. [Google Scholar]
- Camerota, L.; Ritelli, M.; Wischmeijer, A.; Majore, S.; Cinquina, V.; Fortugno, P.; Monetta, R.; Gigante, L.; Marfan Syndrome Study Group Tor Vergata University Hospital; Sangiuolo, F.C.; et al. Genotypic Categorization of Loeys-Dietz Syndrome Based on 24 Novel Families and Literature Data. Genes 2019, 10, 764. [Google Scholar] [CrossRef] [PubMed]
- Van De Laar, I.M.B.H.; Oldenburg, R.A.; Pals, G.; Roos-Hesselink, J.W.; De Graaf, B.M.; Verhagen, J.M.A.; Hoedemaekers, Y.M.; Willemsen, R.; Severijnen, L.-A.; Venselaar, H.; et al. Mutations in SMAD3 cause a syndromic form of aortic aneurysms and dissections with early-onset osteoarthritis. Nat. Genet. 2011, 43, 121–126. [Google Scholar] [CrossRef] [PubMed]
- Loeys, B.L.; Schwarze, U.; Holm, T.; Callewaert, B.L.; Thomas, G.H.; Pannu, H.; De Backer, J.F.; Oswald, G.L.; Symoens, S.; Manouvrier, S.; et al. Aneurysm syndromes caused by mutations in the TGF-beta receptor. N. Engl. J. Med. 2006, 355, 788–798. [Google Scholar] [CrossRef] [PubMed]
- Boileau, C.; Guo, D.C.; Hanna, N.; Regalado, E.S.; Detaint, D.; Gong, L.; Milewicz, D.M. TGFB2 mutations cause familial thoracic aortic aneurysms and dissections associated with mild systemic features of Marfan syndrome. Nat. Genet. 2012, 44, 916–921. [Google Scholar] [CrossRef] [PubMed]
- Braverman, A.C.; Blinder, K.J.; Khanna, S.; Willing, M. Ectopia lentis in Loeys-Dietz syndrome type 4. Am. J. Med. Genet. A 2020, 182, 1957–1959. [Google Scholar] [CrossRef] [PubMed]
- Lewis, C.W.; Jacob, L.S.; Lehmann, C.U.; Krol, D.; Gereige, R.; Karp, J.; Fisher-Owens, S.; Braun, P.; Segura, A.; SECTION ON ORAL HEALTH. The Primary Care Pediatrician and the Care of Children with Cleft Lip and/or Cleft Palate. Pediatrics 2017, 139, e20170628. [Google Scholar] [CrossRef] [PubMed]
- Viassolo, V.; Lituania, M.; Marasini, M.; Dietz, H.; Benelli, F.; Forzano, F.; Faravelli, F. Fetal aortic root dilation: A prenatal feature of the Loeys-Dietz syndrome. Prenat. Diagn. 2006, 26, 1081–1083. [Google Scholar] [CrossRef] [PubMed]
- Kawazu, Y.; Inamura, N.; Kayatani, F.; Okamoto, N.; Morisaki, H. Prenatal complex congenital heart disease with Loeys–Dietz syndrome. Cardiol. Young 2011, 22, 116–119. [Google Scholar] [CrossRef] [PubMed]
- Baldo, F.; Morra, L.; Feresin, A.; Faletra, F.; Al Naber, Y.; Memo, L.; Travan, L. Neonatal presentation of Loeys-Dietz syndrome: Two case reports and review of the literature. Ital. J. Pediatr. 2022, 48, 85. [Google Scholar] [CrossRef] [PubMed]
- Yetman, A.T.; Beroukhim, R.S.; Ivy, D.D.; Manchester, D. Importance of the Clinical Recognition of Loeys-Dietz Syndrome in the Neonatal Period. Pediatrics 2007, 119, e1199–e1202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muramatsu, Y.; Kosho, T.; Magota, M.; Yokotsuka, T.; Ito, M.; Yasuda, A.; Kito, O.; Suzuki, C.; Nagata, Y.; Kawai, Y.; et al. Progressive aortic root and pulmonary artery aneurysms in a neonate with Loeys-Dietz syndrome type 1B. Am. J. Med. Genet. A 2010, 152A, 417–421. [Google Scholar] [CrossRef] [PubMed]
- MacCarrick, G.; Black, J.H.; Bowdin, S.; El-Hamamsy, I.; Frischmeyer-Guerrerio, P.A.; Guerrerio, A.L.; Sponseller, P.D.; Loeys, B.; Dietz, H.C. Loeys-Dietz syndrome: A primer for diagnosis and management. Genet. Med. 2014, 16, 576–587. [Google Scholar] [CrossRef] [PubMed]
- Frischmeyer-Guerrerio, P.A.; Guerrerio, A.L.; Oswald, G. TGFbeta receptor mutations impose a strong predisposition for human allergic disease. Sci. Transl. Med. 2013, 5, 195ra94. [Google Scholar] [CrossRef] [PubMed]
- Valenzuela, I.; Fernández-Alvarez, P.; Munell, F.; Sanchez-Montanez, A.; Giralt, G.; Vendrell, T.; Tizzano, E.F. Arthrogryposis as neonatal presentation of Loeys-Dietz syndrome due to a novel TGFBR2 mutation. Eur. J. Med. Genet. 2017, 60, 303–307. [Google Scholar] [CrossRef] [PubMed]
- Dueppers, P.; Prêtre, R.; Hofmann, M.; Bettex, D.; Huber, F.A.; Zimmermann, A. Complex Multi-Stage Total Aortic and Subclavian Artery Replacement in a 9-year old boy with Loeys-Dietz-Syndrome. Ann. Vasc. Surg. 2022, 80, 396.e1–396.e6. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zaza, P.; Indrio, F.; Fracchiolla, A.; Rinaldi, M.; Meliota, G.; Salatto, A.; Bonacaro, A.; Maffei, G. Cleft Palate and Aortic Dilatation as Clues for Loeys–Dietz Syndrome. Children 2022, 9, 1290. https://doi.org/10.3390/children9091290
Zaza P, Indrio F, Fracchiolla A, Rinaldi M, Meliota G, Salatto A, Bonacaro A, Maffei G. Cleft Palate and Aortic Dilatation as Clues for Loeys–Dietz Syndrome. Children. 2022; 9(9):1290. https://doi.org/10.3390/children9091290
Chicago/Turabian StyleZaza, Pierluigi, Flavia Indrio, Annalisa Fracchiolla, Matteo Rinaldi, Giovanni Meliota, Alessia Salatto, Antonio Bonacaro, and Gianfranco Maffei. 2022. "Cleft Palate and Aortic Dilatation as Clues for Loeys–Dietz Syndrome" Children 9, no. 9: 1290. https://doi.org/10.3390/children9091290
APA StyleZaza, P., Indrio, F., Fracchiolla, A., Rinaldi, M., Meliota, G., Salatto, A., Bonacaro, A., & Maffei, G. (2022). Cleft Palate and Aortic Dilatation as Clues for Loeys–Dietz Syndrome. Children, 9(9), 1290. https://doi.org/10.3390/children9091290