
Tubulinopathy Presenting as Developmental and Epileptic Encephalopathy


Abstract
:1. Introduction
2. Case Presentation
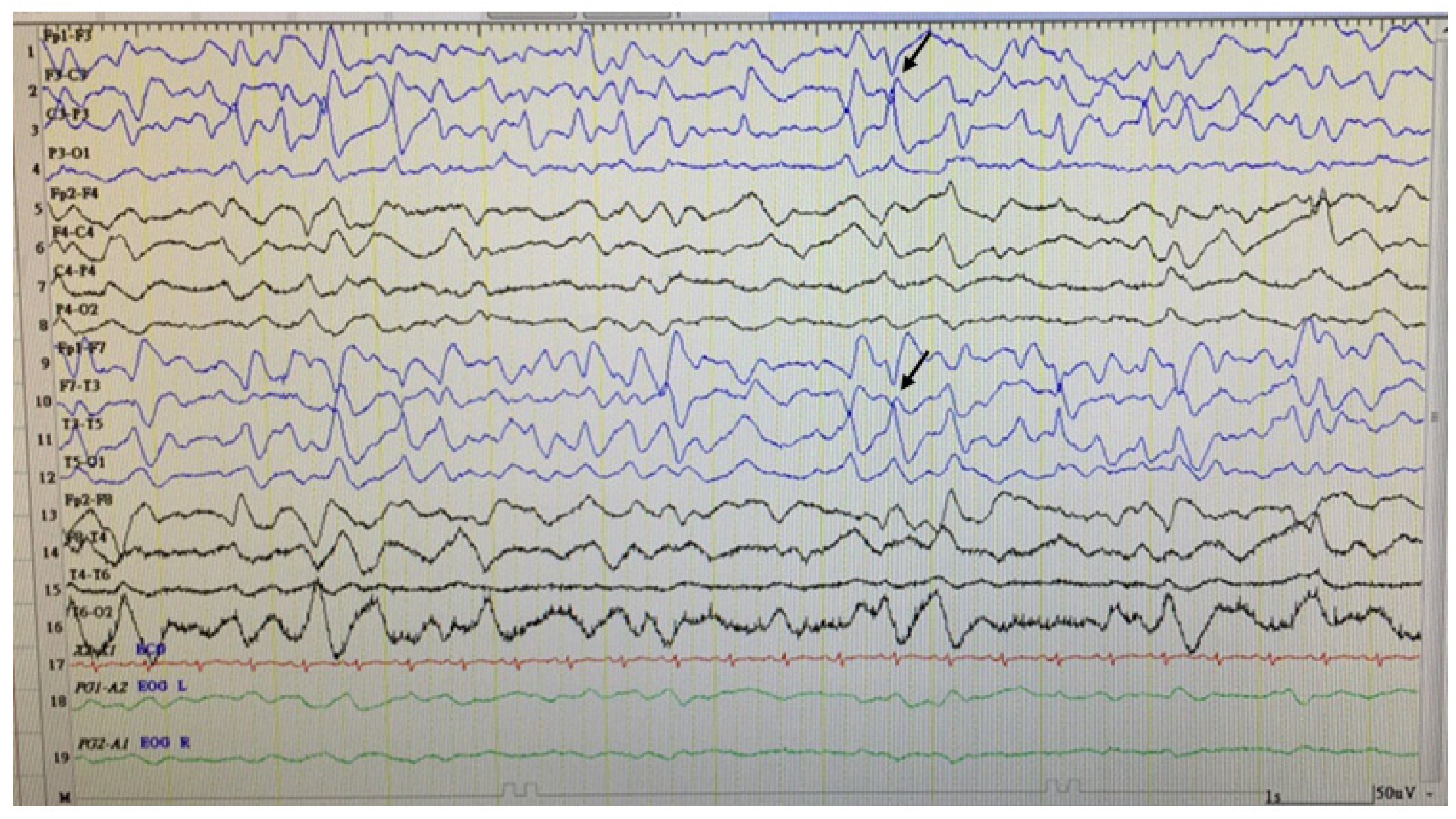
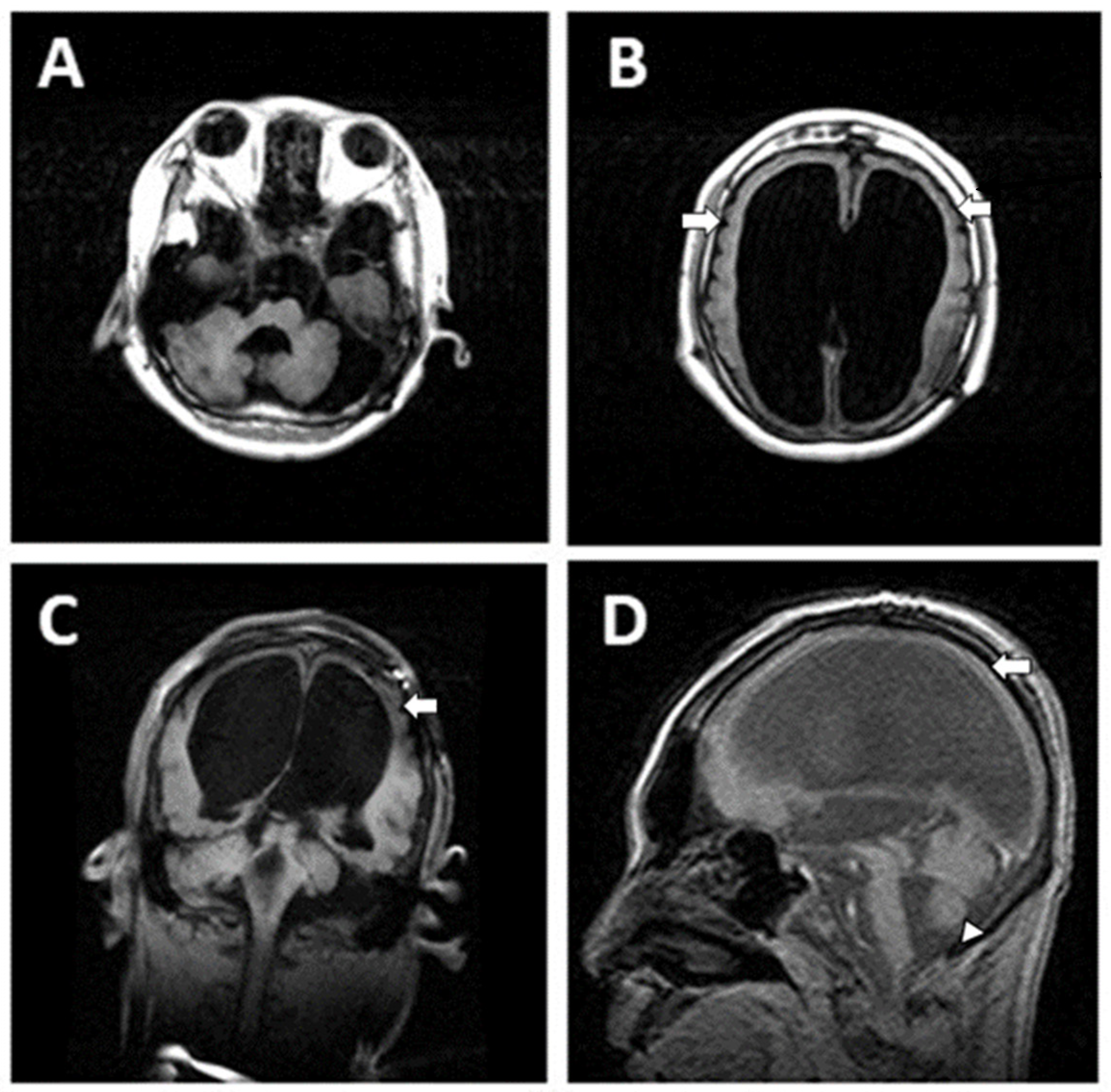
2.1. Case 1
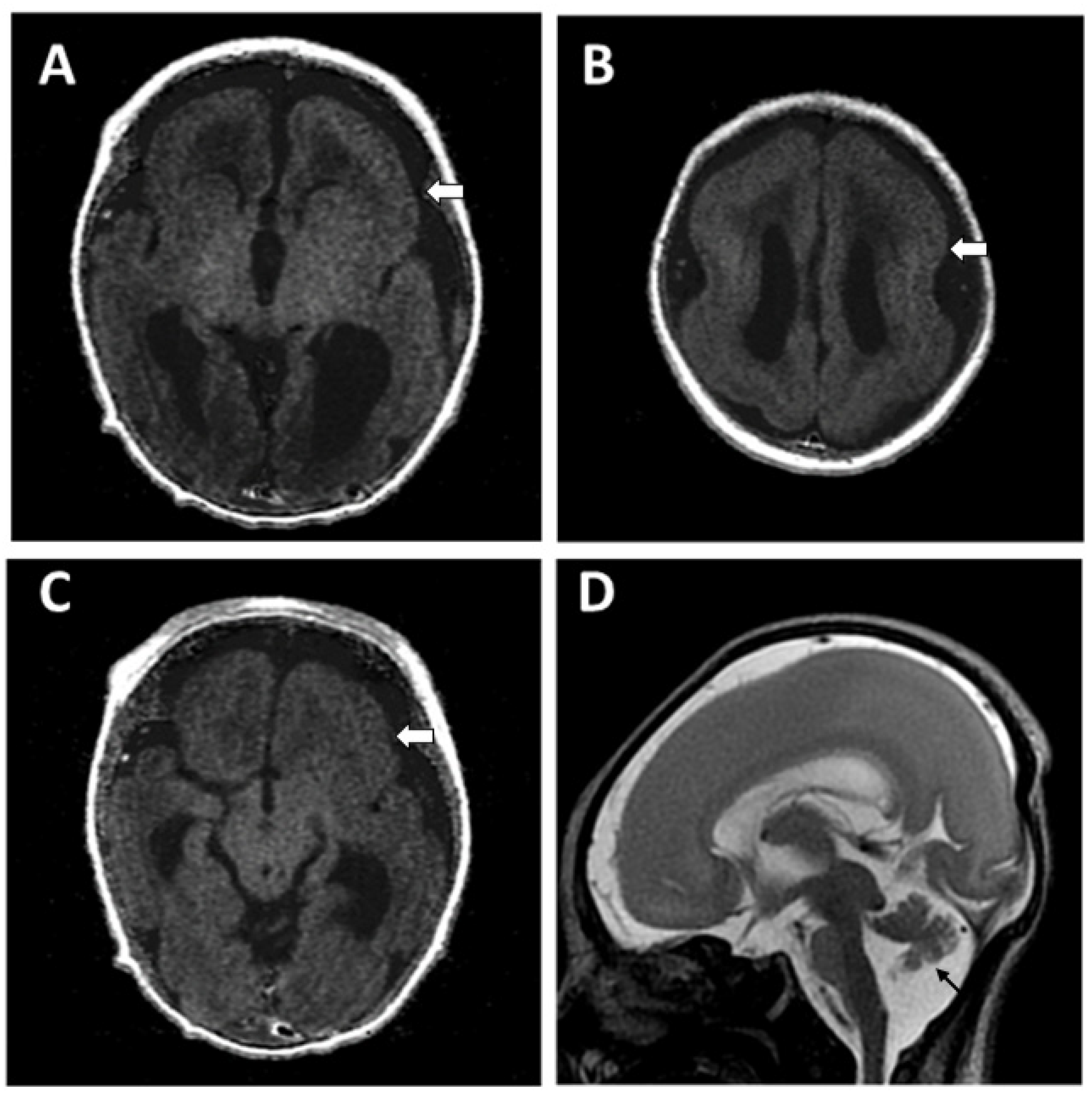
Patient Information
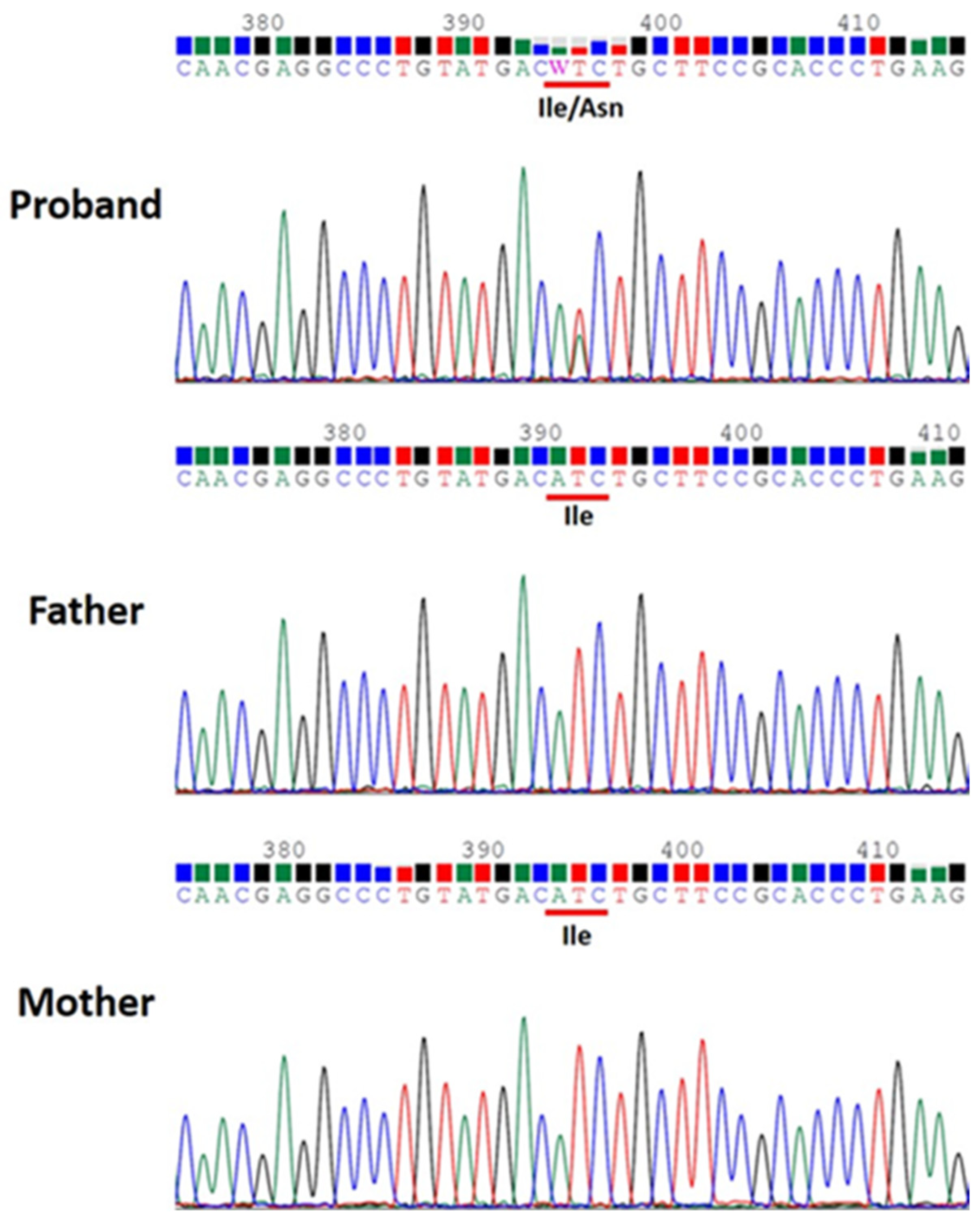
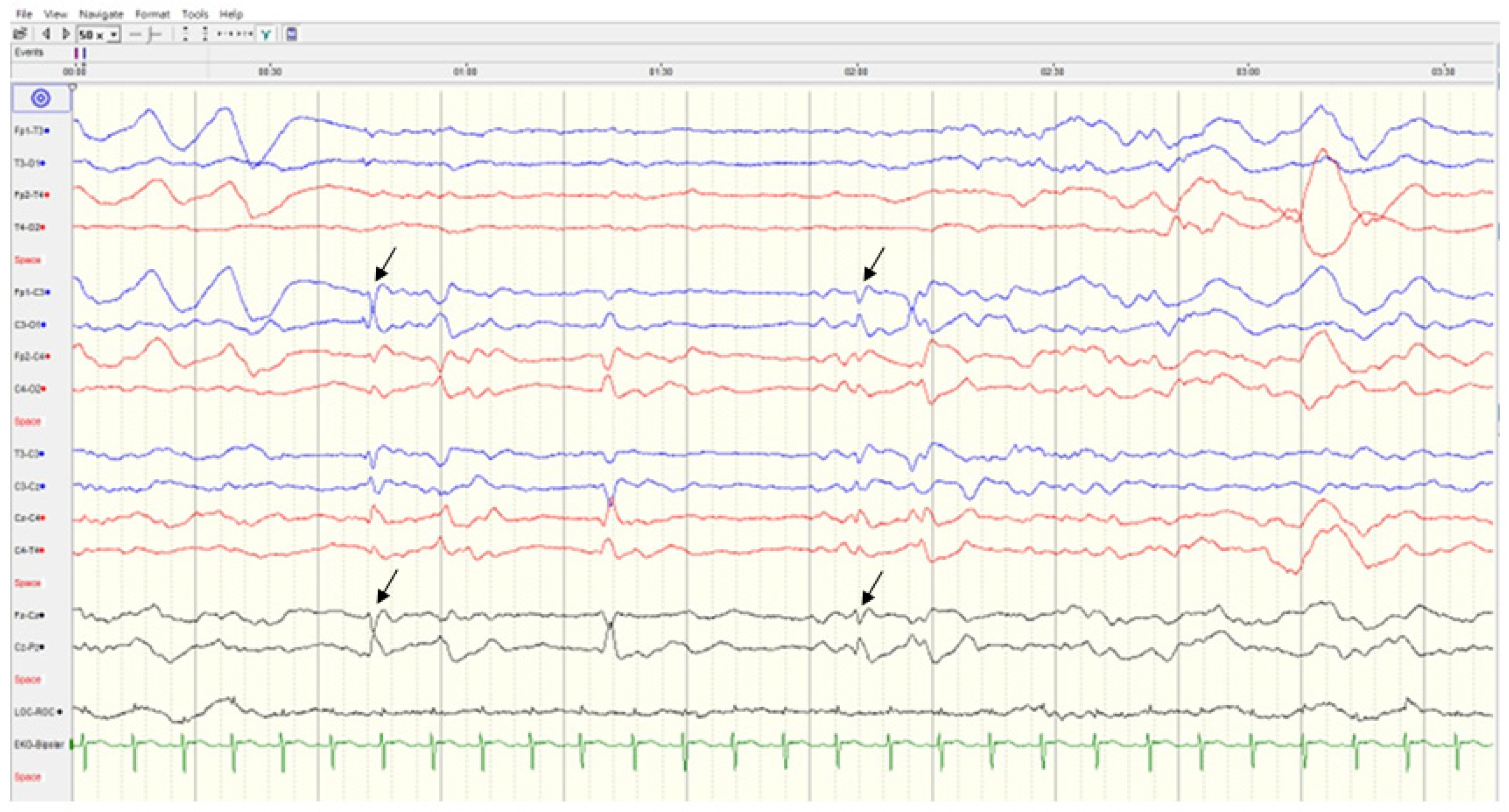
2.2. Case 2
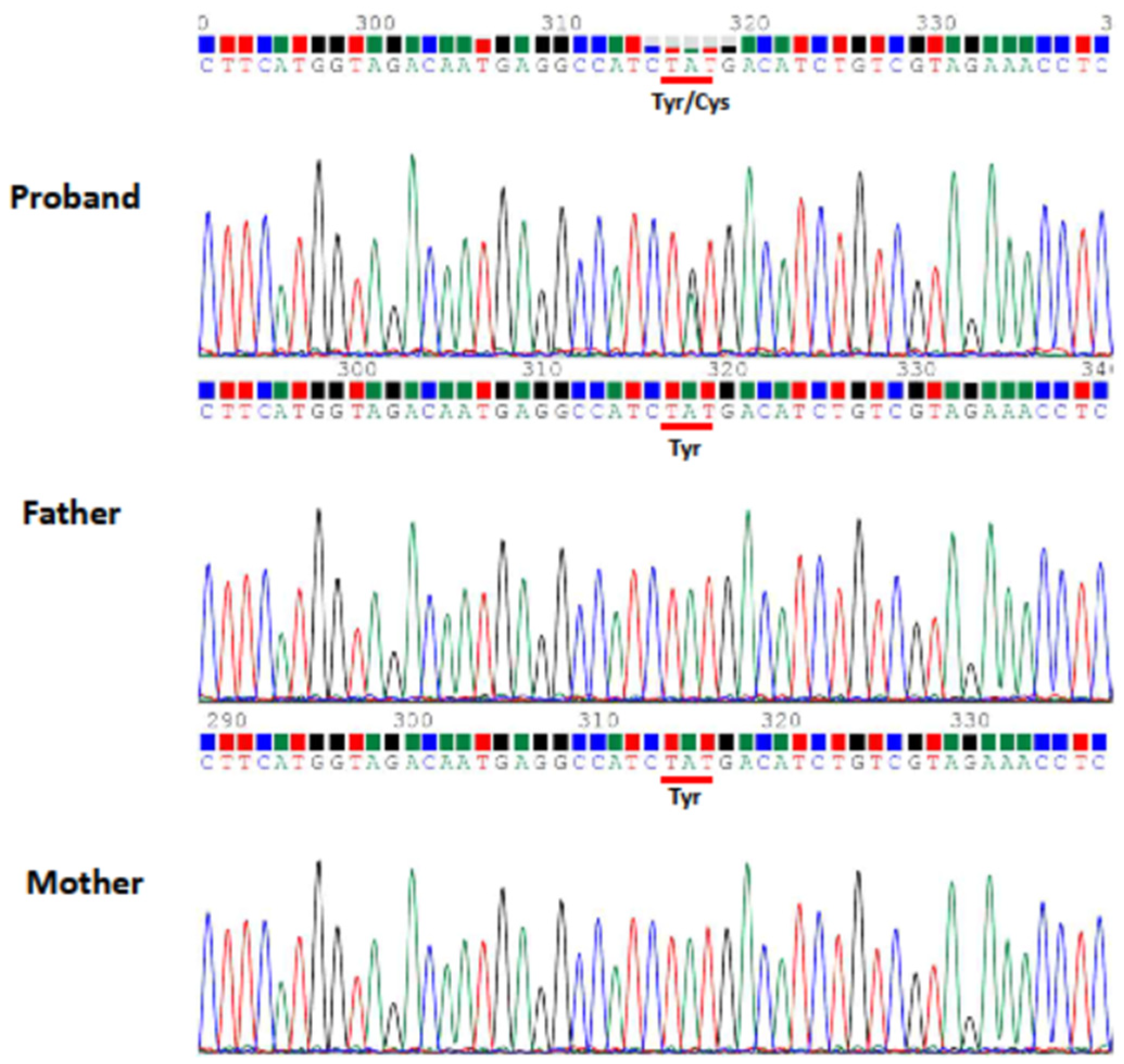
Patient Information
3. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Scheffer, I.E.; Berkovic, S.; Capovilla, G.; Connolly, M.B.; French, J.; Guilhoto, L.; Hirsch, E.; Jain, S.; Mathern, G.W.; Moshé, S.L.; et al. ILAE classification of the epilepsies: Position Paper of the ILAE Commission for Classification and Terminology. Epilepsia 2017, 58, 512–521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McTague, A.; Howell, K.B.; Cross, J.H.; Kurian, M.A.; Scheffer, I.E. The genetic landscape of the epileptic encephalopathies of infancy and childhood. Lancet Neurol. 2016, 15, 304–316. [Google Scholar] [CrossRef]
- Bartolini, E. Inherited developmental and epileptic encephalopathies. Neurol. Int. 2021, 13, 555–568. [Google Scholar] [CrossRef] [PubMed]
- Specchio, N.; Curatolo, P. Developmental and epileptic encephalopathies: What we do and do not know. Brain 2021, 144, 32–43. [Google Scholar] [CrossRef] [PubMed]
- Romaniello, R.; Arrigoni, F.; Fry, A.E.; Bassi, M.T.; Rees, M.I.; Borgatti, R.; Pilz, D.T.; Cushion, T.D. Tubulin genes and malformations of cortical development. Eur. J. Med. Genet. 2018, 61, 744–754. [Google Scholar] [CrossRef] [PubMed]
- Romaniello, R.; Arrigoni, F.; Bassi, M.T.; Borgatti, R. Mutations in α- and β-tubulin encoding genes: Implications in brain malformations. Brain Dev. 2015, 37, 273–280. [Google Scholar] [CrossRef] [PubMed]
- Ayala, R.; Shu, T.; Tsai, L.H. Trekking across the brain: The journey of neuronal migration. Cell 2007, 128, 29–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaglin, X.H.; Chelly, J. Tubulin-related cortical dysgeneses: Microtubule dysfunction underlying neuronal migration defects. Trends Genet. 2009, 25, 555–566. [Google Scholar] [CrossRef] [PubMed]
- Bahi-Buisson, N.; Poirier, K.; Fourniol, F.; Saillour, Y.; Valence, S.; Lebrun, N.; Hully, M.; Bianco, C.F.; Boddaert, N.; Elie, C.; et al. The wide spectrum of tubulinopathies: What are the key features for the diagnosis? Brain 2014, 137, 1676–1700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keays, D.A.; Tian, G.; Poirier, K.; Huang, G.J.; Siebold, C.; Cleak, J.; Oliver, P.L.; Fray, M.; Harvey, R.J.; Molnár, Z.; et al. Mutations in alpha-tubulin cause abnormal neuronal migration in mice and lissencephaly in humans. Cell 2007, 128, 45–47. [Google Scholar] [CrossRef] [PubMed]
- Schröter, J.; Döring, J.H.; Garbade, S.F.; Hoffmann, G.F.; Kölker, S.; Ries, M.; Syrbe, S. Cross-sectional quantitative analysis of the natural history of TUBA1A and TUBB2B tubulinopathies. Genet. Med. 2021, 23, 516–523. [Google Scholar] [CrossRef] [PubMed]
- Mutch, C.A.; Poduri, A.; Sahin, M.; Barry, B.; Walsh, C.A.; Barkovich, A.J. Disorders of microtubule function in neurons: Imaging correlates. Am. J. Neuroradiol. 2016, 37, 528–535. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.A.; Pilz, D.T.; Babatz, T.D.; Cushion, T.D.; Harvey, K.; Topf, M.; Yates, L.; Robb, S.; Uyanik, G.; Mancini, G.M.; et al. TUBA1A mutations cause wide spectrum lissencephaly (smooth brain) and suggest that multiple neuronal migration pathways converge on alpha tubulins. Hum. Mol. Genet. 2010, 19, 2817–2827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaglin, X.H.; Poirier, K.; Saillour, Y.; Buhler, E.; Tian, G.; Bahi-Buisson, N.; Fallet-Bianco, C.; Phan-Dinh-Tuy, F.; Kong, X.; Bomont, P. Mutations in the beta-tubulin gene TUBB2B result in asymmetrical polymicrogyria. Nat. Genet. 2009, 41, 746–752. [Google Scholar] [CrossRef] [PubMed]
- Cabet, S.; Karl, K.; Garel, C.; Delius, M.; Hartung, J.; Lesca, G.; Chaoui, R.; Guibaud, L. Two different prenatal imaging cerebral patterns of tubulinopathy. Ultrasound Obstet. Gynecol. 2021, 57, 493–497. [Google Scholar] [CrossRef] [PubMed]
- Romaniello, R.; Zucca, C.; Arrigoni, F.; Bonanni, P.; Panzeri, E.; Bassi, M.T.; Borgatti, R. Epilepsy in Tubulinopathy: Personal series and literature review. Cells 2019, 8, 669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E. ACMG Laboratory Quality Assurance Committee Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jansen, A.C.; Oostra, A.; Desprechins, B.; De Vlaeminck, Y.; Verhelst, H.; Régal, L.; Verloo, P.; Bockaert, N.; Keymolen, K.; Seneca, S.; et al. TUBA1A mutations: From isolated lissencephaly to familial polymicrogyria. Neurology 2011, 76, 988–992. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Case 1 | Case 2 | |
|---|---|---|
| Gender, age | Male, 23 years | Male, 1 years 2 months |
| Age of seizure onset | 1 h | 4 h |
| Consanguinity | no | no |
| Prenatal history | malformation of brain | unremarkable |
| Epilepsy | multifocal, refractory | focal to generalized, recurrent |
| Hypotonia | yes | yes |
| Developmental delay | global | global |
| Skeletal involvement | scoliosis | unknown |
| Feeding | gastrostomy | oral |
| Mutation | TUBB2B | TUBA1A |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hung, K.-L.; Lu, J.-F.; Su, D.-J.; Hsu, S.-J.; Wang, L.-C. Tubulinopathy Presenting as Developmental and Epileptic Encephalopathy. Children 2022, 9, 1105. https://doi.org/10.3390/children9081105
Hung K-L, Lu J-F, Su D-J, Hsu S-J, Wang L-C. Tubulinopathy Presenting as Developmental and Epileptic Encephalopathy. Children. 2022; 9(8):1105. https://doi.org/10.3390/children9081105
Chicago/Turabian StyleHung, Kun-Long, Jyh-Feng Lu, Da-Jyun Su, Su-Jin Hsu, and Lee-Chin Wang. 2022. "Tubulinopathy Presenting as Developmental and Epileptic Encephalopathy" Children 9, no. 8: 1105. https://doi.org/10.3390/children9081105
APA StyleHung, K.-L., Lu, J.-F., Su, D.-J., Hsu, S.-J., & Wang, L.-C. (2022). Tubulinopathy Presenting as Developmental and Epileptic Encephalopathy. Children, 9(8), 1105. https://doi.org/10.3390/children9081105