
The Current States, Challenges, Ongoing Efforts, and Future Perspectives of Pharmaceutical Excipients in Pediatric Patients in Each Country and Region

 , ,
, ,
Abstract
:1. Introduction
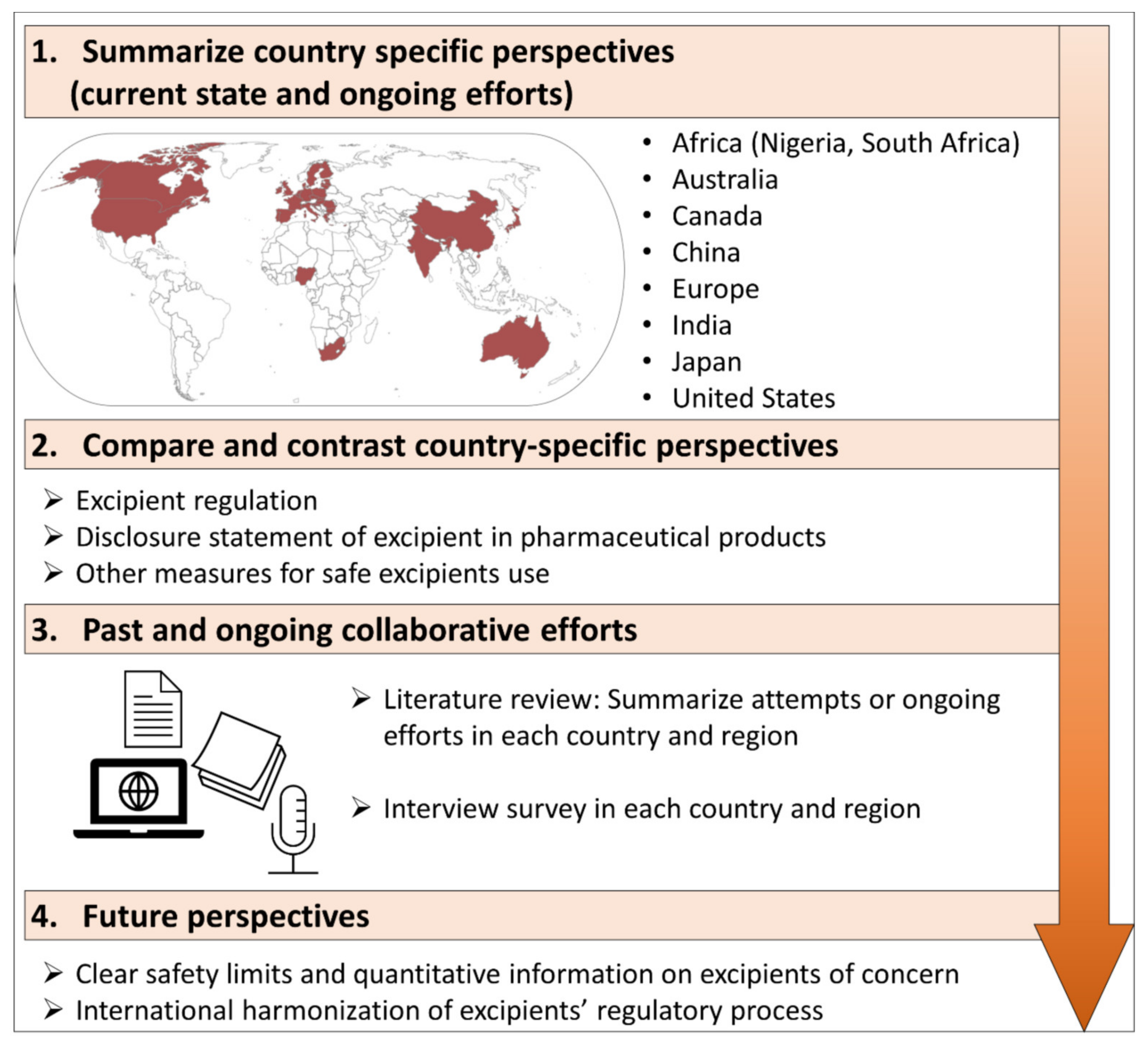
2. Country-Specific Perspectives
2.1. Africa
2.1.1. Current State and Challenges in Excipients Regulation
2.1.2. Ongoing Efforts
2.2. Australia
2.2.1. Current State and Challenges in Excipients Regulation
2.2.2. Ongoing Efforts
2.3. Canada
2.3.1. Current State and Challenges in Excipients Regulation
2.3.2. Ongoing Efforts
2.4. China
2.4.1. Current State and Challenges in Excipients Regulation
2.4.2. Ongoing Efforts
2.5. Europe
2.5.1. Current State and Challenges in Excipients Regulation
2.5.2. Ongoing Efforts
2.6. India
2.6.1. Current State and Challenges in Excipients Regulation
2.6.2. Ongoing Efforts
2.7. Japan
2.7.1. Current State and Challenges in Excipients Regulation
2.7.2. Ongoing Efforts
2.8. United States of America
2.8.1. Current State and Challenges in Excipients Regulation
2.8.2. Ongoing Efforts
3. Compare and Contrast Country-Specific Perspectives
4. Past and Ongoing Collaborative Efforts
4.1. Workshop for the Safety Qualification of Excipients
4.2. The Safe Excipient Exposure in Neonates and Small Children (SEEN) Project
4.3. The European Study of Neonatal Excipient Exposure (ESNEE)
4.4. Paediatric Excipient Risk Assessment (PERA) Framework
4.5. Harmonization of Pharmacopoeia
4.6. Other Efforts
5. Discussion
6. Future Perspectives of Pharmaceutical Excipients in Pediatrics Authors
7. Limitations
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Disclaimer
References
- Van den Anker, J.; Reed, M.D.; Allegaert, K.; Kearns, G.L. Developmental Changes in Pharmacokinetics and Pharmacodynamics. J. Clin. Pharmacol. 2018, 58, S10–S25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walsh, J.; Ranmal, S.R.; Ernest, T.B.; Liu, F. Patient acceptability, safety and access: A balancing act for selecting age-appropriate oral dosage forms for paediatric and geriatric populations. Int. J. Pharm. 2018, 536, 547–562. [Google Scholar] [CrossRef] [PubMed]
- European Medicines Agency (EMA). Guideline on Pharmaceutical Development of Medicines for Paediatric Use; EMA: London, UK, 2013; Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-pharmaceutical-development-medicines-paediatric-use_en.pdf (accessed on 2 February 2022).
- Allegaert, K. Propylene Glycol in Neonates: Never Prescribed, Frequently Administered, Hardly Evaluated. J. Clin. Toxicol. 2012, 2, 9. [Google Scholar] [CrossRef]
- European Medicines Agency (EMA). Questions and Answers on Propylene Glycol Used as an Excipient in Medicinal Products for Human Use; EMA: London, UK, 2017; Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/questions-answers-propylene-glycol-used-excipient-medicinal-products-human-use_en.pdf (accessed on 2 February 2022).
- European Medicines Agency (EMA). Questions and Answers on Benzoic Acid and Benzoates Used as Excipients in Medicinal Products for Human Use; EMA: London, UK, 2014; Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/questions-answers-benzoic-acid-benzoates-used-excipients-medicinal-products-human-use_en.pdf (accessed on 2 February 2022).
- Ohio Northern University. Pharmacist Role in Managing Special Patient Needs Related to Excipients; Medical Association; Ohio Northern University: Ada, OH, USA, 2009; Available online: https://apps.who.int/medicinedocs/en/d/Jh2995e/1.6.2.html (accessed on 2 February 2022).
- Andersen, A. Final amended report on the safety assessment of methylparaben, ethylparaben, propylparaben, isopropylparaben, butylparaben, isobutylparaben, and benzylparaben as used in cosmetic products. Int. J. Toxicol. 2014, 27, 1–82. [Google Scholar]
- Lee, B.H.; Kim, S.H. Benzalkonium chloride induced bronchoconstriction in patients with stable bronchial asthma. Korean J. Intern. Med. 2007, 22, 244–248. [Google Scholar] [CrossRef] [PubMed]
- Zuccotti, G.V.; Fabiano, V. Safety issues with ethanol as an excipient in drugs intended for pediatric use. Expert Opin. Drug Saf. 2011, 10, 499–502. [Google Scholar] [CrossRef] [PubMed]
- Walsh, J.; Cram, A.; Woertz, K.; Breitkreutz, J.; Winzenburg, G.; Turner, R.; Tuleu, C. Playing hide and seek with poorly tasting paediatric medicines: Do not forget the excipients. Adv. Drug Deliv. Rev. 2014, 73, 14–33. [Google Scholar] [CrossRef] [Green Version]
- Rouaz, K.; Chiclana-Rodríguez, B.; Nardi-Ricart, A.; Suñé-Pou, M.; Mercadé-Frutos, D.; Suñé-Negre, J.M.; Pérez-Lozano, P.; García-Montoya, E. Excipients in the Paediatric Population: A Review. Pharmaceutics 2021, 13, 387. [Google Scholar] [CrossRef]
- Pawar, S.; Kumar, A. Issues in the formulation of drugs for oral use in children: Role of excipients. Paediatr. Drugs 2002, 4, 371–379. [Google Scholar] [CrossRef]
- Belayneh, A.; Tadese, E.; Molla, F. Safety and biopharmaceutical challenges of excipients in off-label pediatric formulations. Int. J. Gen. Med. 2020, 13, 1051–1066. [Google Scholar] [CrossRef]
- Groll, A.H.; Wood, L.; Roden, M.; Mickiene, D.; Chiou, C.C.; Townley, E.; Dad, L.; Piscitelli, S.C.; Walsh, T.J. Safety, pharmacokinetics, and pharmacodynamics of cyclodextrin itraconazole in pediatric patients with oropharyngeal candidiasis. Antimicrob. Agents Chemother. 2002, 46, 2554–2563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- British Medical Association and Royal Pharmaceutical Society of Great Britain. General Guidance: Excipients. In Joint Formulary Committee; British National Formulary 2013–2014 Edition; British Medical Association and Royal Pharmaceutical Society of Great Britain: London, UK, 2014. [Google Scholar]
- National Agency for Food and Drug Administration and Control (NAFDAC). Quality Guidelines for the Registration of Pharmaceutical Products for Human Use in Nigeria; NAFDAC: Abuja, Nigeria, 2020. Available online: http://www.nafdac.gov.ng/wp-content/uploads/Files/Resources/Guidelines/R_and_R_Guidelines/GENERAL/Quality-Guidelines-for-Registration-of-Pharmaceutical-Products26381.pdf (accessed on 2 February 2022).
- National Agency for Food and Drug Administration and Control (NAFDAC). Label Guidance; NAFDAC: Abuja, Nigeria, 2021. Available online: https://www.nafdac.gov.ng/wp-content/uploads/Files/Resources/Guidelines/R_and_R_Guidelines/More_On_R_and_R/Guidelines-for-Labelling-26375.pdf (accessed on 2 February 2022).
- National Agency for Food and Drug Administration and Control (NAFDAC). Good Clinical Practice Guidelines 2020; NAFDAC: Abuja, Nigeria, 2020. Available online: https://www.nafdac.gov.ng/wp-content/uploads/Files/Resources/Guidelines/CTD_Guidelines/NAFDAC-Good-Clinical-Practices-Guidelines-2020.pdf (accessed on 2 February 2022).
- National Agency for Food and Drug Administration and Control (NAFDAC). Guidelines for Clinical Trial in Paediatric Populations; NAFDAC: Abuja, Nigeria, 2021. Available online: https://www.nafdac.gov.ng/wp-content/uploads/Files/Resources/Guidelines/DRUG_GUIDELINES/GUIDELINES-FOR-CLINICAL-TRIAL-IN-PAEDIATRIC-POPULATIONS-1.pdf (accessed on 2 February 2022).
- National Agency for Food and Drug Administration and Control (NAFDAC). Non-Nutritive Sweeteners Prohibition in Drugs Regulations 2019; NAFDAC: Abuja, Nigeria, 2019. Available online: https://www.nafdac.gov.ng/wp-content/uploads/Files/Resources/Regulations/DRUG_REGULATIONS/Non-nutritive-sweeteners-Prohibition-in-Drugs-Regulations-2019.pdf (accessed on 2 February 2022).
- Akinyandenu, O. Counterfeit drug in Nigeria: A threat to public health. Afr. J. Pharm. Pharmacol. 2013, 7, 2571–2576. [Google Scholar] [CrossRef] [Green Version]
- Soremekun, R.; Ogbuefi, I.; Aderemi-Williams, R. Prevalence of ethanol and other potentially harmful excipients in pediatric oral medicines: Survey of community pharmacies in a Nigerian City. BMC Res. Notes 2019, 12, 460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- South African Health Products Regulatory Authority (SAHPRA). Guideline for Professional Information for Human Medicine (Categories A and D); SAHPRA: Pretoria, South Africa, 2020. Available online: https://www.sahpra.org.za/wp-content/uploads/2020/02/2.16_Guideline-for-Professional-Information-for-Human-Medicines-Categories-A-and-D_Jul19_v2-1.pdf (accessed on 2 February 2022).
- South African Health Products Regulatory Authority (SAHPRA). Reliance Guideline; SAHPRA: Pretoria, South Africa, 2020. Available online: https://www.sahpra.org.za/wp-content/uploads/2021/11/Reliance-Guideline_v2_23-Oct-2021.pdf (accessed on 2 February 2022).
- South African Health Products Regulatory Authority (SAHPRA). Guideline for Patient Information Leaflet for Human Medicines (Categories A and D); SAHPRA: Pretoria, South Africa, 2020. Available online: https://www.sahpra.org.za/wp-content/uploads/2020/02/2.14_Guideline-for-Patient-Information-Leaflet-for-Human-Medicines-Categories-A-and-D_Jul19_v2-1.pdf (accessed on 2 February 2022).
- South African Health Products Regulatory Authority (SAHPRA). Quality and Bioequivalence Guideline; SAHPRA: Pretoria, South Africa, 2020. Available online: https://www.sahpra.org.za/wp-content/uploads/2020/02/2.02_Quality-and-Bioequivalence-Guideline_Jul19_v7-1.pdf (accessed on 2 February 2022).
- South African Health Products Regulatory Authority (SAHPRA). Interim Variations Addendum for Human and Veterinary Medicines; SAHPRA: Pretoria, South Africa, 2020. Available online: https://www.sahpra.org.za/wp-content/uploads/2020/10/Interim-Variations-Addendum-for-Human-and-Veterinary-Medicines_-Final.docx.pdf (accessed on 2 February 2022).
- Oshikoya, K.A.; Senbanjo, I.O. Providing safe medicines for children in Nigeria: The impediments and remedies. Ann. Afr. Med. 2010, 9, 203–212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ndomondo-Sigonda, M.; Miot, J.; Naidoo, S.; Dodoo, A.; Kaale, E. Medicines Regulation in Africa: Current State and Opportunities. Pharm. Med. 2017, 31, 383–397. [Google Scholar] [CrossRef] [Green Version]
- Australian Government, Department of Health Therapeutic Goods Administration (TGA). Evidence of GMP for Pre-Scription Medicines; TGA: Woden, Australia, 2019. Available online: https://www.tga.gov.au/evidence-gmp-prescription-medicines (accessed on 2 February 2022).
- Australian Government, Department of Health Therapeutic Goods Administration (TGA). Colourings Used in Medicines for Topical and Oral Use; TGA: Woden, Australia, 2018. Available online: https://www.tga.gov.au/publication/colourings-used-medicines-topical-and-oral-use (accessed on 2 February 2022).
- Commission Regulation No. 231/2012 of 9 March 2012. Laying down specifications for food additives listed in Annexes II and III to Regulation No 1333/2008 of the European Parliament and of the Council. Off. J. Eur. Union 2012, 55, 1–295.
- European Medicines Agency. Excipients in the Dossier for Application for Marketing Authorisation of a Medicinal Product; EMA: London, UK, 2008; Available online: https://www.ema.europa.eu/en/excipients-dossier-application-marketing-authorisation-medicinal-product (accessed on 2 February 2022).
- Australian Government, Department of Health Therapeutic Goods Administration (TGA). Guideline for the Labelling of Medicines; TGA: Woden, Australia, 2014. Available online: https://www.tga.gov.au/sites/default/files/consult-labelling-medicines-140822-guideline.pdf (accessed on 2 February 2022).
- Australian Government, Department of Health Therapeutic Goods Administration (TGA). Australian Register of Therapeutic Goods; TGA: Woden, Australia, 2019. Available online: https://www.tga.gov.au/australian-register-therapeutic-goods (accessed on 2 February 2022).
- Government of Canada. Food and Drug Regulations (C.R.C., c. 870) Part C.01.001; Government of Canada: Ottawa, ON, Canada. Available online: https://laws-lois.justice.gc.ca/eng/regulations/c.r.c.,_c._870/page-1.html (accessed on 2 February 2022).
- Health Canada. Guidance Document: Quality (Chemistry and Manufacturing) Guidance: New Drug Submissions (NDSs) and Abbreviated New Drug Submissions (ANDSs); Health Canada: Ottawa, ON, Canada, 2018. Available online: https://www.canada.ca/en/health-canada/services/drugs-health-products/drug-products/announcements/notice-quality-chemistry-manufacturing-guidance-new-drug-submissions-ndss-abbreviated-new-drug-submissions.html (accessed on 2 February 2022).
- Health Canada. Labelling of Pharmaceutical Drugs for Human Use; Health Canada: Ottawa, ON, Canada, 2015. Available online: https://www.canada.ca/en/health-canada/services/drugs-health-products/drug-products/announcements/updates-guidance-document-labelling-pharmaceutical-drugs-human-use.html (accessed on 2 February 2022).
- Health Canada. International Council for Harmonisation (ICH); Health Canada: Ottawa, ON, Canada, 2021. Available online: https://www.canada.ca/en/health-canada/services/drugs-health-products/drug-products/applications-submissions/guidance-documents/international-conference-harmonisation.html (accessed on 2 February 2022).
- Mennella, J.A.; Beauchamp, G.K. Optimizing oral medications for children. Clin. Ther. 2008, 30, 2120–2132. [Google Scholar] [CrossRef] [Green Version]
- Fatima, B.; Ladislav, N. Artificial sweeteners and sugar substitutes: Some properties and potential health benefits and risks. Res. J. Pharm. Biol. Chem. Sci. 2014, 5, 638–649. [Google Scholar]
- Ivanovska, V.; Rademaker, C.M.; Van Dijk, L.; Mantel-Teeuwisse, A.K. Pediatric drug formulations: A review of challenges and progress. Pediatrics 2014, 134, 361–372. [Google Scholar] [CrossRef] [Green Version]
- Buckley, L.A.; Salunke, S.; Thompson, K.; Baer, G.; Fegley, D.; Turner, M.A. Challenges and strategies to facilitate formulation development of pediatric drug products: Safety qualification of excipients. Int. J. Pharm. 2018, 536, 563–569. [Google Scholar] [CrossRef] [Green Version]
- Mei, M.; Xu, H.; Wang, L.; Huang, G.; Gui, Y.; Zhang, X. Current practice and awareness of pediatric off-label drug use in Shanghai, China—A questionnaire-based study. BMC Pediatr. 2019, 19, 281. [Google Scholar] [CrossRef] [Green Version]
- Batchelor, H.K.; Marriott, J.F. Paediatric pharmacokinetics: Key considerations. Br. J. Clin. Pharmacol. 2015, 79, 395–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Health Canada. Policy on Manufacturing and Compounding Drug Products in Canada (POL-0051); Health Canada: Ottawa, ON, Canada, 2009. Available online: https://www.canada.ca/en/health-canada/services/drugs-health-products/compliance-enforcement/good-manufacturing-practices/guidance-documents/policy-manufacturing-compounding-drug-products.html (accessed on 2 February 2022).
- National Association of Pharmacy Regulatory Authorities (NAPRA). Companion to the Model Standards for Pharmacy Compounding of Non-Sterile Preparations; NAPRA: Ottawa, ON, Canada, 2018. Available online: https://napra.ca/sites/default/files/documents/Mdl_Stnds_Pharmacy_Compounding_Nonsterile_Preparations_Guidance_June2018_FINAL.pdf (accessed on 2 February 2022).
- Government of Canada. Forward Regulatory Plan 2021–2023: Amendments to the Food and Drug Regulations—Commercial Compounding; Government of Canada: Ottawa, ON, Canada. Available online: https://www.canada.ca/en/health-canada/corporate/about-health-canada/legislation-guidelines/acts-regulations/forward-regulatory-plan/plan/regulatory-initiative-amendments-food-drug-regulations-commercial-compounding.html (accessed on 11 February 2022).
- Salunke, S.; Brandys, B.; Giacoia, G.; Tuleu, C. The STEP (Safety and Toxicity of Excipients for Paediatrics) database: Part 2—The pilot version. Int. J. Pharm. 2013, 457, 310–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyers, R.S.; Thackray, J.; Matson, K.L.; McPherson, C.; Lubsch, L.; Hellinga, R.C.; Hoff, D.S. Key potentially inap-propriate drugs in pediatrics: The KIDs List. J. Pediatr. Pharmacol. Ther. 2020, 25, 175–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yochana, S.; Yu, M.; Alvi, M.; Varenya, S.; Chatterjee, P. Pharmaceutical excipients and pediatric formulations. Chem. Today 2012, 30, 56–61. Available online: https://www.teknoscienze.com/tks_article/pharmaceutical-excipients-and-pediatric-formulations (accessed on 11 February 2022).
- Government of Canada. Drug and Medical Device Post Market Surveillance; Government of Canada: Ottawa, ON, Canada. Available online: https://www.canada.ca/en/services/health/drug-health-products/drug-medical-device-highlights-2017/post-market-surveillance.html (accessed on 11 February 2022).
- Salunke, S.; Giacoia, G.; Tuleu, C. The STEP (safety and toxicity of excipients for paediatrics) database. Part 1—A need assessment study. Int. J. Pharm. 2012, 435, 101–111. [Google Scholar] [CrossRef]
- Salunke, S.; Tuleu, C. The STEP database through the end-user’s eyes—Usability Study. Int. J. Pharm. 2015, 492, 316–331. [Google Scholar] [CrossRef]
- Challener, C.A. Novel Excipients Needed More Than Ever Before. Pharm. Technol. 2021, 45, 24–29. Available online: https://www.pharmtech.com/view/novel-excipients-needed-more-than-ever-before (accessed on 2 February 2022).
- Kingwell, K. Excipient developers call for regulatory facelift. Nat. Rev. 2020, 19, 823–824. [Google Scholar] [CrossRef]
- Elder, D.P.; Kuentz, M.; Holm, R. Pharmaceutical excipients—Quality, regulatory and biopharmaceutical considerations. Eur. J. Pharm. Sci. 2016, 87, 88–99. [Google Scholar] [CrossRef]
- European Medicines Agency (EMA). ICH Guideline S11 on Nonclinical Safety Testing in Support of Development of Paediatric Pharmaceuticals Step 5; EMA: London, UK, 2020; Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/ich-guideline-s11-nonclinical-safety-testing-support-development-paediatric-pharmaceuticals-step-5_en.pdf (accessed on 2 February 2022).
- The People’s Republic of China. Drug Administration Law of the People’s Republic of China; The People’s Republic of China: Beijing, China, 2020. Available online: http://www.npc.gov.cn/englishnpc/c23934/202012/3c19c24f9ca04d1ba0678c6f8f8a4a8a.shtml (accessed on 2 February 2022).
- The China Food and Drug Administration (CFDA). Chinese Pharmacopoeia; CFDA: Beijing, China, 2019. Available online: http://wp.chp.org.cn/front/chpint/en/ (accessed on 2 February 2022).
- National Law Review. China Amends DMF System on Drug Packaging; Keller and Heckman LLP: Shanghai, China, 2022. Available online: https://www.natlawreview.com/article/china-amends-dmf-system-drug-packaging (accessed on 2 February 2022).
- European Commission (EC). Excipients in the Labelling and Package Leaflet of Medicinal Products for Human Use; EC: Brussels, Belgium, 2018; Available online: https://ec.europa.eu/health/sites/default/files/files/eudralex/vol-2/c/guidelines_excipients_march2018_en.pdf (accessed on 2 February 2022).
- European Medicines Agency (EMA). Annex to the European Commission Guideline on ‘Excipients in the Labelling and Package Leaflet of Medicinal Products for Human Use’; EMA: London, UK, 2019; Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/annex-european-commission-guideline-excipients-labelling-package-leaflet-medicinal-products-human_en.pdf (accessed on 2 February 2022).
- The European parliament and the council of the European Union. Directive 2004/27/EC of the European parliament and of the council of 31 March 2004. Off. J. Eur. Union 2004, L311, 34–57. Available online: https://ec.europa.eu/health/sites/default/files/files/eudralex/vol-1/dir_2004_27/dir_2004_27_en.pdf (accessed on 2 February 2022).
- European Medicines Agency (EMA). Information for the Package Leaflet Regarding Fructose and Sorbitol Used as Excipients in Medicinal Products for Human Use; EMA: London, UK, 2017; Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/information-package-leaflet-regarding-fructose-sorbitol-used-excipients-medicinal-products-human-use_en.pdf (accessed on 2 February 2022).
- Christiansen, N. Ethanol exposure through medicines commonly used in paediatrics. Arch. Dis. Child. 2015, 100, 101–104. [Google Scholar] [CrossRef] [PubMed]
- National Health Service (NHS). The Truth about Sweeteners; National Health Service (NHS): London, UK. Available online: https://www.nhs.uk/live-well/eat-well/are-sweeteners-safe (accessed on 2 February 2022).
- National Health Service (NHS). Saccharin Link to Cancer Discredited; National Health Service (NHS): London, UK. Available online: http://www.nhs.uk/Livewell/Goodfood/Pages/the-truth-about-saccharin.aspx (accessed on 2 February 2022).
- De Schaepdrijver, L.; Mariën, D.; Rhimi, C.; Voets, M.; Van Heerden, M.; Lammens, L. Juvenile animal testing of hydroxypropyl-β-cyclodextrin in support of pediatric drug development. Reprod. Toxicol. 2015, 56, 87–96. [Google Scholar] [CrossRef] [PubMed]
- European Medicines Agency (EMA). Questions and Answers on Cyclodextrins Used as Excipients in Medicinal Products for Human Use; EMA: London, UK, 2013; Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/questions-answers-cyclodextrins-used-excipients-medicinal-products-human-use_en.pdf (accessed on 2 February 2022).
- O’Brien, F.; Pawar, R.; Upadhaya, P.; Syed, S.; Patravale, V.; Salunke, S. Excipients in parenteral formulations used in Paediatric and neonatal patients: A systematic review. In Proceedings of the 12th EuPFI Virtual Conference, 9–10 September 2020. [Google Scholar]
- UNICEF. Children in India; UNICEF: New York, NY, USA, 2019; Available online: https://www.unicef.org/india/children-in-india (accessed on 2 February 2022).
- Nasrollahi, S.; Meera, N.K.; Boregowda, S. Pharmaceutical Excipient Exposure in a Neonatal Intensive Care Unit. Indian Pediatr. 2020, 57, 801–804. [Google Scholar] [CrossRef]
- World Population Prospects—Population Division. Available online: https://population.un.org/wpp/dataquery (accessed on 2 February 2022).
- Statista. India-Age Distribution. 2019. Available online: https://www.statista.com/statistics/271315/age-distribution-in-india (accessed on 2 February 2022).
- Galande, A.D.; Khurana, N.A.; Mutalik, S. Pediatric dosage forms—Challenges and recent developments: A critical review. J. Appl. Pharm. Sci. 2020, 10, 155–166. [Google Scholar] [CrossRef]
- Jain, A.; Venkatesh, M.P.; Pramod Kumar, T.M.; Naveen Kumar, S. Regulation for paediatric drug development in India: Need of the hour. J. Clin. Stud. 2014, 6, 14–17. [Google Scholar]
- Imran, M.; Najmi, A.K.; Rashid, M.F.; Tabrez, S.; Shah, M.A. Clinical research regulation in India-history, development, initiatives, challenges and controversies: Still long way to go. J. Pharm. Bioallied Sci. 2013, 5, 2–9. [Google Scholar] [CrossRef]
- Saluja, V.; Sekhon, B. The regulation of pharmaceutical excipients. J. Excip. Food Chem. 2013, 4, 95–106. [Google Scholar]
- Pharmaceuticals and Medical Devices Agency (PMDA). Notifications and Administrative Notices; Notification No. 853 of the PAB Dated 1 October 1988; PMDA: Tokyo, Japan, 1988. [Google Scholar]
- Pharmaceuticals and Medical Devices Agency (PMDA). Notifications and Administrative Notices; Notification No. 0608-(1) of the Safety Division, PSEHB Dated 8 June 2017; PMDA: Tokyo, Japan, 2017; Available online: https://www.pmda.go.jp/files/000218446.pdf (accessed on 2 February 2022).
- Pharmaceuticals and Medical Devices Agency (PMDA). Notifications and Administrative Notices; PFSB/ELD Notification No. 0210001, 10 February, 2005; PMDA: Tokyo, Japan, 2005; Available online: https://www.pmda.go.jp/files/000153677.pdf (accessed on 2 February 2022).
- Saito, J.; Akabane, M.; Ishikawa, Y.; Nakamura, H.; Yamatani, A. Potentially Harmful Excipients in Neonatal Medications: An Observational and Cross-Regional Comparison of Japan and Europe. Neonatal. Pediatr. Med. 2018, 4, 172. [Google Scholar] [CrossRef]
- Saito, J.; Nadatani, N.; Setoguchi, M.; Nakao, M.; Kimura, H.; Sameshima, M.; Kobayashi, K.; Matsumoto, H.; Yoshikawa, N.; Yokoyama, T.; et al. Potentially harmful excipients in neonatal medications: A multicenter nationwide observational study in Japan. J. Pharm. Health Care Sci. 2021, 7, 23. [Google Scholar] [CrossRef]
- Food and Drug Administration (FDA). Guidance for Industry: Nonclinical Studies for the Safety Evaluation of Pharmaceutical Excipients; FDA: Rockville, MD, USA, 2005. Available online: https://www.fda.gov/media/72260/download (accessed on 2 February 2022).
- Food and Drug Administration (FDA). List of Drug Master Files (DMFs); FDA: Silver Spring, MD, USA, 2021. Available online: https://www.fda.gov/drugs/drug-master-files-dmfs/list-drug-master-files-dmfs (accessed on 2 February 2022).
- Food and Drug Administration (FDA). Guidance for Industry: Nonclinical Safety Evaluation of Pediatric Drug Products; FDA: Rockville, MD, USA, 2006. Available online: https://www.fda.gov/media/119658/download (accessed on 2 February 2022).
- Schmitt, G. Safety of Excipients in Pediatric Formulations-A Call for Toxicity Studies in Juvenile Animals? Children 2015, 2, 191–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- USP 35. In United States Pharmacopeial Convention: United States Pharmacopeia and the National Formulary (USP 35-NF 30); The United States Pharmacopeial Convention: Rockville, MD, USA, 2012.
- The International Pharmaceutical Excipients Council (IPEC). The IPEC Excipient Composition Guide; IPEC: Brussels, Belgium, 2009; Available online: https://ipec-federation.org/ipec-federation-revises-its-excipient-composition-guide (accessed on 2 February 2022).
- Collins, G.; Ulman, K.; Muse, D.G.; Zeleznik, J.; Zawislak, P.; Tocce, E.; Moreton, R.C. Additives and Processing Aids in Pharmaceutical Excipients. Pharm. Technol. APIs Excip. Manuf. 2019, 5, s16–s19. [Google Scholar]
- Food and Drug Administration (FDA). Pediatric Information Incorporated into Human Prescription Drug and Biological Product Labeling Guidance for Industry; FDA: Silver Spring, MD, USA, 2019. Available online: https://www.fda.gov/media/84949/download (accessed on 2 February 2022).
- Challener, C.A. Exploring a new approval process for continued excipient innovation. Pharmaceutical Technology, 1 September 2014. Available online: https://www.pharmtech.com/view/exploring-new-approval-process-continued-excipient-innovation (accessed on 2 February 2022).
- Sheehan, C. Highlights of FDA-USP workshop on “Critical Importance of Excipients in Drug Development–Why Excipients are Important Now and, In the Future,” The Need to update USP Excipient Standards. In Proceedings of the AAM Fall Tech Conference 2017, Rockville, MD, USA, 6–8 November 2017; Available online: https://accessiblemeds.org/sites/default/files/2017-11/John%20Giannone-Catherine%20Sheehan.pdf (accessed on 2 February 2022).
- Food and Drug Administration (FDA). Novel excipient review program proposal; Request for information and comments. Fed. Regist. 2019; 84, pp. 66669–666671. Available online: https://www.govinfo.gov/content/pkg/FR-2019-12-05/pdf/2019-26266.pdf (accessed on 2 February 2022).
- Valeur, K.S.; Holst, H.; Allegaert, K. Excipients in Neonatal Medicinal Products: Never Prescribed, Commonly Administered. Pharm. Med. 2018, 32, 251–258. [Google Scholar] [CrossRef] [Green Version]
- Turner, M.A.; Duncan, J.; Shah, U.; Metsvaht, T.; Varendi, H.; Nellis, G.; Lutsar, I.; Vaconsin, P.; Storme, T.; Rieutord, A.; et al. European Study of Neonatal Exposure to Excipients: An update. Int. J. Pharm. 2013, 457, 357–358. [Google Scholar] [CrossRef]
- Nellis, G.; Metsvaht, T.; Varendi, H.; Toompere, K.; Lass, J.; Mesek, I.; Nunn, A.J.; Turner, M.A.; Lutsar, I. Potentially harmful excipients in neonatal medicines: A pan-European observational study. Arch. Dis. Child. 2015, 100, 694–699. [Google Scholar] [CrossRef] [PubMed]
- Shehab, N.; Lewis, C.L.; Streetman, D.D.; Donn, S.M. Exposure to the pharmaceutical excipients benzyl alcohol and propylene glycol among critically ill neonates. Pediatr. Crit. Care Med. 2009, 10, 256–259. [Google Scholar] [CrossRef] [PubMed]
- Pharmacopoeial Discussion Group. Statement of Harmonization Policy; Pharmaceuticals and Medical Devices Agency: Tokyo, Japan, 2017; Available online: https://www.pmda.go.jp/files/000152959.pdf (accessed on 21 March 2022).
- Council of Europe. Harmonisasion Status of Excipient Monograph (PDG); Council of Europe: Strasbourg, France. Available online: https://www.edqm.eu/en/harmonisation-status-excipient-monographs-pdg (accessed on 2 February 2022).
- Pandya, H.C.; Mulla, H.; Hubbard, M.; Cordell, R.L.; Monks, P.S.; Yakkundi, S.; McElnay, J.C.; Nunn, A.J.; Turner, M.A. Essential medicines containing ethanol elevate blood acetaldehyde concentrations in neonates. Eur. J. Pediatr. 2016, 175, 841–847. [Google Scholar] [CrossRef] [Green Version]
- Whittaker, A.; Currie, A.E.; Turner, M.A.; Field, D.J.; Mulla, H.; Pandya, H.C. Toxic additives in medication for preterm infants. Arch. Dis. Child. Fetal Neonatal Ed. 2009, 94, F236–F240. [Google Scholar] [CrossRef] [Green Version]
- Akinmboni, T.O.; Davis, N.L.; Falck, A.J.; Bearer, C.F.; Mooney, S.M. Excipient exposure in very low birth weight preterm neonates. J. Perinatol. 2018, 38, 169–174. [Google Scholar] [CrossRef] [Green Version]
- Sridharan, K.; Hasan, H.M.; Al Jufairi, M.; Al Daylami, A.; Al Ansari, E.; Qader, A.M.; Pasha, S. Possible effects of excipients used in the parenteral drugs administered in critically ill adults, children, and neonates. Expert Opin. Drug Saf. 2020, 19, 1625–1640. [Google Scholar] [CrossRef]
- De Cock, R.F.; Allegaert, K.; Vanhaesebrouck, S.; De Hoon, J.; Verbesselt, R.; Danhof, M.; Knibbe, C.A. Low but inducible contribution of renal elimination to clearance of propylene glycol in preterm and term neonates. Ther. Drug Monit. 2014, 36, 278–287. [Google Scholar] [CrossRef] [PubMed]
- Ursino, M.G.; Poluzzi, E.; Caramella, C.; De Ponti, F. Excipients in medicinal products used in gastroenterology as a possible cause of side effects. Regul. Toxicol. Pharmacol. 2011, 60, 93–105. [Google Scholar] [CrossRef] [PubMed]
- Kogermann, K.; Lass, J.; Nellis, G.; Metsvaht, T.; Lutsar, I. Age-Appropriate Formulations Including Pharmaceutical Excipients in Neonatal Medicines. Curr. Pharm. Des. 2017, 23, 5779–5789. [Google Scholar] [CrossRef] [PubMed]
- Soni, M.G.; Burdock, G.A.; Taylor, S.L.; Greenberg, N.A. Safety assessment of propyl paraben: A review of the published literature. Food Chem. Toxicol. 2001, 39, 513–532. [Google Scholar] [CrossRef]
- Sviestina, I.; Mozgis, D. A retrospective and observational analysis of harmful excipients in medicines for hospitalised neonates in Latvia. Eur. J. Hosp. Pharm. 2018, 25, 176–182. [Google Scholar] [CrossRef]
- Lass, J.; Naelapää, K.; Shah, U.; Käär, R.; Varendi, H.; Turner, M.A.; Lutsar, I. Hospitalised neonates in Estonia commonly receive potentially harmful excipients. BMC Pediatr. 2012, 12, 136. [Google Scholar] [CrossRef] [Green Version]
- De Souza, A.S., Jr.; Dos Santos, D.B.; Rey, L.C.; Medeiros, M.G.; Vieira, M.G.; Coelho, H. Off-label use and harmful potential of drugs in a NICU in Brazil: A descriptive study. BMC Pediatr. 2016, 16, 13. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Palop, B.; Movilla Polanco, E.; Cañete Ramirez, C.; Cabañas Poy, M.J. Harmful excipients in medicines for neonates in Spain. Int. J. Clin. Pharm. 2016, 38, 238–242. [Google Scholar] [CrossRef]
- Fister, P.; Urh, S.; Karner, A.; Krzan, M.; Paro-Panjan, D. The prevalence and pattern of pharmaceutical and excipient exposure in a neonatal unit in Slovenia. J. Matern. Fetal Neonatal Med. 2015, 28, 2053–2061. [Google Scholar] [CrossRef]
- Mulla, H.; Yakkundi, S.; McElnay, J.; Lutsar, I.; Metsvaht, T.; Varendi, H.; Nellis, G.; Nunn, A.; Duncan, J.; Pandya, H.; et al. An observational study of blood concentrations and kinetics of methyl- and propyl-parabens in neonates. Pharm. Res. 2015, 32, 1084–1093. [Google Scholar] [CrossRef]
- Kulo, A.; De Hoon, J.N.; Allegaert, K. The propylene glycol research project to illustrate the feasibility and difficulties to study toxicokinetics in neonates. Int. J. Pharm. 2012, 435, 112–114. [Google Scholar] [CrossRef] [PubMed]
- Yakkundi, S.; Mulla, H.; Pandya, H.; Turner, M.A.; McElnay, J. Quantitative analysis of methyl and propyl parabens in neonatal DBS using LC-MS/MS. Bioanalysis 2016, 8, 1173–1182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allegaert, K.; Vanhaesebrouck, S.; Kulo, A.; Cosaert, K.; Verbesselt, R.; Debeer, A.; De Hoon, J. Prospective assessment of short-term propylene glycol tolerance in neonates. Arch. Dis. Child. 2010, 95, 1054–1058. [Google Scholar] [CrossRef] [PubMed]
- Graham, S.; Turner, M.A. European Study of. Neonatal Exposure to Excipients (ESNEE). Infant 2011, 7, 196–199. Available online: https://www.infantjournal.co.uk/pdf/inf_042_ien.pdf (accessed on 2 February 2022).
- Schrier, L.; Hadjipanayis, A.; Stiris, T.; Ross-Russell, R.I.; Valiulis, A.; Turner, M.A.; Zhao, W.; De Cock, P.; de Wildt, S.N.; Allegaert, K.; et al. Off-label use of medicines in neonates, infants, children, and adolescents: A joint policy statement by the European Academy of Paediatrics and the European society for Developmental Perinatal and Pediatric Pharmacology. Eur. J. Pediatr. 2020, 179, 839–847. [Google Scholar] [CrossRef]
- Tandel, K.R. Sugar substitutes: Health controversy over perceived benefits. J. Pharmacol. Pharmacother. 2011, 2, 236–243. [Google Scholar] [CrossRef] [Green Version]
- Kameyama, Y.; Matsuhama, M.; Mizumaru, C.; Saito, R.; Ando, T.; Miyazaki, S. Comparative Study of Pharmacopoeias in Japan, Europe, and the United States: Toward the Further Convergence of International Pharmacopoeial Standards. Chem. Pharm. Bull. 2019, 67, 1301–1313. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
| Country/Region | Regulation on Pharmaceutical Excipients | Disclosure of Excipients Information | Quantitative Information | Any other Special Measures for Safety of Excipients Use for Pediatrics |
|---|---|---|---|---|
| Africa |
|
|
|
|
| Australia |
|
|
|
|
| Canada |
|
|
|
|
| China |
|
|
| |
| EU |
|
|
|
|
| India |
|
|
|
|
| Japan |
|
|
|
|
| US |
|
|
|
|
| Country/Region | Other Attempts or Ongoing Efforts |
|---|---|
| Africa | Study on excipients exposure in pediatrics [23] |
| Australia | None |
| Canada | None |
| China | None |
| EU | Study on excipient exposure in pediatrics [67,97,99,111,112,115,119] ESNEE project [120] SEEN project [97] Workshop for the Safety Qualification of Excipients [44] |
| India | Study on excipients exposure in pediatrics [74] |
| Japan | A nationwide study on excipients exposure in neonates [85] |
| US | Study on excipients exposure in pediatrics [100,105] Incude pediatric safety in FDA IID Nove excipient review pilot program by CDER Workshops, survey, and pediatric excipient risk assessment framework development by IQ pediatric consortium |
| Challenges |
|
| |
| |
| |
| Solution |
|
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saito, J.; Agrawal, A.; Patravale, V.; Pandya, A.; Orubu, S.; Zhao, M.; Andrews, G.P.; Petit-Turcotte, C.; Landry, H.; Croker, A.; et al. The Current States, Challenges, Ongoing Efforts, and Future Perspectives of Pharmaceutical Excipients in Pediatric Patients in Each Country and Region. Children 2022, 9, 453. https://doi.org/10.3390/children9040453
Saito J, Agrawal A, Patravale V, Pandya A, Orubu S, Zhao M, Andrews GP, Petit-Turcotte C, Landry H, Croker A, et al. The Current States, Challenges, Ongoing Efforts, and Future Perspectives of Pharmaceutical Excipients in Pediatric Patients in Each Country and Region. Children. 2022; 9(4):453. https://doi.org/10.3390/children9040453
Chicago/Turabian StyleSaito, Jumpei, Anjali Agrawal, Vandana Patravale, Anjali Pandya, Samuel Orubu, Min Zhao, Gavin P. Andrews, Caroline Petit-Turcotte, Hannah Landry, Alysha Croker, and et al. 2022. "The Current States, Challenges, Ongoing Efforts, and Future Perspectives of Pharmaceutical Excipients in Pediatric Patients in Each Country and Region" Children 9, no. 4: 453. https://doi.org/10.3390/children9040453
APA StyleSaito, J., Agrawal, A., Patravale, V., Pandya, A., Orubu, S., Zhao, M., Andrews, G. P., Petit-Turcotte, C., Landry, H., Croker, A., Nakamura, H., Yamatani, A., & Salunke, S. (2022). The Current States, Challenges, Ongoing Efforts, and Future Perspectives of Pharmaceutical Excipients in Pediatric Patients in Each Country and Region. Children, 9(4), 453. https://doi.org/10.3390/children9040453