Abstract
Hypertriglyceridemia induced acute pancreatitis is a rare cause of pancreatitis in children. Hepatic lipase deficiency is an extremely rare cause of hypertriglyceridemia, reported in only a few families to date. Hepatic lipase is the enzyme involved in the hydrolysis of triglycerides and phospholipids in remnants of triglyceride-rich lipoproteins that have a role in the conversion of very low density lipoprotein remnants to low density lipoproteins. Hepatic lipase deficiency is inherited in an autosomal recessive pattern. Detection of heterozygous carriers of hepatic lipase mutations remains accidental at the population level, as affected persons with a heterozygous state of hepatic lipase mutation do not display specific lipoprotein abnormalities and also patients with complete hepatic lipase deficiency have inconstant phenotype. The proximal promoter of the LIPC gene consists of four polymorphic sites in complete linkage disequilibrium. Five missense mutations in encoding exons have been described and proved to be responsible for hepatic lipase deficiency to date: S267F, T383M, L334F, A174T, and R186H, affecting the activity and secretion of hepatic lipase. We identified a primary disorder of the lipid metabolism as the cause of the acute episode of pancreatitis in a four years old patient, consisting of hepatic lipase deficiency caused by a novel genetic variant of the LIPC gene, a gross deletion of the genomic region encompassing exon 1. This variant was not previously described in the literature in persons with LIPC-related disorders and its significance is currently uncertain, but in the presented clinical and paraclinical context, it has the characteristics of a pathological variant inducing a hepatic lipase deficiency phenotype.
1. Introduction
Primary diseases of lipid metabolism causing hypertriglyceridemia derive from genetic anomalies in triglyceride synthesis and metabolism [1]. Hypertriglyceridemia induced acute pancreatitis is an extremely rare, but important cause of pancreatitis in the pediatric population because it leads to significant morbidity and mortality [2]. An extremely rare cause of hypertriglyceridemia is hepatic lipase deficiency, an autosomal recessive transmitted disease that has been reported only in a few families to date [3]. Hepatic lipase is a lipolytic serine hydrolase synthesized and secreted by the hepatic cells, attached to the liver sinusoidal surface by heparin sulphate proteoglycans [4]. Only a few mutations and gene polymorphisms of hepatic lipase gene were reported [5]. We present a hypertriglyceridemia induced acute pancreatitis case in a pediatric patient caused by a previously unreported LIPC gene variant, a gross deletion of the genomic region encompassing exon 1 of the LIPC gene.
The aim of this paper is to present the rare case of acute pancreatitis in a pediatric patient determined by an extremely rare cause of hypertriglyceridemia, a novel variant of the LIPC gene, reported in only a few families to date.
2. Case Report
A 4 years and 9 months old boy presented to our Emergency Care Unit after 24 h of abdominal pain, anorexia, nausea, and bilious vomiting at home. Initial monitoring of the patient revealed a temperature of 36.5 C, heart rate 97 bpm, blood pressure 119/83 mmHg, respiratory rate 22 breaths/min, and an oxygen saturation of 98% on room air. Initial blood analysis showed leukocytosis with neutrophilia (22.14 × 109/L and 18.73 × 109/L) and elevated inflammatory markers (ESR 60 mm/1 h, CRP 2.4 mg/dL). Blood gas analysis and biochemical analysis could not be initially achieved due to the high lactescent serum. Lipid profile was highly modified, with total lipids counting 3441 mg/dL with total cholesterol of 923 mg/dL and triglycerides level of 1274 mg/dL. Initial abdominal ultrasound identified a hyperechoic pancreas without any other pathological aspects. Thoraco-abdominal X-ray was normal.
The patient was admitted to the Pediatric Department of our hospital for management and further investigations.
His physical examination identified no xanthelasma or skin eruptions. The patient has a BMI of 20.1 (29 Kg, 118 cm), above 95th percentile according to CDC (Center for Disease Control and Prevention). Abdominal examination revealed a non-distended abdomen with mild tenderness and diffuse sensibility, accentuated in the periumbilical region. The patient was previously healthy without any medical or surgical history. The patient has a defective diet, with more than six meals every day, including fast-food and other high-processed foods and drinks. There was no history of medication, infection, recent surgeries, gastrointestinal endoscopic procedures, or abdominal trauma. There were no similar episodes previously. There was no family history of pancreatitis or gallstones. His mother, 32 years, suffers from migraines. Apart from that, she has no medical history and her lipidic profile is normal. His father, 38 years old, has mixt dyslipidemia. He has an 11-year-old brother without any significant medical history. His maternal grandfather has type 2 diabetes mellitus.
The complete blood analyses obtained after admission in the Pediatric Department also revealed a mild anemia (Hb 11.8 g/dL) and elevated pancreatic enzymes (amylase 76 U/L, lipase 69 U/L), that reached 153 U/l and, respectively 176 U/L within a week from admission. Liver enzymes including ALT, AST, gamma-GT, ALP, and total bilirubin levels were within referenced ranges. Random glucose level was 124 mg/dL. A fasting lipid panel was collected, and the most striking abnormality was a marked elevation of triglycerides (1359 mg/dL) and total cholesterol (538 mg mg/dL) with HDL- 35.6 mg/dL, LDL- 214.8 mg/dL, and VLDL- 54 mg/dL. Blood gas analysis identified a pH level of 7.56, PCO2- 24 mm Hg, PO2- 152 mmHg, and HCO3- 24.7 mmol/L. Serum electrolytes and coagulation tests were within normal ranges. An abdominal computed tomography was performed and was compatible with the diagnosis of acute pancreatitis involving the entire pancreas, with significant fat stranding, but without any systematized fluid collections. The other abdominal organs were within normal limits (Figure 1). Cardiovascular exam was unremarkable.
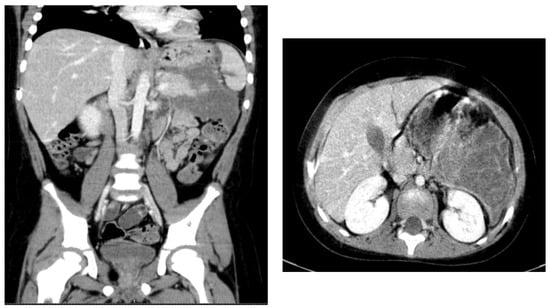
Figure 1.
Computed tomography aspect- pancreatic pseudocyst.
As the general condition of the patient continued to deteriorate under medical treatment and there was increasing patient distress, he was transferred into the Intensive Care Unit for supportive measures and management. Patient management included non-oral intake, intravenous rehydration, antibiotics, antiemetics, antacids, pain medication, statins, fibrates, and pancreatin supplement. Clinical and biologic status of the patient slowly improved, and he was readmitted to the Pediatric Department. Periodical abdominal ultrasonography identified the evolution of the pancreatic inflammation into a pancreatic pseudocyst supplementary characterized by computed tomography 13 days after admission as a large peripancreatic collection of 88/85/131 mm (T/AP/CC) (Figure 1). Endoscopic and laparoscopic drainage were attempted, but without satisfactory evacuation of the collection, a reintervention for external drainage of the pseudocyst being required with favorable evolution. The drainage was removed on postoperative day 10. Computed tomography reevaluation after 30 days showed the collection measuring 23/33/35 mm (T/AP/CC). Ultrasonographic reevaluation 60 days postoperatively did not identify any collection.
As the acute event was successfully treated, it was mandatory to focus on its cause to prevent recurrent episodes of pancreatitis.
Initial complete lipid profile identified important hypertriglyceridemia (1359 mg/dL) and hypercholesterolemia (538 mg mg/dL) with LDL predominance (214.8 mg/dL). Lipoprotein electrophoresis revealed hyper-β-lipoproteinemia and hypo-α-lipoproteinemia. Triglyceride levels decreased under diet and fibrate treatment and reached 214 mg/dL after 30 days. As these values were too elevated to be only secondary to the acute episode of pancreatitis, a primary dyslipidemia was considered. Sequence analysis and deletion/duplication testing using Next-Generation Sequencing technologies was performed on a panel of 36 genes involved in lipid metabolism (Table 1). DNA was extracted from peripheral blood. A gross deletion of the genomic region encompassing exon 1 of the LIPC gene, which includes the initial codon, was identified. The 5′ end of this event is unknown, as it extends beyond the assayed region for this gene and therefore may encompass additional genes. The 3′ boundary is likely confined to intron 1 of the LIPC gene. It is anticipated to produce a missing or disrupted protein product. However, the available clinical and genetic data is not adequate to demonstrate whether loss-of-function variants in LIPC cause disease. This variant was not previously reported in the literature in persons with LIPC-related disorders, and its significance is currently uncertain, but in the presented clinical and paraclinical context, it has the characteristics of a pathological variant inducing a hepatic lipase deficiency phenotype.

Table 1.
List of lipid metabolism involved genes analyzed for our patient.
3. Discussion
Primary disorders of lipid metabolism inducing hypertriglyceridemia derive from genetic errors in triglyceride production and metabolism. Triglyceride levels above 500 mg/dL are encountered in less than 0.2% of cases, but when detected, an immediate concern of a primary triglyceride metabolism disorder should be considered [1].
The global incidence of acute pancreatitis in pediatric population is ascending, having an estimated incidence of 1/10,000 children annually for pancreatitis determined by all causes [6]. In the pediatric population, etiologies of acute pancreatitis are varied, including genetic disorders, systemic conditions, infectious diseases, autoimmune disorders, metabolic causes, drugs and toxins, obstructive causes, and trauma [7]. The specific incidence of hypertriglyceridemia induced acute pancreatitis is currently unknown due to its rarity, being mostly presented as case reports [2]. Although the precise mechanism of pancreatitis is not currently established, it is thought that triglyceride-rich chylomicrons decrease circulatory stream in capillary beds of the pancreas leading to ischemia and generating an inflammatory response [8]. Pancreatitis usually appears when triglyceride levels are above 1000 to 1500 mg/dL, but triglyceride levels from 200 to 1000 mg/dL can be detected in the initial period of acute pancreatitis of all etiologies [2,9]. In our case, the patient had an initial triglyceride level of 1274 mg/dL.
Once the diagnosis is established and the acute episode treated, prevention of pancreatitis is essential as mortality caused by pancreatitis can reach 11% [10]. Prevention of pancreatitis of this etiology is based on triglyceride lowering below 500 mg/dL. In our case, we managed to obtain a triglyceride level of 214 mg/dL after 30 days of diet and fibrate treatment. Usually, recurrent episodes of pancreatitis appear because the compliance with a low-fat diet is challenging and pharmacologic therapy is not constantly efficient. Additionally, pediatric patients with constant moderate-to-high levels of triglyceride can be at high risk of early cardiovascular disease during maturity [1]. In our patient, the cardiovascular exam was normal, but he will be periodically reevaluated for the occurrence of any signs of cardiovascular disease.
We identified a primary disorder of the lipid metabolism as the cause of the acute episode of pancreatitis in our patient, consisting of a hepatic lipase deficiency caused by a novel genetic variant of the LIPC gene, a gross deletion of the genomic region encompassing exon 1. Hepatic lipase (HL) is an essential enzyme in the metabolism of triglyceride and phospholipids [11]. HL is a lipolytic serine hydrolase encrypt by the LIPC gene, produced and secreted by liver cells and attached to the liver sinusoidal area by heparin sulfate proteoglycans [12]. Establishment of the role of HL in human lipid metabolism has been simplified by the recognition of individuals with HL deficiency. HL is involved in the hydrolysis of triglycerides and phospholipids in remnants of triglyceride-rich lipoproteins that have a role in the transformation of VLDL remnants to LDL [13]. HL also promotes the incorporation of these particles, acting as their ligand in liver cells. Additionally, HL plays a role in the transformation of large buoyant high-density lipoprotein (HDL) to small HDL influencing the phospholipids load of these remnants [13]. HL is discharged from the surface of the hepatocytes into circulation by the intravenous injection of heparin and usually evaluated in humans as enzyme activity in post heparin plasma [14,15].
The LIPC gene is found on chromosome 15q21-q23 lengthening 35 kb and it consists of a total of nine exons and eight introns encoding a 476 amino acid glycoprotein lipolytic serine hydrolase [14,16]. The HL gene, or LIPC gene, is a member of the lipase gene family [17]. The three distinct lipases, lipoprotein lipase (LPL), hepatic lipase (HL), and pancreatic lipase (PL), have a considerable amount of similarities in their protein sequences, especially proximal to the three amino acids that form the catalytic triad involved in enzyme activity [18,19]. LIPC gene is expressed, even if to variable levels, in nearly all species [20]. Human HL has appreciable DNA and protein sequence homology with HL of rat and rabbit [21].
The proximal promoter of the LIPC gene consists of four polymorphic sites in complete linkage disequilibrium: G-250A, C-514T, T-710C, and A-763G, as established by the nomenclature of Amesis et al. There have been reported correlations between variations in plasma biochemical traits and variations in the promoter sequence of the LIPC gene [22]. Several variations of the LIPC gene have been reported (Table 2). Polymorphisms in the promoter sequence of LIPC gene can cause or not cause amino acid substitutions. Polymorphisms causing amino acid substitutions are V73M, N193S, S267F, T383M, L334F, and R186H. Polymorphisms that do not lead to amino acid change were reported in codons V133V, T202T, T457T, G175G, and T344T. Moreover, a C-toT substitution in nucleotide -480 in the promoter region of the LIPC gene and a mutation in intron 1 have been reported [5]. The polymorphism C-514T in the promoter region explains about 38% of the inconsistency of the HL activity [23]. There are also fewer common variants, and their frequency was reported to be between 0.15 to 0.21 among Caucasians, 0.45 to 0.53 among African Americans, and 0.47 among Japanese Americans [23,24].
Table 2.
LIPC gene variants [25].
Five missense mutations in encoding exons were reported and proved to determine HL deficiency to date: S267F, T383M, L334F, A174T, and R186H [3,26,27,28], Al [29].
The practical importance of these mutations was established by site directed mutagenesis and in vitro expression analysis. The S276F mutation seems to critically influence the activity and releasing of HL, while the T383M mutant variant maintains partial activity, but is inadequately secreted [30]. The L334F mutation determines almost normal production of a HL product that has only around 30% of the wild type protein function [27]. The R186H mutation determines arginine to histidine substitution in the mature protein producing an inactive HL protein [27]. The A174T mutation was associated with T383M mutation in three individuals of the Quebec family who had extremely low to undetectable HL activity [4].
The T383M (thr383-to-met) substitution in the mature enzyme caused by a 1221C-T transition in exon 8 of the LIPC gene was described by Hegele et al. in 1991 in six individuals with complete HL deficiency from two unrelated families, one of French descent living in Quebec and a second of Irish and English descent living in Ontario. Hegele et al. in 1991 identified in three affected individuals of the Ontario family another mutation in the LIPC gene, an 873C-T transition in exon 8 resulting in S267F (ser-267-to-phe) substitution. Ruel et al. identified in 2003 a second mutation in affected members of the Quebec family, a G-to-A transition in exon 5 resulting in A174T (ala174-to-thr) substitution in a highly conserved region of the mature product. Knudsen et al. described in 1996, in a Finnish man with HL deficiency, a heterozygous mutation in the LIPC gene: R186H and L334F substitution in exons 5 and 7 of the HL gene. In 2010 Al Riyami et al. identified homozygosity for the L334F (leu334-to-phr) mutation in the LIPC gene in an Arab patient born with consanguineous parents with HL deficiency; post heparin plasma HL activity in this patient was zero [3,26,27,28,29].
This extremely rare genetic disease, which seems to be transmitted in an autosomal recessive manner, was described only in five families to date [31,32,33,34,35]. HL deficiency is transmitted in an autosomal recessive manner; thereby, both copies of the gene in each cell have mutations. Both parents of a person with an autosomal recessive disorder are carriers of one copy of the mutated gene, but they usually do not present signs of the disease [36]. Its real prevalence might be significantly higher due to the struggle in diagnosis by conventional clinical and biochemical criteria [11]. In clinical practice, the usual lipid markers such as serum total LDL and HDL cholesterol and triglycerides do not reveal any specific aberrations. Therefore, detection of heterozygous carriers of HL mutations is incidental at population level [27]. In the case of heterozygous carriers, functional mutations can determine a clinical impact only if there are interactions between environment factors and other genes [27]. Affected persons that have a heterozygous state for HL mutations do not develop characteristic lipoprotein anomalies, and even individuals with complete HL deficiency show an inconstant phenotype [3]. Data from HL deficient patients indicate that secondary factors such as age, gender, and visceral obesity with insulin resistance can modify the phenotypic manifestations of HL mutations [3].
By influencing the phospholipid and triglyceride load of IDL, LDL, and HDL, HL plays and important role in establishing their lipid composition, density, size and by that their metabolic fate [3]. The most frequent consequences of HL deficiency on lipoprotein profiles have been triglyceride enrichment of LDL and HDL, presence of beta-VLDL and modified catabolism of chylomicrons [37]. This phenotype is similar to type III hyperlipoproteinemia [11]. Our patient presented with hypertriglyceridemia and hypercholesterolemia with LDL predominance. The limitations of our study include that we present a singular case and our report may therefore be taken with reservations when extrapolated to other patients and long term consequences of this variant in our patient are not available to report at the moment and can only be predicted based on other studies.
4. Conclusions
We report a pediatric case of acute pancreatitis caused by a previously unknown variant of the LIPC gene, a gross deletion of the genomic region encompassing exon 1 of the LIPC gene resulting in hypertriglyceridemia and hypercholesterolemia.
Author Contributions
Conceptualization, L.B. and R.B.; methodology, L.B.; software, A.C.; validation, L.B., A.M. and R.B.; formal analysis, A.C.; investigation, A.C. and G.S.; resources, A.C., C.M. and D.P.; data curation, A.C.; writing—original draft preparation, A.C.; writing—review and editing, L.B. and A.M.; visualization, L.B.; supervision, L.B.; project administration, L.B. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Data Availability Statement
Data is available at cardoneanu.anca@gmail.com if needed.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Shah, A.S.; Wilson, D.P. Primary hypertriglyceridemia in children and adolescents. J. Clin. Lipidol. 2015, 9, S20–S28. [Google Scholar] [CrossRef] [PubMed]
- Ippisch, H.M.; Alfaro-Cruz, L.; Fei, L.; Zou, Y.; Thompson, T.; Abu-El-Haija, M. Hypertriglyceridemia Induced Pancreatitis: Inpatient Management at a Single Pediatric Institution. Pancreas 2020, 49, 429–434. [Google Scholar] [CrossRef] [PubMed]
- Ruel, I.L.; Couture, P.; Gagné, C.; Deshaies, Y.; Simard, J.; Hegele, R.A.; Lamarche, B. Characterization of a novel mutation causing hepatic lipase deficiency among French Canadians. J. Lipid Res. 2003, 44, 1508–1514. [Google Scholar] [CrossRef] [PubMed]
- Ruel, I.L.; Couture, P.; Cohn, J.S.; Bensadoun, A.; Marcil, M.; Lamarche, B. Evidence that hepatic lipase deficiency in humans is not associated with proatherogenic changes in HDL composition and metabolism. J Lipid Res. 2004, 45, 1528–1537. [Google Scholar] [CrossRef]
- Murtomäki, S.; Tahvanainen, E.; Antikainen, M.; Tiret, L.; Nicaud, V.; Jansen, H.; Ehnholm, C. Hepatic lipase gene polymorphisms influence plasma HDL levels: Results from Finnish EARS participants. Arterioscler. Thromb. Vasc. Biol. 1997, 17, 1879–1884. [Google Scholar] [CrossRef]
- Morinville, V.D.; Barmada, M.M.; Lowe, M.E. Increasing incidence of acute pancreatitis at an American pediatric tertiary care center: Is greater awareness among physicians responsible? Pancreas 2010, 39, 5–8. [Google Scholar] [CrossRef]
- Majbar, A.A.; Cusick, E.; Johnson, P.; Lynn, R.M.; Hunt, L.P.; Shield, J.P.H. Incidence and Clinical Associations of Childhood Acute Pancreatitis. Pediatrics 2016, 138, e20161198. [Google Scholar] [CrossRef]
- Gilbert, B.; Rouis, M.; Griglio, S.; de Lumley, L.; Laplaud, P. Lipoprotein lipase (LPL) deficiency: A new patient homozygote for the preponderant mutation Gly188Glu in the human LPL gene and review of reported mutations: 75% are clustered in exons 5 and 6. Ann. Genet. 2001, 44, 25–32. [Google Scholar] [CrossRef]
- Scherer, J.; Singh, V.P.; Pitchumoni, C.S.; Yadav, D. Issues in hypertriglyceridemic pancreatitis: An update. J. Clin. Gastroenterol. 2014, 48, 195–203. [Google Scholar] [CrossRef]
- Werlin, S.L.; Kungathasan, S.; Frautschy, B.C. Pancreatitis in children. J. Pediatr. Gastroenterol. Nutr. 2003, 37, 591–595. [Google Scholar] [CrossRef]
- Huff, M.W.; Sawyez, C.G.; Connelly, P.W.; Maguire, G.F.; Little, J.A.; Hegele, R.A. β-VLDL in hepatic lipase deficiency induces apoE-mediated cholesterol ester accumulation in macrophages. Arterioscler. Thromb. 1993, 13, 1282–1290. [Google Scholar] [CrossRef]
- Oliva, C.P.; Pisciotta, L.; Volti, G.L.; Sambataro, M.P.; Cantafora, A.; Bellocchio, A.; Catapano, A.L.; Tarugi, P.; Bertolini, S.; Calandra, S. Inherited apolipoprotein A-V deficiency in severe hypertriglyceridemia. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 411–417. [Google Scholar] [CrossRef]
- Buonuomo, P.S.; Rabacchi, C.; Macchiaiolo, M.; Trenti, C.; Fasano, T.; Tarugi, P.; Bartuli, A.; Bertolini, S.; Calandra, S. Incidental finding of severe hypertriglyceridemia in children. Role of multiple rare variants in genes affecting plasma triglyceride. J. Clin. Lipidol. 2017, 11, 1329–1337.e3. [Google Scholar] [CrossRef]
- Connelly, P.W.; Ranganathan, S.; Maguire, G.F.; Lee, M.; Myher, J.J.; Kottke, B.A.; Kuksis, A.; Little, J.A. The β very low density lipoprotein present in hepatic lipase deficiency competitively inhibits low density lipoprotein binding fibroblasts and stimulates fibroblast acyl-Coa:cholesterol acyltransferase. J. Biol. Chem. 1988, 263, 14184–14188. [Google Scholar] [CrossRef]
- Huttunen, J.K.; Ehnholm, C.; Kekki, M.; Nikkilä, E.A. Post-heparin plasma lipoprotein lipase and hepatic lipase in normal subjects and in patients with hypertriglyceridaemia: Correlations to sex, age and various parameters of triglyceride metabolism. Clin. Sci. Mol. Med. 1976, 50, 249–260. [Google Scholar] [CrossRef]
- Perret, B.; Mabile, L.; Martinez, L.; Tercé, F.; Barbaras, R.; Collet, X. Hepatic lipase: Structure/function relationship, synthesis, and regulation. J. Lipid Res. 2002, 43, 1163–1169. [Google Scholar] [CrossRef]
- Pirim, D.; Bunker, C.H.; Hokanson, J.E.; Hamman, R.F.; Demirci, F.Y.; Kamboh, M.I. Hepatic lipase (LIPC) sequencing in individuals with extremely high and low high-density lipoprotein cholesterol levels. PLoS ONE 2020, 15, e0243919. [Google Scholar] [CrossRef]
- Hide, W.A.; Chan, L.; Li, W.H. Structure and evolution of the lipase superfamily. J. Lipid Res. 1992, 33, 167–178. [Google Scholar] [CrossRef]
- Sparkes, R.S.; Zollman, S.; Klisak, I.; Kirchgessner, T.G.; Komaromy, M.C.; Mohandas, T.; Schotz, M.C.; Lusis, A.J. Human genes involved in lipolysis of plasma lipoproteins: Mapping of loci for lipoprotein lipase to 8p22 and hepatic lipase to 15q21. Genomics 1987, 1, 138–144. [Google Scholar] [CrossRef]
- Jansen, H.; Verhoeven, A.J.M.; Sijbrands, E.J.G. Hepatic lipase: A pro- or anti-atherogenic protein? J. Lipid Res. 2002, 43, 1352–1362. [Google Scholar] [CrossRef]
- Datta, S.; Luo, C.C.; Li, W.H.; Vantuinen, P.; Ledbetter, D.H.; Brown, M.A.; Chen, S.H.; Liu, S.W.; Chan, L. Human hepatic lipase. Cloned cDNA sequence, restriction fragment length polymorphisms, chromosomal localization, and evolutionary relationships with lipoprotein lipase and pancreatic lipase. J. Biol. Chem. 1988, 263, 1107–1110. [Google Scholar] [CrossRef]
- Tahvanainen, E.; Syvänne, M.; Frick, M.H.; Murtomäki-Repo, S.; Antikainen, M.; Kesäniemi, Y.A.; Kauma, H.; Pasternak, A.; Taskinen, M.R.; Ehnholm, C. Association of variation in hepatic lipase activity with promoter variation in the hepatic lipase gene. J. Clin. Investig. 1998, 101, 956–960. [Google Scholar] [CrossRef] [PubMed]
- Deeb, S.S.; Zambon, A.; Carr, M.C.; Ayyobi, A.F.; Brunzell, J.D. Hepatic lipase and dyslipidemia: Interactions among genetic variants, obesity, gender, and diet. J. Lipid Res. 2003, 44, 1279–1286. [Google Scholar] [CrossRef] [PubMed]
- Vega, G.L.; Gao, J.; Bersot, T.P.; Mahley, R.W.; Verstraete, R.; Grundy, S.M.; White, A.; Cohen, J.C. The -514 polymorphism in the hepatic lipase gene (LIPC) does not influence androgen-mediated stimulation of hepatic lipase activity. J. Lipid Res. 1998, 39, 1520–1524. [Google Scholar] [CrossRef]
- Variations for Hepatic Lipase Deficiency. 2022. Available online: https://www.malacards.org/card/hepatic_lipase_deficiency?limit[ClinVarVariations]=58#sources (accessed on 7 December 2021).
- Hegele, R.A.; Tu, L.; Connelly, P.W. Human hepatic lipase mutations and polymorphisms. Hum. Mutat. 1992, 1, 320–324. [Google Scholar] [CrossRef] [PubMed]
- Knudsen, P.; Antikainen, M.; Ehnholm, S.; Uusi-Oukari, M.; Tenkanen, H.; Lahdenperä, S.; Kahri, J.; Tilly-Kiesi, M.; Bensadoun, A.; Taskinen, M.R.; et al. A compound heterozygote for hepatic lipase gene mutations Leu334 → Phe and Thr383 → Met: Correlation between hepatic lipase activity and phenotypic expression. J. Lipid Res. 1996, 37, 825–834. [Google Scholar] [CrossRef]
- Knudsen, P.; Antikainen, M.; Uusi-Oukari, M.; Ehnholm, S.; Lahdenperä, S.; Bensadoun, A.; Funke, H.; Wiebusch, H.; Ass-mann, G.; Taskinen, M.-R.; et al. Heterozygous hepatic lipase deficiency, due to two missense mutations R186H and L334F, in the HL gene. Atherosclerosis 1997, 128, 165–174. [Google Scholar] [CrossRef]
- Al Riyami, N.; Al-Ali, A.M.; Al-Sarraf, A.J.; Hill, J.; Sachs-Barrable, K.; Hegele, R.; Wasan, K.M.; Frohlich, J. Hepatic lipase deficiency in a Middle-Eastern-Arabic male. BMJ Case Rep. 2010, 2010, 2009–2011. [Google Scholar] [CrossRef]
- Durstenfeld, A.; Ben-zeev, O.; Reue, K.; Stahnke, G.; Doolittle, M.H. Molecular Characterization of Human Hepatic Lipase Deficiency. In vitro expression of two naturally occurring mutations. Arterioscler. Thromb. J. Vasc. Biol. 1994, 14, 381–385. [Google Scholar] [CrossRef][Green Version]
- Hegele, R.A. The molecular basis of hepatic lipase deficiency. CMAJ 1991, 145, 1277. [Google Scholar]
- Breckenridge, W.; Little, J.; Alaupovic, P.; Wang, C.; Kuksis, A.; Kakis, G.; Lindgren, F.; Gardiner, G. Lipoprotein abnormalities associated with a familial deficiency of hepatic lipase. Atherosclerosis 1982, 45, 161–179. [Google Scholar] [CrossRef]
- Carlson, L.A.; Holmquist, L.; Nilsson-Ehle, P. Deficiency of Hepatic Lipase Activity in Post-heparin Plasma in Familial Hyper-α-Triglyceridemia. Acta Med. Scand. 1986, 219, 435–447. [Google Scholar] [CrossRef]
- Auwerx, J.H.; Marzetta, C.A.; Hokanson, J.E.; Brunzell, J.D. Large buoyant LDL-like particles in hepatic lipase deficiency. Arteriosclerosis 1989, 9, 319–325. [Google Scholar] [CrossRef]
- Sheriff, D.S.; El Fakhri, M.; Ghwarsha, K. Libyan family with hypercholesterolemia and increased high-density lipoprotein cholesterol in plasma. Clin. Chem. 1994, 40, 2313–2316. [Google Scholar] [CrossRef]
- Kobayashi, J.; Miyashita, K.; Nakajima, K.; Mabuchi, H. Hepatic Lipase: A Comprehensive View of its Role on Plasma Lipid and Lipoprotein Metabolism. J. Atheroscler. Thromb. 2015, 22, 1001–1011. [Google Scholar] [CrossRef]
- Brand, K.; Dugi, K.A.; Brunzell, J.D.; Nevin, D.N.; Santamarina-Fojo, S. A novel A → G mutation in intron I of the hepatic lipase gene leads to alternative splicing resulting in enzyme deficiency. J. Lipid Res. 1996, 37, 1213–1223. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).