

Endocrine Disorders in Children with Brain Tumors: At Diagnosis, after Surgery, Radiotherapy and Chemotherapy

Abstract
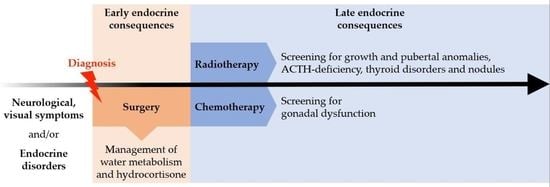
1. Introduction
- -
- Infratentorial tumors involving the cerebellum, brain stem, and fourth ventricle;
- -
- Supratentorial tumors involving the hypothalamus, thalamic nuclei, corpus callosum, basal ganglia, lateral ventricles, and cerebral hemispheres.
2. Brain Tumors and Endocrine Manifestations
- -
- Infratentorial tumors are divided into (1) brain stem tumors, (2) tumors of the posterior fossa and (3) spinal cord tumors;
- -
- Supratentorial tumors are divided into (4) supratentorial tumors (except diencephalic tumors) and (5) central tumors (of the diencephalon).
3. Peri and Post-Surgery Endocrine Complications and Their Management
3.1. Alterations of Water Homeostasis
- -
- Tablets—25–500 mcg/day in three doses;
- -
- Intranasal—2.5–20 mcg/day in one–three doses;
- -
- Sublingual—1–4 mcg/kg/day.
3.2. Central Hypocortisolism and Adrenal Crisis
3.3. Central Hypothyroidism
4. Late Endocrine Consequences according to Treatment Modalities
4.1. Post-Surgical Consequences
4.2. Post-Radiotherapy Consequences
4.3. Post-Chemotherapy Consequences
4.4. Consequences of New-Therapeutic Agents
5. Late Endocrine Consequences and Their Management
- ▪
- Clinical—height velocity, pubertal development;
- ▪
- Biochemical—free thyroxin (FT4), TSH, and morning cortisol;
- ▪
- Frequency—every six months in pre- and peri-pubertal patients;
- ▪
- Warning signs—decline in growth velocity, precocious or delayed pubertal development, general clinical signs, and symptoms or laboratory tests suggestive of hypothalamic–pituitary deficiency.
- ▪
- Clinical—general physical exam;
- ▪
- Biochemical—FT4, TSH, morning cortisol, estradiol, follicle-stimulating hormone (FSH), and luteinizing hormone (LH) in females, morning testosterone and LH in males;
- ▪
- Frequency—every 12 months in post-pubertal and adult patients;
- ▪
- Warning signs—general clinical signs and symptoms or laboratory tests suggestive of hypothalamic–pituitary deficiency.
5.1. Central and Peripheral Hypothyroidism
5.2. Thyroid Nodules and Secondary Differentiated Thyroid Carcinoma
- 1.
- Clinical screening—neck palpation (high specificity, but low sensitivity);
- Frequency—every 1–2 years;
- 2.
- Radiological screening—thyroid ultrasound (high specificity and sensitivity);
- Frequency—every 3–5 years.
5.3. GH Deficiency
5.4. Delayed Puberty and Disorders of Gonadal Function
5.5. Precocious Puberty
5.6. Central Diabetes Insipidus
5.7. Bone Fragility
5.8. Hypothalamic Obesity
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ostrom, Q.T.; Cioffi, G.; Gittleman, H.; Patil, N.; Waite, K.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2012–2016. Neuro-Oncology 2019, 21, v1–v100. [Google Scholar] [CrossRef] [PubMed]
- Linabery, A.; Ross, J.A. Trends in childhood cancer incidence in the U.S. (1992–2004). Cancer 2008, 112, 416–432. [Google Scholar] [CrossRef] [PubMed]
- Cooksey, R.; Wu, S.Y.; Klesse, L.; Oden, J.D.; Bland, R.E.; Hodges, J.C.; Gargan, L.; Vega, G.L.; Bowers, D. Metabolic syndrome is a sequela of radiation exposure in hypothalamic obesity among survivors of childhood brain tumors. J. Investig. Med. 2019, 67, 295–302. [Google Scholar] [CrossRef] [PubMed]
- Udaka, Y.T.; Packer, R.J. Pediatric Brain Tumors. Neurol. Clin. 2018, 36, 533–556. [Google Scholar] [CrossRef] [PubMed]
- Pollack, I.F.; Agnihotri, S.; Broniscer, A. Childhood brain tumors: Current management, biological insights, and future directions: JNSPG 75th Anniversary Invited Review Article. J. Neurosurg. Pediatr. 2019, 23, 261–273. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.P.; Fisher, J.L.; Nichols, E.; Abd-Allah, F.; Abdela, J.; Abdelalim, A.; Abraha, H.N.; Agius, D.; Alahdab, F.; Alam, T.; et al. Global, regional, and national burden of brain and other CNS cancer, 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2019, 18, 376–393. [Google Scholar] [CrossRef]
- Miller, K.D.; Ostrom, Q.T.; Kruchko, C.; Patil, N.; Tihan, T.; Cioffi, G.; Fuchs, H.E.; Waite, K.A.; Jemal, A.; Siegel, R.L.; et al. Brain and other central nervous system tumor statistics, 2021. CA A Cancer J. Clin. 2021, 71, 381–406. [Google Scholar] [CrossRef]
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO Classification of Tumors of the Central Nervous System: A summary. Neuro-Oncology 2021, 23, 1231–1251. [Google Scholar] [CrossRef]
- Malbari, F. Pediatric Neuro-Oncology. Neurol. Clin. 2021, 39, 829–845. [Google Scholar] [CrossRef]
- Pfister, S.M.; Reyes-Múgica, M.; Chan, J.K.; Hasle, H.; Lazar, A.J.; Rossi, S.; Ferrari, A.; Jarzembowski, J.A.; Pritchard-Jones, K.; Hill, D.A.; et al. A Summary of the Inaugural WHO Classification of Pediatric Tumors: Transitioning from the Optical into the Molecular Era. Cancer Discov. 2022, 12, 331–355. [Google Scholar] [CrossRef]
- Fangusaro, J. Pediatric High Grade Glioma: A Review and Update on Tumor Clinical Characteristics and Biology. Front. Oncol. 2012, 2, 105. [Google Scholar] [CrossRef] [PubMed]
- Fetcko, K.; Dey, M. Primary Central Nervous System Germ Cell Tumors: A Review and Update. Med. Res. Arch. 2018, 6, 1719. [Google Scholar] [CrossRef] [PubMed]
- Wilne, S.; Collier, J.; Kennedy, C.; Koller, K.; Grundy, R.; Walker, D. Presentation of childhood CNS tumours: A systematic review and meta-analysis. Lancet Oncol. 2007, 8, 685–695. [Google Scholar] [CrossRef]
- Müller, H.L. Diagnostics, treatment, and follow-up in craniopharyngioma. Front. Endocrinol. 2011, 2, 70. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Roberts, P.; Dhaliwal, S.; Della Rn, P. Transitioning adolescent and young adults with chronic disease and/or disabilities from paediatric to adult care services—An integrative review. J. Clin. Nurs. 2016, 25, 3113–3130. [Google Scholar] [CrossRef] [PubMed]
- Shanmugavadivel, D.; Liu, J.-F.; Murphy, L.; Wilne, S.; Walker, D. Accelerating diagnosis for childhood brain tumours: An analysis of the HeadSmart UK population data. Arch. Dis. Child. 2020, 105, 355–362. [Google Scholar] [CrossRef]
- HeadSmart: Be Brain Tumour Aware; Walker, D.; Wilne, S.; Grundy, R.; Kennedy, C.; Neil Dickson, A.; Lindsell, S.; Trusler, J.; Evans, A. A new clinical guideline from the Royal College of Paediatrics and Child Health with a national awareness campaign accelerates brain tumor diagnosis in UK children—“HeadSmart: Be Brain Tumour Aware”. Neuro-Oncology 2015, 18, 445–454. [Google Scholar] [CrossRef]
- Wilne, S.H.; Ferris, R.C.; Nathwani, A.; Kennedy, C.R. The presenting features of brain tumours: A review of 200 cases. Arch. Dis. Child. 2006, 91, 502–506. [Google Scholar] [CrossRef]
- Taylor, M.; Couto-Silva, A.-C.; Adan, L.; Trivin, C.; Sainte-Rose, C.; Zerah, M.; Valteau-Couanet, D.; Doz, F.; Chalumeau, M.; Brauner, R. Hypothalamic-Pituitary Lesions in Pediatric Patients: Endocrine Symptoms Often Precede Neuro-Ophthalmic Presenting Symptoms. J. Pediatr. 2012, 161, 855–863.e3. [Google Scholar] [CrossRef]
- Müller, H.L.; Tauber, M.; Lawson, E.A.; Özyurt, J.; Bison, B.; Martinez-Barbera, J.-P.; Puget, S.; Merchant, T.E.; van Santen, H.M. Hypothalamic syndrome. Nat. Rev. Dis. Prim. 2022, 8, 24. [Google Scholar] [CrossRef]
- van Roessel, I.M.A.A.; van Schaik, J.; Meeteren, A.Y.N.S.-V.; Boot, A.M.; der Grinten, H.L.C.-V.; Clement, S.C.; van Iersel, L.; Han, K.S.; van Trotsenburg, A.S.P.; Vandertop, W.P.; et al. Body mass index at diagnosis of a childhood brain tumor; a reflection of hypothalamic-pituitary dysfunction or lifestyle? Support. Care Cancer 2022, 30, 6093–6102. [Google Scholar] [CrossRef] [PubMed]
- van Schaik, J.; van Roessel, I.M.A.A.; Meeteren, N.A.Y.N.S.-V.; van Iersel, L.; Clement, S.C.; Boot, A.M.; der Grinten, H.L.C.-V.; Fiocco, M.; Janssens, G.O.; van Vuurden, D.G.; et al. High Prevalence of Weight Gain in Childhood Brain Tumor Survivors and Its Association With Hypothalamic-Pituitary Dysfunction. J. Clin. Oncol. 2021, 39, 1264–1273. [Google Scholar] [CrossRef] [PubMed]
- Maghnie, M.; Cosi, G.; Genovese, E.; Manca-Bitti, M.L.; Cohen, A.; Zecca, S.; Tinelli, C.; Gallucci, M.; Bernasconi, S.; Boscherini, B.; et al. Central Diabetes Insipidus in Children and Young Adults. N. Engl. J. Med. 2000, 343, 998–1007. [Google Scholar] [CrossRef]
- Grois, N.; Prosch, H.; Waldhauser, F.; Minkov, M.; Strasser, G.; Steiner, M.; Unger, E.; Prayer, D. Pineal gland abnormalities in Langerhans cell histiocytosis. Pediatr. Blood Cancer 2004, 43, 261–266. [Google Scholar] [CrossRef] [PubMed]
- Marchand, I.; Barkaoui, M.A.; Garel, C.; Polak, M.; Donadieu, J.; for the Writing Committee. Central Diabetes Insipidus as the Inaugural Manifestation of Langerhans Cell Histiocytosis: Natural History and Medical Evaluation of 26 Children and Adolescents. J. Clin. Endocrinol. Metab. 2011, 96, E1352–E1360. [Google Scholar] [CrossRef]
- Korkmaz, H.A.; Kapoor, R.R.; Kalitsi, J.; Aylwin, S.J.; Buchanan, C.R.; Arya, V.B. Central Diabetes Insipidus in Children and Adolescents: Twenty-Six Year Experience from a Single Centre. Int. J. Endocrinol. 2022, 2022, 9397130. [Google Scholar] [CrossRef]
- White, A.; de Andrade, E.J.; Kshettry, V.R.; Sindwani, R.; Recinos, P.F. Preoperative Workup for Patients with Pituitary Lesions. Otolaryngol. Clin. North Am. 2022, 55, 233–246. [Google Scholar] [CrossRef]
- Houdemont, S.P.; De Carli, E.; DeLion, M.; Ringuier, B.; Chapotte, C.; Jeudy, C.; Mercier, P.; Granry, J.-C.; Rialland, X. Short-term neurological outcome of children after surgery for brain tumors: Incidence and characteristics in a pediatric intensive care unit. Child’s Nerv. Syst. 2011, 27, 933–941. [Google Scholar] [CrossRef]
- Prete, A.; Corsello, S.M.; Salvatori, R. Current best practice in the management of patients after pituitary surgery. Ther. Adv. Endocrinol. Metab. 2017, 8, 33–48. [Google Scholar] [CrossRef]
- Bereket, A. Postoperative and Long-Term Endocrinologic Complications of Craniopharyngioma. Horm. Res. Paediatr. 2020, 93, 497–509. [Google Scholar] [CrossRef]
- Finken, M.J.; Zwaveling-Soonawala, N.; Walenkamp, M.J.; Vulsma, T.; van Trotsenburg, A.P.; Rotteveel, J. Frequent Occurrence of the Triphasic Response (Diabetes Insipidus/Hyponatremia/Diabetes Insipidus) after Surgery for Craniopharyngioma in Childhood. Horm. Res. Paediatr. 2011, 76, 22–26. [Google Scholar] [CrossRef] [PubMed]
- Mak, D.; Schaller, A.L.; Storgion, S.A.; Lahoti, A. Evaluating a standardized protocol for the management of diabetes insipidus in pediatric neurosurgical patients. J. Pediatr. Endocrinol. Metab. 2022, 35, 197–203. [Google Scholar] [CrossRef] [PubMed]
- Hannon, M.J.; Finucane, F.M.; Sherlock, M.; Agha, A.; Thompson, C.J. Disorders of Water Homeostasis in Neurosurgical Patients. J. Clin. Endocrinol. Metab. 2012, 97, 1423–1433. [Google Scholar] [CrossRef] [PubMed]
- Castle-Kirszbaum, M.; Kyi, M.; Wright, C.; Goldschlager, T.; Danks, R.A.; Parkin, W.G. Hyponatraemia and hypernatraemia: Disorders of Water Balance in Neurosurgery. Neurosurg. Rev. 2021, 44, 2433–2458. [Google Scholar] [CrossRef] [PubMed]
- Edate, S.; Albanese, A. Management of Electrolyte and Fluid Disorders after Brain Surgery for Pituitary/Suprasellar Tumours. Horm. Res. Paediatr. 2015, 83, 293–301. [Google Scholar] [CrossRef]
- Winzeler, B.; Zweifel, C.; Nigro, N.; Arici, B.; Bally, M.; Schuetz, P.; Blum, C.A.; Kelly, C.; Berkmann, S.; Huber, A.; et al. Postoperative Copeptin Concentration Predicts Diabetes Insipidus After Pituitary Surgery. J. Clin. Endocrinol. Metab. 2015, 100, 2275–2282. [Google Scholar] [CrossRef]
- Dabrowski, E.; Kadakia, R.; Zimmerman, D. Diabetes insipidus in infants and children. Best Pract. Res. Clin. Endocrinol. Metab. 2016, 30, 317–328. [Google Scholar] [CrossRef]
- Elder, C.J.; Dimitri, P.J. Diabetes insipidus and the use of desmopressin in hospitalised children. Arch. Dis. Child.-Educ. Pract. Ed. 2017, 102, 100–104. [Google Scholar] [CrossRef][Green Version]
- Driano, J.E.; Lteif, A.N.; Creo, A.L. Vasopressin-Dependent Disorders: What Is New in Children? Pediatrics 2021, 147, e2020022848. [Google Scholar] [CrossRef]
- Di Iorgi, N.; Morana, G.; Napoli, F.; Allegri, A.E.M.; Rossi, A.; Maghnie, M. Management of diabetes insipidus and adipsia in the child. Best Pract. Res. Clin. Endocrinol. Metab. 2015, 29, 415–436. [Google Scholar] [CrossRef]
- Williams, C.N.; Riva-Cambrin, J.; Bratton, S.L. Etiology of postoperative hyponatremia following pediatric intracranial tumor surgery. J. Neurosurg. Pediatr. 2016, 17, 303–309. [Google Scholar] [CrossRef] [PubMed]
- Oh, J.Y.; Shin, J.I. Syndrome of Inappropriate Antidiuretic Hormone Secretion and Cerebral/Renal Salt Wasting Syndrome: Similarities and Differences. Front. Pediatr. 2015, 2, 146. [Google Scholar] [CrossRef]
- Cuesta, M.; Thompson, C. The syndrome of inappropriate antidiuresis (SIAD). Best Pract. Res. Clin. Endocrinol. Metab. 2016, 30, 175–187. [Google Scholar] [CrossRef] [PubMed]
- Jameel, P.Z.; Lohiya, S.; Vagha, K.; Ahmed, T.; Pujari, D.; Vagha, J.; Varma, A. Concurrent central diabetes insipidus and cerebral salt wasting disease in a post-operative case of craniopharyngioma: A case report. BMC Pediatr. 2021, 21, 502. [Google Scholar] [CrossRef] [PubMed]
- Alghamdi, K.; Albakri, L.A.; Alotaibi, Y.; Alghamdi, A.H.; Alaidarous, S. Coexistence of Triphasic Diabetes Insipidus and Cerebral Salt Wasting Syndrome Following Extraction of Sellar/Suprasellar Grade I Pilocytic Astrocytoma. Cureus 2021, 13, e17661. [Google Scholar] [CrossRef]
- Bornstein, S.R.; Allolio, B.; Arlt, W.; Barthel, A.; Don-Wauchope, A.; Hammer, G.D.; Husebye, E.S.; Merke, D.P.; Murad, M.H.; Stratakis, C.A.; et al. Diagnosis and Treatment of Primary Adrenal Insufficiency: An Endocrine Society Clinical Practice Guideline. J. Clin. Endocrinol. Metab. 2016, 101, 364–389. [Google Scholar] [CrossRef]
- Husebye, E.S.; Pearce, S.H.; Krone, N.P.; Kämpe, O. Adrenal insufficiency. Lancet 2021, 397, 613–629. [Google Scholar] [CrossRef]
- Woodcock, T.; Barker, P.; Daniel, S.; Fletcher, S.; W.ass, J.A.H.; Tomlinson, J.W.; Misra, U.; Dattani, M.; Arlt, W.; Vercueil, A. Guidelines for the management of glucocorticoids during the peri-operative period for patients with adrenal insufficiency. Anaesthesia 2020, 75, 654–663. [Google Scholar] [CrossRef]
- Persani, L.; Brabant, G.; Dattani, M.; Bonomi, M.; Feldt-Rasmussen, U.; Fliers, E.; Gruters, A.; Maiter, D.; Schoenmakers, N.; van Trotsenburg, A.P. 2018 European Thyroid Association (ETA) Guidelines on the Diagnosis and Management of Central Hypothyroidism. Eur. Thyroid J. 2018, 7, 225–237. [Google Scholar] [CrossRef]
- Filges, I.; Bischof-Renner, A.; Röthlisberger, B.; Potthoff, C.; Glanzmann, R.; Günthard, J.; Schneider, J.; Huber, A.R.; Zumsteg, U.; Miny, P.; et al. Panhypopituitarism Presenting as Life-Threatening Heart Failure Caused by an Inherited Microdeletion in 1q25 Including LHX4. Pediatrics 2012, 129, e529–e534. [Google Scholar] [CrossRef]
- van Trotsenburg, P.; Stoupa, A.; Léger, J.; Rohrer, T.; Peters, C.; Fugazzola, L.; Cassio, A.; Heinrichs, C.; Beauloye, V.; Pohlenz, J.; et al. Congenital Hypothyroidism: A 2020–2021 Consensus Guidelines Update—An ENDO-European Reference Network Initiative Endorsed by the European Society for Pediatric Endocrinology and the European Society for Endocrinology. Thyroid 2021, 31, 387–419. [Google Scholar] [CrossRef] [PubMed]
- van Iersel, L.; Mulder, R.L.; Denzer, C.; Cohen, L.E.; Spoudeas, H.A.; Meacham, L.R.; Sugden, E.; Meeteren, A.Y.N.S.-V.; Hoving, E.W.; Packer, R.J.; et al. Hypothalamic-Pituitary and Other Endocrine Surveillance Among Childhood Cancer Survivors. Endocr. Rev. 2022, 43, 794–823. [Google Scholar] [CrossRef] [PubMed]
- Sklar, C.A.; Antal, Z.; Chemaitilly, W.; Cohen, L.E.; Follin, C.; Meacham, L.R.; Murad, M.H. Hypothalamic–Pituitary and Growth Disorders in Survivors of Childhood Cancer: An Endocrine Society* Clinical Practice Guideline. J. Clin. Endocrinol. Metab. 2018, 103, 2761–2784. [Google Scholar] [CrossRef] [PubMed]
- Clement, S.C.; Meeteren, A.Y.S.-V.; Boot, A.M.; der Grinten, H.L.C.-V.; Granzen, B.; Han, K.S.; Janssens, G.O.; Michiels, E.M.; van Trotsenburg, A.P.; Vandertop, W.P.; et al. Prevalence and Risk Factors of Early Endocrine Disorders in Childhood Brain Tumor Survivors: A Nationwide, Multicenter Study. J. Clin. Oncol. 2016, 34, 4362–4370. [Google Scholar] [CrossRef] [PubMed]
- Lebbink, C.A.; Ringers, T.P.; Meeteren, A.Y.N.S.-V.; van Iersel, L.; Clement, S.C.; Boot, A.M.; der Grinten, H.L.C.-V.; Janssens, G.O.; van Vuurden, D.G.; Michiels, E.M.; et al. Prevalence and risk factors of hypothalamic–pituitary dysfunction in infant and toddler childhood brain tumor survivors. Eur. J. Endocrinol. 2021, 185, 597–606. [Google Scholar] [CrossRef] [PubMed]
- Laughton, S.J.; Merchant, T.E.; Sklar, C.A.; Kun, L.E.; Fouladi, M.; Broniscer, A.; Morris, E.B.; Sanders, R.P.; Krasin, M.J.; Shelso, J.; et al. Endocrine Outcomes for Children With Embryonal Brain Tumors After Risk-Adapted Craniospinal and Conformal Primary-Site Irradiation and High-Dose Chemotherapy With Stem-Cell Rescue on the SJMB-96 Trial. J. Clin. Oncol. 2008, 26, 1112–1118. [Google Scholar] [CrossRef]
- Darzy, K.H. Radiation-induced hypopituitarism after cancer therapy: Who, how and when to test. Nat. Clin. Pract. Endocrinol. Metab. 2009, 5, 88–99. [Google Scholar] [CrossRef]
- Chemaitilly, W.; Li, Z.; Huang, S.; Ness, K.K.; Clark, K.L.; Green, D.M.; Barnes, N.; Armstrong, G.T.; Krasin, M.J.; Srivastava, D.K.; et al. Anterior Hypopituitarism in Adult Survivors of Childhood Cancers Treated With Cranial Radiotherapy: A Report From the St Jude Lifetime Cohort Study. J. Clin. Oncol. 2015, 33, 492–500. [Google Scholar] [CrossRef]
- DeNunzio, N.J.; Yock, T.I. Modern Radiotherapy for Pediatric Brain Tumors. Cancers 2020, 12, 1533. [Google Scholar] [CrossRef]
- Eaton, B.R.; Esiashvili, N.; Kim, S.; Patterson, B.; Weyman, E.A.; Thornton, L.T.; Mazewski, C.; MacDonald, T.J.; Ebb, D.; MacDonald, S.M.; et al. Endocrine outcomes with proton and photon radiotherapy for standard risk medulloblastoma. Neuro-Oncology 2016, 18, 881–887. [Google Scholar] [CrossRef]
- Aldrich, K.D.; Horne, V.E.; Bielamowicz, K.; Sonabend, R.Y.; Scheurer, M.E.; Paulino, A.C.; Mahajan, A.; Chintagumpala, M.; Okcu, M.F.; Brown, A.L. Comparison of hypothyroidism, growth hormone deficiency, and adrenal insufficiency following proton and photon radiotherapy in children with medulloblastoma. J. Neuro-Oncol. 2022, 155, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Vatner, R.E.; Niemierko, A.; Misra, M.; Weyman, E.A.; Goebel, C.P.; Ebb, D.H.; Jones, R.M.; Huang, M.S.; Mahajan, A.; Grosshans, D.R.; et al. Endocrine Deficiency As a Function of Radiation Dose to the Hypothalamus and Pituitary in Pediatric and Young Adult Patients With Brain Tumors. J. Clin. Oncol. 2018, 36, 2854–2862. [Google Scholar] [CrossRef] [PubMed]
- Briceño, L.G.G.; Kariyawasam, D.; Samara-Boustani, D.; Giani, E.; Beltrand, J.; Bolle, S.; Fresneau, B.; Puget, S.; Sainte-Rose, C.; Alapetite, C.; et al. High Prevalence of Early Endocrine Disorders After Childhood Brain Tumors in a Large Cohort. J. Clin. Endocrinol. Metab. 2022, 107, e2156–e2166. [Google Scholar] [CrossRef] [PubMed]
- Merchant, T.E.; Rose, S.R.; Bosley, C.; Wu, S.; Xiong, X.; Lustig, R.H. Growth Hormone Secretion After Conformal Radiation Therapy in Pediatric Patients With Localized Brain Tumors. J. Clin. Oncol. 2011, 29, 4776–4780. [Google Scholar] [CrossRef]
- Beckers, D.; Thomas, M.; Jamart, J.; Francois, I.; Maes, M.; Lebrethon, M.C.; De Waele, K.; Tenoutasse, S.; De Schepper, J. Adult final height after GH therapy for irradiation-induced GH deficiency in childhood survivors of brain tumors: The Belgian experience. Eur. J. Endocrinol. 2010, 162, 483–490. [Google Scholar] [CrossRef]
- Maciel, J.; Dias, D.; Cavaco, D.; Donato, S.; Pereira, M.C.; Simões-Pereira, J. Growth hormone deficiency and other endocrinopathies after childhood brain tumors: Results from a close follow-up in a cohort of 242 patients. J. Endocrinol. Investig. 2021, 44, 2367–2374. [Google Scholar] [CrossRef]
- Thorbinson, C.; Kilday, J.-P. Childhood Malignant Brain Tumors: Balancing the Bench and Bedside. Cancers 2021, 13, 6099. [Google Scholar] [CrossRef]
- Cochrane, A.M.; Cheung, C.; Rangan, K.; Freyer, D.; Nahata, L.; Dhall, G.; Finlay, J.L. Long-term follow-up of endocrine function among young children with newly diagnosed malignant central nervous system tumors treated with irradiation-avoiding regimens. Pediatr. Blood Cancer 2017, 64, e26616. [Google Scholar] [CrossRef]
- Rose, S.R.; Horne, V.E.; Howell, J.; Lawson, S.A.; Rutter, M.M.; Trotman, G.E.; Corathers, S.D. Late endocrine effects of childhood cancer. Nat. Rev. Endocrinol. 2016, 12, 319–336. [Google Scholar] [CrossRef]
- Hwang, E.I.; Sayour, E.J.; Flores, C.T.; Grant, G.; Wechsler-Reya, R.; Hoang-Minh, L.B.; Kieran, M.W.; Salcido, J.; Prins, R.M.; Figg, J.W.; et al. The current landscape of immunotherapy for pediatric brain tumors. Nat. Cancer 2022, 3, 11–24. [Google Scholar] [CrossRef]
- Robson, H.; Siebler, T.; Shalet, S.M.; Williams, G.R. Interactions between GH, IGF-I, Glucocorticoids, and Thyroid Hormones during Skeletal Growth. Pediatr. Res. 2002, 52, 137–147. [Google Scholar] [CrossRef] [PubMed]
- Lebbink, C.A.; Waguespack, S.G.; van Santen, H.M. Thyroid Dysfunction and Thyroid Cancer in Childhood Cancer Survivors: Prevalence, Surveillance and Management. Endocr. Metab. Late Eff. Cancer Surviv. 2021, 54, 140–153. [Google Scholar] [CrossRef]
- Bhatti, P.; Veiga, L.H.S.; Ronckers, C.M.; Sigurdson, A.J.; Stovall, M.; Smith, S.A.; Weathers, R.; Leisenring, W.; Mertens, A.C.; Hammond, S.; et al. Risk of Second Primary Thyroid Cancer after Radiotherapy for a Childhood Cancer in a Large Cohort Study: An Update from the Childhood Cancer Survivor Study. Radiat. Res. 2010, 174, 741–752. [Google Scholar] [CrossRef] [PubMed]
- van Santen, H.M.; Tytgat, G.A.; van de Wetering, M.D.; van Eck-Smit, B.L.; Hopman, S.M.; van der Steeg, A.F.; van Dijkum, E.J.N.; van Trotsenburg, A.P. Differentiated Thyroid Carcinoma After 131I-MIBG Treatment for Neuroblastoma During Childhood: Description of the First Two Cases. Thyroid Off. J. Am. Thyroid Assoc. 2012, 22, 643–646. [Google Scholar] [CrossRef] [PubMed]
- Sfeir, J.G.; Kittah, N.E.N.; Tamhane, S.; Jasim, S.; Chemaitilly, W.; Cohen, L.E.; Murad, M.H. Diagnosis of GH Deficiency as a Late Effect of Radiotherapy in Survivors of Childhood Cancers. J. Clin. Endocrinol. Metab. 2018, 103, 2785–2793. [Google Scholar] [CrossRef] [PubMed]
- Boguszewski, M.C.S.; Boguszewski, C.L.; Chemaililly, W.; Cohen, L.E.; Gebauer, J.; Higham, C.; Hoffman, A.R.; Polak, M.; Yuen, K.C.J.; Alos, N.; et al. Safety of growth hormone replacement in survivors of cancer and intracranial and pituitary tumours: A consensus statement. Eur. J. Endocrinol. 2022, 186, P35–P52. [Google Scholar] [CrossRef]
- Pollock, N.I.; Cohen, L.E. Growth Hormone Deficiency and Treatment in Childhood Cancer Survivors. Front. Endocrinol. 2021, 12, 745932. [Google Scholar] [CrossRef]
- Thomas-Teinturier, C.; Oliver-Petit, I.; Pacquement, H.; Fresneau, B.; Allodji, R.; Veres, C.; Bolle, S.; Berchery, D.; Demoor-Goldschmidt, C.; Haddy, N.; et al. Influence of growth hormone therapy on the occurrence of a second neoplasm in survivors of childhood cancer. Eur. J. Endocrinol. 2020, 183, 471–480. [Google Scholar] [CrossRef]
- Wang, Z.-F.; Chen, H.-L. Growth hormone treatment and risk of recurrence or development of secondary neoplasms in survivors of pediatric brain tumors. J. Clin. Neurosci. 2014, 21, 2155–2159. [Google Scholar] [CrossRef]
- Chemaitilly, W.; Liu, Q.; Van Iersel, L.; Ness, K.K.; Li, Z.; Wilson, C.L.; Brinkman, T.M.; Klosky, J.L.; Barnes, N.; Clark, K.L.; et al. Leydig Cell Function in Male Survivors of Childhood Cancer: A Report From the St Jude Lifetime Cohort Study. J. Clin. Oncol. 2019, 37, 3018–3031. [Google Scholar] [CrossRef]
- Metzger, M.L.; Meacham, L.R.; Patterson, B.; Casillas, J.S.; Constine, L.S.; Hijiya, N.; Kenney, L.B.; Leonard, M.; Lockart, B.A.; Likes, W.; et al. Female Reproductive Health After Childhood, Adolescent, and Young Adult Cancers: Guidelines for the Assessment and Management of Female Reproductive Complications. J. Clin. Oncol. 2013, 31, 1239–1247. [Google Scholar] [CrossRef] [PubMed]
- Lew, R. Natural history of ovarian function including assessment of ovarian reserve and premature ovarian failure. Best Pract. Res. Clin. Obstet. Gynaecol. 2019, 55, 2–13. [Google Scholar] [CrossRef] [PubMed]
- Anderson, R.A.; Mitchell, R.T.; Kelsey, T.W.; Spears, N.; Telfer, E.E.; Wallace, W.H.B. Cancer treatment and gonadal function: Experimental and established strategies for fertility preservation in children and young adults. Lancet Diabetes Endocrinol. 2015, 3, 556–567. [Google Scholar] [CrossRef]
- Van Dorp, W.; Mulder, R.L.; Kremer, L.C.; Hudson, M.M.; van den Heuvel-Eibrink, M.M.; van Dulmen-den Berg, M.H.; Levine, J.M.; Broeder, E.V.D.-D.; Di Iorgi, N.; Albanese, A.; et al. Recommendations for Premature Ovarian Insufficiency Surveillance for Female Survivors of Childhood, Adolescent, and Young Adult Cancer: A Report From the International Late Effects of Childhood Cancer Guideline Harmonization Group in Collaboration With the PanCareSurFup Consortium. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2016, 34, 3440–3450. [Google Scholar] [CrossRef]
- Grinspon, R.P.; Ropelato, M.G.; Gottlieb, S.; Keselman, A.; Martínez, A.; Ballerini, M.G.; Domené, H.M.; Rey, R.A. Basal Follicle-Stimulating Hormone and Peak Gonadotropin Levels after Gonadotropin-Releasing Hormone Infusion Show High Diagnostic Accuracy in Boys with Suspicion of Hypogonadotropic Hypogonadism. J. Clin. Endocrinol. Metab. 2010, 95, 2811–2818. [Google Scholar] [CrossRef]
- Resende, E.; Lara, B.H.J.; Reis, J.D.; Ferreira, B.P.; Pereira, G.A.; Borges, M.F. Assessment of Basal and Gonadotropin-Releasing Hormone-Stimulated Gonadotropins by Immunochemiluminometric and Immunofluorometric Assays in Normal Children. J. Clin. Endocrinol. Metab. 2007, 92, 1424–1429. [Google Scholar] [CrossRef]
- Norjavaara, E.; Ankarberg-Lindgren, C.; Kriström, B. Sex Steroid Replacement Therapy in Female Hypogonadism from Childhood to Young Adulthood. Endocr. Dev. 2016, 29, 198–213. [Google Scholar] [CrossRef]
- Dunkel, L.; Quinton, R. Transition in endocrinology: Induction of puberty. Eur. J. Endocrinol. 2014, 170, R229–R239. [Google Scholar] [CrossRef]
- Palmert, M.R.; Dunkel, L. Delayed Puberty. N. Engl. J. Med. 2012, 366, 443–453. [Google Scholar] [CrossRef]
- Nordenström, A.; Ahmed, S.F.; Akker, E.V.D.; Blair, J.; Bonomi, M.; Brachet, C.; Broersen, L.H.A.; der Grinten, H.L.C.-V.; Dessens, A.B.; Gawlik, A.; et al. Pubertal induction and transition to adult sex hormone replacement in patients with congenital pituitary or gonadal reproductive hormone deficiency: An Endo-ERN clinical practice guideline. Eur. J. Endocrinol. 2022, 186, G9–G49. [Google Scholar] [CrossRef]
- Oberfield, S.E.; Soranno, D.; Nirenberg, A.; Heller, G.; Allen, J.C.; David, R.; Levine, L.S.; Sklar, C.A. Age at Onset of Puberty Following High-Dose Central Nervous System Radiation Therapy. Arch. Pediatr. Adolesc. Med. 1996, 150, 589–592. [Google Scholar] [CrossRef] [PubMed]
- Chemaitilly, W.; Merchant, T.E.; Li, Z.; Barnes, N.; Armstrong, G.T.; Ness, K.K.; Pui, C.-H.; Kun, L.E.; Robison, L.L.; Hudson, M.M.; et al. Central precocious puberty following the diagnosis and treatment of paediatric cancer and central nervous system tumours: Presentation and long-term outcomes. Clin. Endocrinol. 2016, 84, 361–371. [Google Scholar] [CrossRef] [PubMed]
- Gan, H.-W.; Phipps, K.; Aquilina, K.; Gaze, M.N.; Hayward, R.; Spoudeas, H.A. Neuroendocrine Morbidity After Pediatric Optic Gliomas: A Longitudinal Analysis of 166 Children Over 30 Years. J. Clin. Endocrinol. Metab. 2015, 100, 3787–3799. [Google Scholar] [CrossRef] [PubMed]
- Gleeson, H.K.; Shalet, S.M. The impact of cancer therapy on the endocrine system in survivors of childhood brain tumours. Endocr.-Relat. Cancer 2004, 11, 589–602. [Google Scholar] [CrossRef]
- Ward, L.M. Part I: Which Child with a Chronic Disease Needs Bone Health Monitoring? Curr. Osteoporos. Rep. 2021, 19, 278–288. [Google Scholar] [CrossRef]
- De Vos, M.; Devroey, P.; Fauser, B.C. Primary ovarian insufficiency. Lancet 2010, 376, 911–921. [Google Scholar] [CrossRef]
- Ward, L.M. Part 2: When Should Bisphosphonates Be Used in Children with Chronic Illness Osteoporosis? Curr. Osteoporos. Rep. 2021, 19, 289–297. [Google Scholar] [CrossRef]
- Gurney, J.G.; Kaste, S.C.; Liu, W.; Srivastava, D.K.; Chemaitilly, W.; Ness, K.K.; Lanctot, J.Q.; Ojha, R.; Nottage, K.A.; Wilson, C.L.; et al. Bone mineral density among long-term survivors of childhood acute lymphoblastic leukemia: Results from the St. Jude Lifetime Cohort Study. Pediatr. Blood Cancer 2014, 61, 1270–1276. [Google Scholar] [CrossRef]
- Kvammen, J.A.; Stensvold, E.; Godang, K.; Bollerslev, J.; Myklebust, T.; Brandal, P.; Henriksen, C.; Bechensteen, A.G. Bone mineral density and nutrition in long-term survivors of childhood brain tumors. Clin. Nutr. ESPEN 2022, 50, 162–169. [Google Scholar] [CrossRef]
- Schündeln, M.M.; Fritzemeier, S.; Goretzki, S.C.; Hauffa, P.K.; Munteanu, M.; Kiewert, C.; Hauffa, B.P.; Fleischhack, G.; Tippelt, S.; Grasemann, C. Prevalence of osteopathologies in a single center cohort of survivors of childhood primary brain tumor. Front. Pediatr. 2022, 10, 913343. [Google Scholar] [CrossRef]
- Van Iersel, L.; Brokke, K.E.; Adan, R.A.H.; Bulthuis, L.C.M.; van den Akker, E.L.T.; Van Santen, H.M. Pathophysiology and Individualized Treatment of Hypothalamic Obesity Following Craniopharyngioma and Other Suprasellar Tumors: A Systematic Review. Endocr. Rev. 2019, 40, 193–235. [Google Scholar] [CrossRef] [PubMed]
- Elowe-Gruau, E.; Beltrand, J.; Brauner, R.; Pinto, G.; Samara-Boustani, D.; Thalassinos, C.; Busiah, K.; Laborde, K.; Boddaert, N.; Zerah, M.; et al. Childhood Craniopharyngioma: Hypothalamus-Sparing Surgery Decreases the Risk of Obesity. J. Clin. Endocrinol. Metab. 2013, 98, 2376–2382. [Google Scholar] [CrossRef] [PubMed]

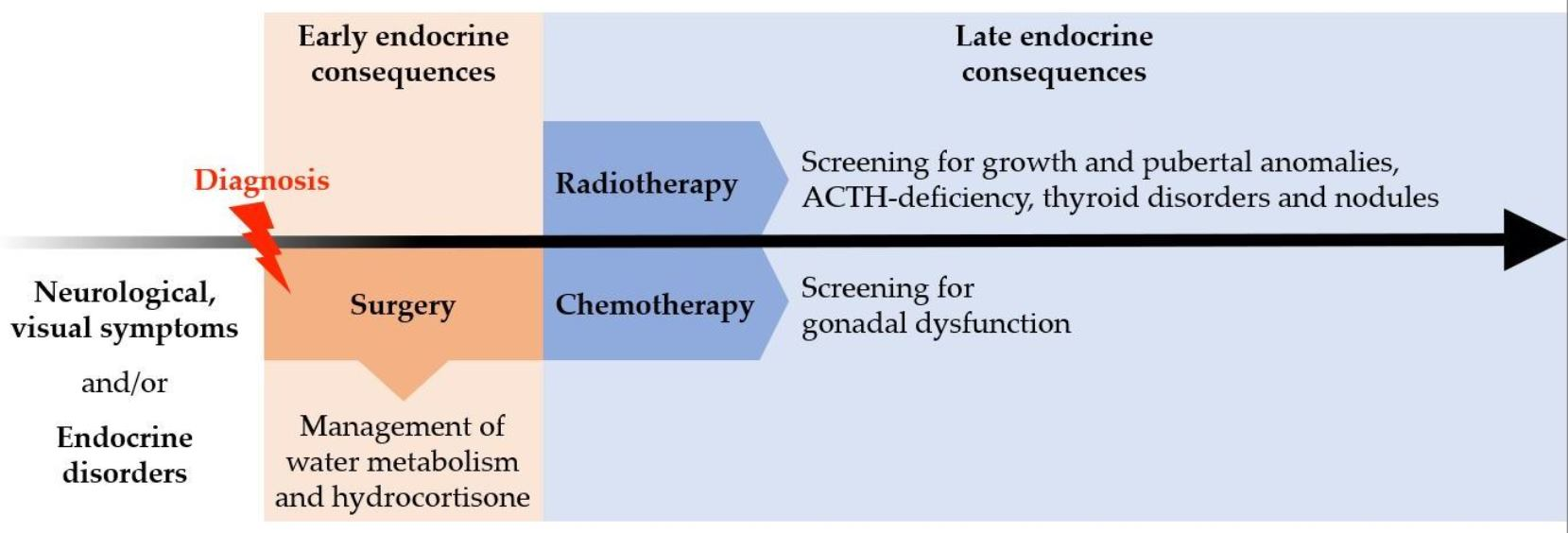{kind=link}
| Age | ||||
|---|---|---|---|---|
| <5 Years | 5–12 Years | 12–18 Years | ||
| Neurological and visual symptoms | Persistent or recurrent vomiting | |||
| Balance, co-ordination, walking problems | ||||
| Abnormal eye movements | ||||
| Suspected loss of vision | ||||
| Blurred or double vision, loss of vision | ||||
| Behavior change | ||||
| Seizures or fits (without fever) | ||||
| Abnormal head position | ||||
| Reduced consciousness | ||||
| Fastly increasing head circumference | ||||
| Persistent or recurrent headache | ||||
| Endocrine symptoms | Diabetes insipidus | |||
| Abnormal growth | ||||
| Early pubertya,b | ||||
| Delayed or suspended puberty b | ||||
Seek for specific medical evaluation if the following are true:
| Seek for emergency evaluation if the following are true:
| |||
| Diagnosis | Clinical Features | Therapy | |||
|---|---|---|---|---|---|
| Fluid Status | Diuresis | Vital Parameters | |||
| Hypernatremia | Diabetes insipidus | Euvolemic | Polyuria | If thirst mechanism intact and/or normal ability to drink: HR normal, BP normal | Free access to oral fluid |
| Hypovolemic | Polyuria | If patient adipsic or not able to drink: HR high, BP low | Fluid replacement, desmopressin | ||
| Hyponatremia | Syndrome of inappropriate ADH secretion | Euvolemic | Oliguria | HR normal, BP normal | Fluid restriction |
| Cerebral salt wasting | Hypovolemic | Polyuria | HR high, BP low | Fluid and sodium replacement | |
| Adrenal crisis | Hypovolemic | Polyuria | HR high, BP low | Hydrocortisone, and fluid and sodium replacement | |
| Therapy | |
|---|---|
| Girls | Estradiol transdermal patches [87]:
|
| Boys | Testosterone esters intramuscular:
|
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Claude, F.; Ubertini, G.; Szinnai, G. Endocrine Disorders in Children with Brain Tumors: At Diagnosis, after Surgery, Radiotherapy and Chemotherapy. Children 2022, 9, 1617. https://doi.org/10.3390/children9111617
Claude F, Ubertini G, Szinnai G. Endocrine Disorders in Children with Brain Tumors: At Diagnosis, after Surgery, Radiotherapy and Chemotherapy. Children. 2022; 9(11):1617. https://doi.org/10.3390/children9111617
Chicago/Turabian StyleClaude, Fabien, Graziamaria Ubertini, and Gabor Szinnai. 2022. "Endocrine Disorders in Children with Brain Tumors: At Diagnosis, after Surgery, Radiotherapy and Chemotherapy" Children 9, no. 11: 1617. https://doi.org/10.3390/children9111617
APA StyleClaude, F., Ubertini, G., & Szinnai, G. (2022). Endocrine Disorders in Children with Brain Tumors: At Diagnosis, after Surgery, Radiotherapy and Chemotherapy. Children, 9(11), 1617. https://doi.org/10.3390/children9111617