
Two Approaches for a Genetic Analysis of Pompe Disease: A Literature Review of Patients with Pompe Disease and Analysis Based on Genomic Data from the General Population


Abstract
1. Introduction
2. Materials and Methods
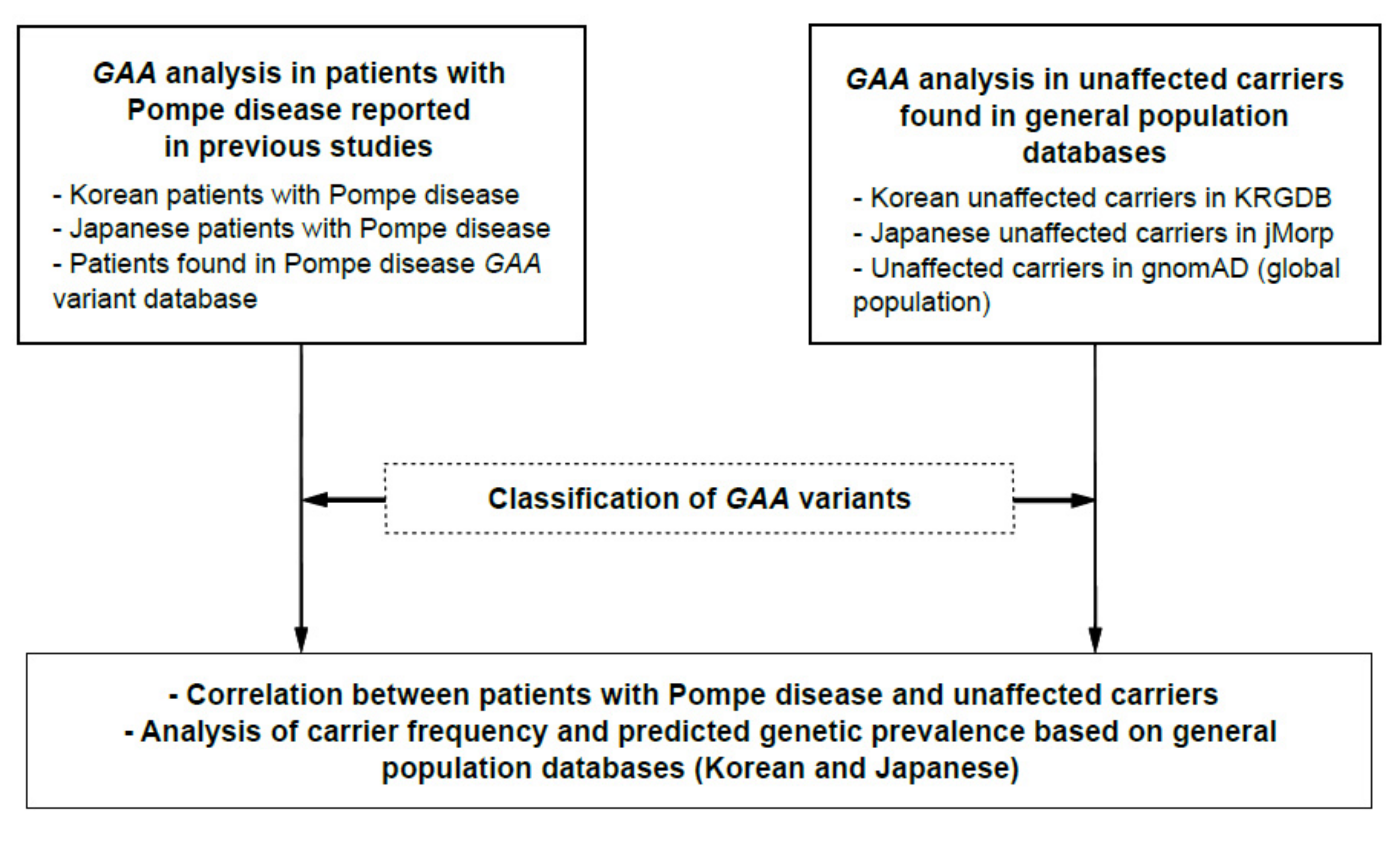
2.1. Analysis Workflow
2.2. GAA Variant Classification
2.3. Analysis of Carrier Frequency and Predicted Genetic Prevalence
3. Results
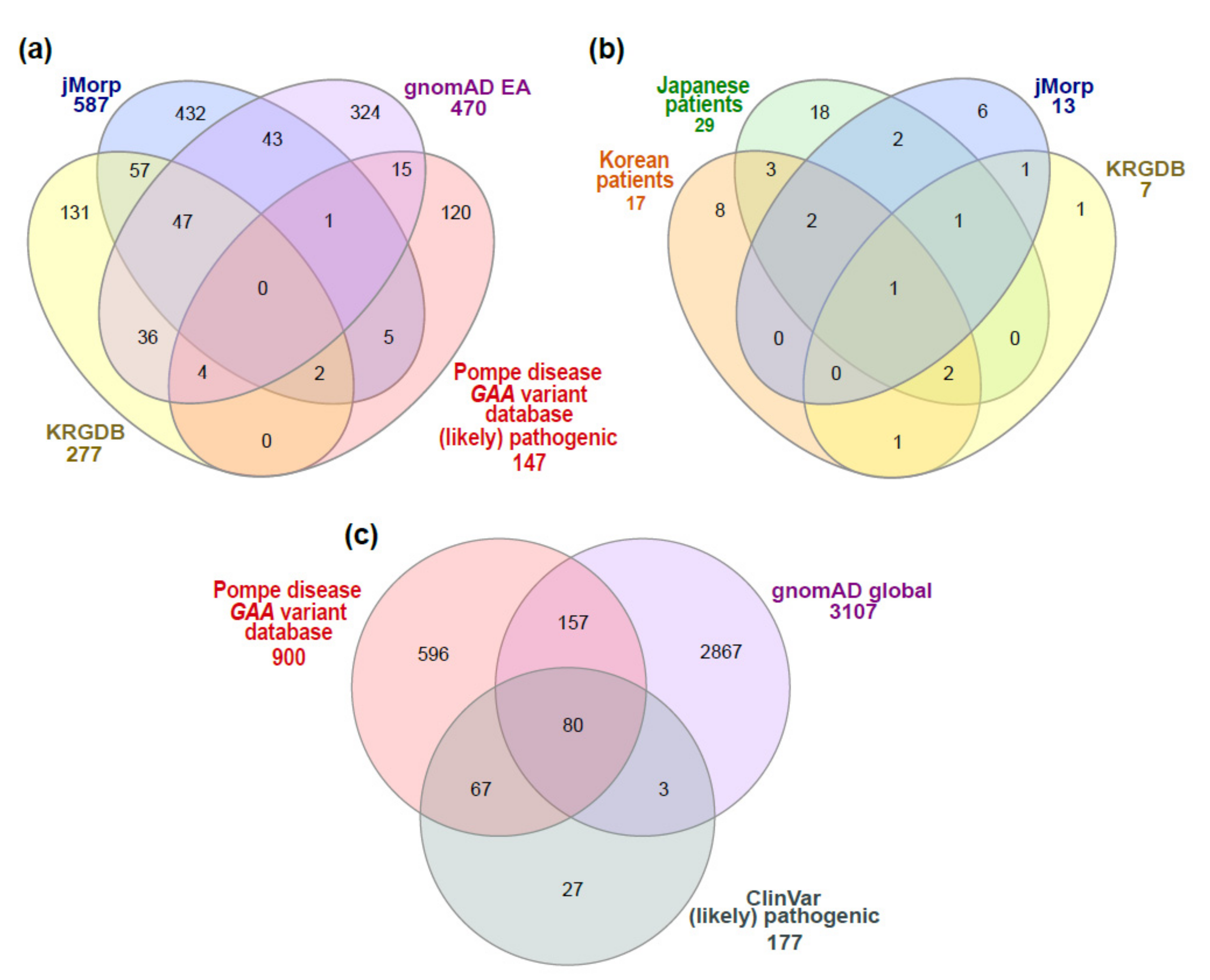
3.1. GAA Variants Found in Patients with Pompe Disease or General Population Databases
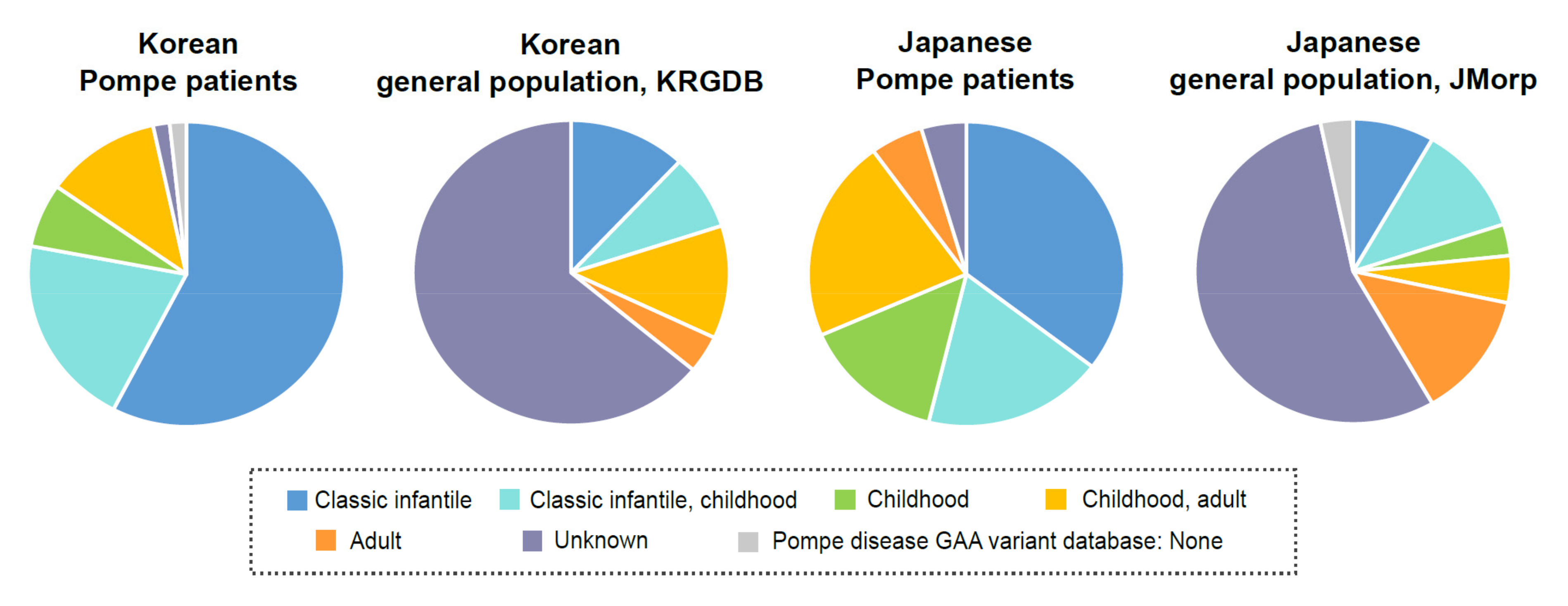
3.2. Correlation between Patients with Pompe Disease and Unaffected Carriers
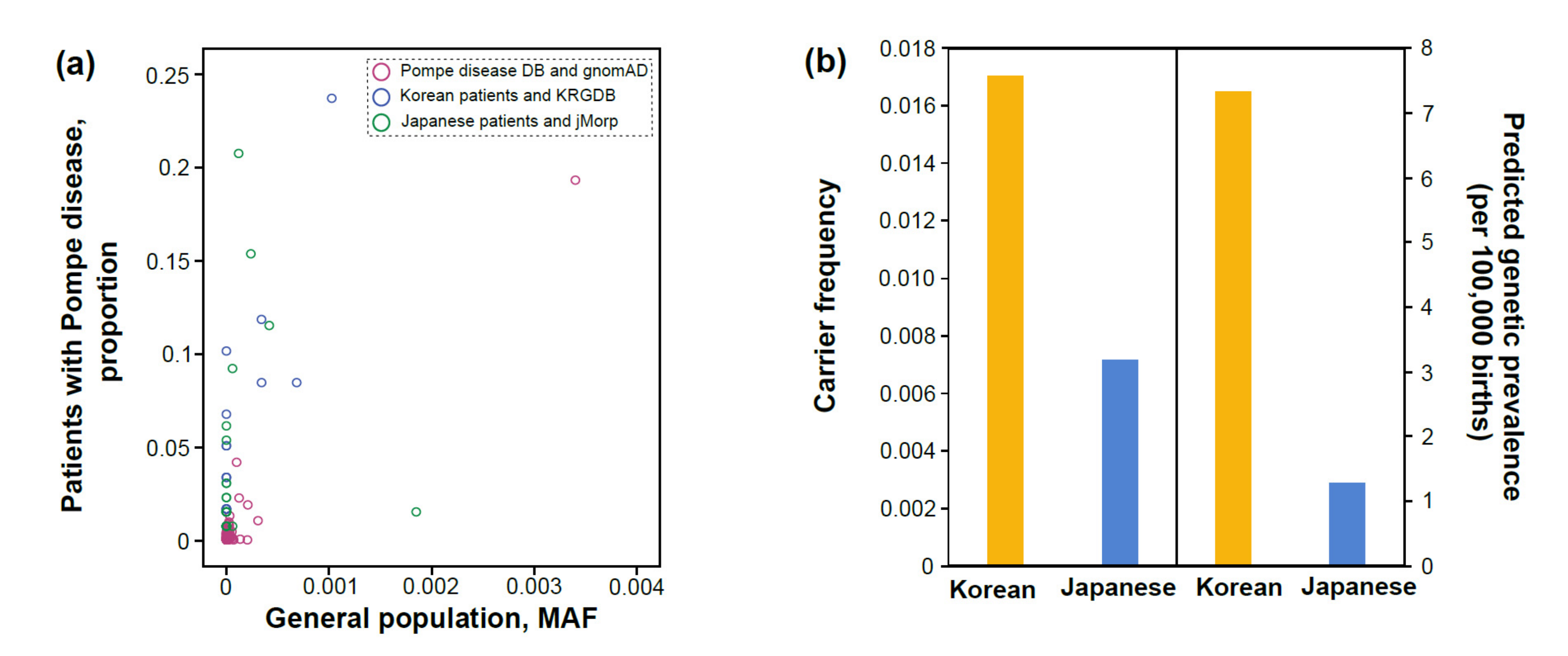
3.3. Carrier Frequency and Predicted Genetic Prevalence Based on General Population Databases
4. Discussion
5. Conclusions
Supplementary Materials
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- van der Ploeg, A.T.; Reuser, A.J. Pompe’s disease. Lancet 2008, 372, 1342–1353. [Google Scholar] [CrossRef]
- Peruzzo, P.; Pavan, E.; Dardis, A. Molecular genetics of Pompe disease: A comprehensive overview. Ann. Transl. Med. 2019, 7, 278. [Google Scholar] [CrossRef]
- Gungor, D.; Reuser, A.J. How to describe the clinical spectrum in Pompe disease? Am. J. Med. Genet. A 2013, 161A, 399–400. [Google Scholar] [CrossRef] [PubMed]
- Bodamer, O.A.; Scott, C.R.; Giugliani, R.; Pompe Disease Newborn Screening Working, G. Newborn Screening for Pompe Disease. Pediatrics 2017, 140, S4–S13. [Google Scholar] [CrossRef]
- Jung, K.S.; Hong, K.W.; Jo, H.Y.; Choi, J.; Ban, H.J.; Cho, S.B.; Chung, M. KRGDB: The large-scale variant database of 1722 Koreans based on whole genome sequencing. Database 2020, 2020. [Google Scholar] [CrossRef] [PubMed]
- Tadaka, S.; Saigusa, D.; Motoike, I.N.; Inoue, J.; Aoki, Y.; Shirota, M.; Koshiba, S.; Yamamoto, M.; Kinoshita, K. jMorp: Japanese Multi Omics Reference Panel. Nucleic Acids Res. 2018, 46, D551–D557. [Google Scholar] [CrossRef]
- Tadaka, S.; Hishinuma, E.; Komaki, S.; Motoike, I.N.; Kawashima, J.; Saigusa, D.; Inoue, J.; Takayama, J.; Okamura, Y.; Aoki, Y.; et al. jMorp updates in 2020: Large enhancement of multi-omics data resources on the general Japanese population. Nucleic Acids Res. 2021, 49, D536–D544. [Google Scholar] [CrossRef]
- Park, K.S. Carrier frequency and predicted genetic prevalence of Pompe disease based on a general population database. Mol. Genet. Metab. Rep. 2021, 27, 100734. [Google Scholar] [CrossRef]
- Nino, M.Y.; In ‘t Groen, S.L.M.; Bergsma, A.J.; van der Beek, N.; Kroos, M.; Hoogeveen-Westerveld, M.; van der Ploeg, A.T.; Pijnappel, W. Extension of the Pompe mutation database by linking disease-associated variants to clinical severity. Hum. Mutat. 2019, 40, 1954–1967. [Google Scholar] [CrossRef] [PubMed]
- Heberle, H.; Meirelles, G.V.; da Silva, F.R.; Telles, G.P.; Minghim, R. InteractiVenn: A web-based tool for the analysis of sets through Venn diagrams. BMC Bioinform. 2015, 16, 169. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Ioannidis, N.M.; Rothstein, J.H.; Pejaver, V.; Middha, S.; McDonnell, S.K.; Baheti, S.; Musolf, A.; Li, Q.; Holzinger, E.; Karyadi, D.; et al. REVEL: An Ensemble Method for Predicting the Pathogenicity of Rare Missense Variants. Am. J. Hum. Genet. 2016, 99, 877–885. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, R.; Oak, N.; Plon, S.E. Evaluation of in silico algorithms for use with ACMG/AMP clinical variant interpretation guidelines. Genome Biol. 2017, 18, 225. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, J.M.; Cooper, D.N.; Schuelke, M.; Seelow, D. MutationTaster2: Mutation prediction for the deep-sequencing age. Nat. Methods 2014, 11, 361–362. [Google Scholar] [CrossRef] [PubMed]
- Yeo, G.; Burge, C.B. Maximum entropy modeling of short sequence motifs with applications to RNA splicing signals. J. Comput. Biol. 2004, 11, 377–394. [Google Scholar] [CrossRef] [PubMed]
- Jaganathan, K.; Kyriazopoulou Panagiotopoulou, S.; McRae, J.F.; Darbandi, S.F.; Knowles, D.; Li, Y.I.; Kosmicki, J.A.; Arbelaez, J.; Cui, W.; Schwartz, G.B.; et al. Predicting Splicing from Primary Sequence with Deep Learning. Cell 2019, 176, 535–548.e524. [Google Scholar] [CrossRef] [PubMed]
- Hanany, M.; Rivolta, C.; Sharon, D. Worldwide carrier frequency and genetic prevalence of autosomal recessive inherited retinal diseases. Proc. Natl. Acad. Sci. USA 2020, 117, 2710–2716. [Google Scholar] [CrossRef]
- Cho, A.; Kim, S.J.; Lim, B.C.; Hwang, H.; Park, J.D.; Kim, G.B.; Jin, D.K.; Lee, J.; Ki, C.S.; Kim, K.J.; et al. Infantile Pompe disease: Clinical and genetic characteristics with an experience of enzyme replacement therapy. J. Child Neurol. 2012, 27, 319–324. [Google Scholar] [CrossRef]
- Kim, M.S.; Song, A.; Im, M.; Huh, J.; Kang, I.S.; Song, J.; Yang, A.; Kim, J.; Kwon, E.K.; Choi, E.J.; et al. Clinical and molecular characterization of Korean children with infantile and late-onset Pompe disease: 10 years of experience with enzyme replacement therapy at a single center. Korean J. Pediatr. 2019, 62, 224–234. [Google Scholar] [CrossRef]
- Ko, J.M.; Park, K.S.; Kang, Y.; Nam, S.H.; Kim, Y.; Park, I.; Chae, H.W.; Lee, S.M.; Lee, K.A.; Kim, J.W. A New Integrated Newborn Screening Workflow Can Provide a Shortcut to Differential Diagnosis and Confirmation of Inherited Metabolic Diseases. Yonsei Med. J. 2018, 59, 652–661. [Google Scholar] [CrossRef]
- Lee, D.H.; Qiu, W.J.; Lee, J.; Chien, Y.H.; Hwu, W.L. Hypertrophic cardiomyopathy in pompe disease is not limited to the classic infantile-onset phenotype. JIMD Rep. 2014, 17, 71–75. [Google Scholar] [CrossRef]
- Lee, J.H.; Shin, J.H.; Park, H.J.; Kim, S.Z.; Jeon, Y.M.; Kim, H.K.; Kim, D.S.; Choi, Y.C. Targeted population screening of late onset Pompe disease in unspecified myopathy patients for Korean population. Neuromuscul. Disord. 2017, 27, 550–556. [Google Scholar] [CrossRef]
- Park, H.D.; Lee, D.H.; Choi, T.Y.; Lee, Y.K.; Lee, S.Y.; Kim, J.W.; Ki, C.S.; Lee, Y.W. Three patients with glycogen storage disease type II and the mutational spectrum of GAA in Korean patients. Ann. Clin. Lab. Sci. 2013, 43, 311–316. [Google Scholar] [PubMed]
- Park, H.J.; Jang, H.; Kim, J.H.; Lee, J.H.; Shin, H.Y.; Kim, S.M.; Park, K.D.; Yim, S.V.; Lee, J.H.; Choi, Y.C. Discovery of pathogenic variants in a large Korean cohort of inherited muscular disorders. Clin. Genet. 2017, 91, 403–410. [Google Scholar] [CrossRef]
- Park, J.S.; Kim, H.G.; Shin, J.H.; Choi, Y.C.; Kim, D.S. Effect of enzyme replacement therapy in late onset Pompe disease: Open pilot study of 48 weeks follow-up. Neurol. Sci. 2015, 36, 599–605. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.E.; Park, K.H.; Lee, C.H.; Kim, C.M.; Kim, D.S. Two new missense mutations of GAA in late onset glycogen storage disease type II. J. Neurol. Sci. 2006, 251, 113–117. [Google Scholar] [CrossRef]
- Kim, E.H.; Ko, J.M.; Lee, B.H.; Kim, G.H.; Choi, J.H.; Yoo, H.W. Two patients with atypical infantile Pompe disesase presenting with hypertrophic cardiomyopathy. J. Genet. Med. 2009, 6, 161–165. [Google Scholar]
- Fukuhara, Y.; Fuji, N.; Yamazaki, N.; Hirakiyama, A.; Kamioka, T.; Seo, J.H.; Mashima, R.; Kosuga, M.; Okuyama, T. A molecular analysis of the GAA gene and clinical spectrum in 38 patients with Pompe disease in Japan. Mol. Genet. Metab. Rep. 2018, 14, 3–9. [Google Scholar] [CrossRef]
- Hermans, M.M.; van Leenen, D.; Kroos, M.A.; Beesley, C.E.; Van Der Ploeg, A.T.; Sakuraba, H.; Wevers, R.; Kleijer, W.; Michelakakis, H.; Kirk, E.P.; et al. Twenty-two novel mutations in the lysosomal alpha-glucosidase gene (GAA) underscore the genotype-phenotype correlation in glycogen storage disease type II. Hum. Mutat. 2004, 23, 47–56. [Google Scholar] [CrossRef] [PubMed]
- Matsuoka, T.; Miwa, Y.; Tajika, M.; Sawada, M.; Fujimaki, K.; Soga, T.; Tomita, H.; Uemura, S.; Nishino, I.; Fukuda, T.; et al. Divergent clinical outcomes of alpha-glucosidase enzyme replacement therapy in two siblings with infantile-onset Pompe disease treated in the symptomatic or pre-symptomatic state. Mol. Genet. Metab. Rep. 2016, 9, 98–105. [Google Scholar] [CrossRef]
- Pipo, J.R.; Feng, J.H.; Yamamoto, T.; Ohsaki, Y.; Nanba, E.; Tsujino, S.; Sakuragawa, N.; Martiniuk, F.; Ninomiya, H.; Oka, A.; et al. New GAA mutations in Japanese patients with GSDII (Pompe disease). Pediatr. Neurol. 2003, 29, 284–287. [Google Scholar] [CrossRef]
- Tsujino, S.; Huie, M.; Kanazawa, N.; Sugie, H.; Goto, Y.; Kawai, M.; Nonaka, I.; Hirschhorn, R.; Sakuragawa, N. Frequent mutations in Japanese patients with acid maltase deficiency. Neuromuscul. Disord. 2000, 10, 599–603. [Google Scholar] [CrossRef]
- Nabatame, S.; Taniike, M.; Sakai, N.; Kato-Nishimura, K.; Mohri, I.; Kagitani-Shimono, K.; Okinaga, T.; Tachibana, N.; Ozono, K. Sleep disordered breathing in childhood-onset acid maltase deficiency. Brain Dev. 2009, 31, 234–239. [Google Scholar] [CrossRef] [PubMed]
- Tsuburaya, R.S.; Monma, K.; Oya, Y.; Nakayama, T.; Fukuda, T.; Sugie, H.; Hayashi, Y.K.; Nonaka, I.; Nishino, I. Acid phosphatase-positive globular inclusions is a good diagnostic marker for two patients with adult-onset Pompe disease lacking disease specific pathology. Neuromuscul. Disord. 2012, 22, 389–393. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, H.; Shimada, Y.; Ikegami, M.; Kawai, T.; Sakurai, K.; Urashima, T.; Ijima, M.; Fujiwara, M.; Kaneshiro, E.; Ohashi, T.; et al. Prognostic factors for the late onset Pompe disease with enzyme replacement therapy: From our experience of 4 cases including an autopsy case. Mol. Genet. Metab. 2010, 100, 14–19. [Google Scholar] [CrossRef] [PubMed]
- Maimaiti, M.; Takahashi, S.; Okajima, K.; Suzuki, N.; Ohinata, J.; Araki, A.; Tanaka, H.; Mukai, T.; Fujieda, K. Silent exonic mutation in the acid-alpha-glycosidase gene that causes glycogen storage disease type II by affecting mRNA splicing. J. Hum. Genet. 2009, 54, 493–496. [Google Scholar] [CrossRef] [PubMed]
- Hossain, M.A.; Miyajima, T.; Akiyama, K.; Eto, Y. A Case of Adult-onset Pompe Disease with Cerebral Stroke and Left Ventricular Hypertrophy. J. Stroke Cerebrovasc. Dis. 2018, 27, 3046–3052. [Google Scholar] [CrossRef]
- Fujimoto, S.; Manabe, Y.; Fujii, D.; Kozai, Y.; Matsuzono, K.; Takahashi, Y.; Narai, H.; Omori, N.; Adachi, K.; Nanba, E.; et al. A novel mutation of the GAA gene in a patient with adult-onset Pompe disease lacking a disease-specific pathology. Intern. Med. 2013, 52, 2461–2464. [Google Scholar] [CrossRef] [PubMed]
- Isayama, R.; Shiga, K.; Seo, K.; Azuma, Y.; Araki, Y.; Hamano, A.; Takezawa, H.; Kuriyama, N.; Takezawa, N.; Mizuno, T.; et al. Sixty six-month follow-up of muscle power and respiratory function in a case with adult-type Pompe disease treated with enzyme replacement therapy. J. Clin. Neuromuscul. Dis. 2014, 15, 152–156. [Google Scholar] [CrossRef]
- Ishigaki, K.; Yoshikawa, Y.; Kuwatsuru, R.; Oda, E.; Murakami, T.; Sato, T.; Saito, T.; Umezu, R.; Osawa, M. High-density CT of muscle and liver may allow early diagnosis of childhood-onset Pompe disease. Brain Dev. 2012, 34, 103–106. [Google Scholar] [CrossRef]
- Tsunoda, H.; Ohshima, T.; Tohyama, J.; Sasaki, M.; Sakuragawa, N.; Martiniuk, F. Acid alpha-glucosidase deficiency: Identification and expression of a missense mutation (S529V) in a Japanese adult phenotype. Hum. Genet. 1996, 97, 496–499. [Google Scholar] [CrossRef] [PubMed]
- Ishigaki, K.; Murakami, T.; Nakanishi, T.; Oda, E.; Sato, T.; Osawa, M. Close monitoring of initial enzyme replacement therapy in a patient with childhood-onset Pompe disease. Brain Dev. 2012, 34, 98–102. [Google Scholar] [CrossRef]
- Huie, M.L.; Tsujino, S.; Sklower Brooks, S.; Engel, A.; Elias, E.; Bonthron, D.T.; Bessley, C.; Shanske, S.; DiMauro, S.; Goto, Y.I.; et al. Glycogen storage disease type II: Identification of four novel missense mutations (D645N, G648S, R672W, R672Q) and two insertions/deletions in the acid alpha-glucosidase locus of patients of differing phenotype. Biochem. Biophys. Res. Commun. 1998, 244, 921–927. [Google Scholar] [CrossRef]
- Muraoka, T.; Murao, K.; Imachi, H.; Kikuchi, F.; Yoshimoto, T.; Iwama, H.; Hosokawa, H.; Nishino, I.; Fukuda, T.; Sugie, H.; et al. Novel mutations in the gene encoding acid alpha-1,4-glucosidase in a patient with late-onset glycogen storage disease type II (Pompe disease) with impaired intelligence. Intern. Med. 2011, 50, 2987–2991. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Chiang, S.C.; Hwu, W.L.; Lee, N.C.; Hsu, L.W.; Chien, Y.H. Algorithm for Pompe disease newborn screening: Results from the Taiwan screening program. Mol. Genet. Metab. 2012, 106, 281–286. [Google Scholar] [CrossRef] [PubMed]
- Momosaki, K.; Kido, J.; Yoshida, S.; Sugawara, K.; Miyamoto, T.; Inoue, T.; Okumiya, T.; Matsumoto, S.; Endo, F.; Hirose, S.; et al. Newborn screening for Pompe disease in Japan: Report and literature review of mutations in the GAA gene in Japanese and Asian patients. J. Hum. Genet. 2019, 64, 741–755. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variant | Korean Patients | Japanese Patients | General Population Databases, MAF | |||||
|---|---|---|---|---|---|---|---|---|
| Allele Count | [Ref] (HT/HM) | Allele Count | [Ref] (HT/HM) | KRGDB 1 | jMorp 2 | gnomAD 3, East Asian | gnomAD 3, Global | |
| c.-32-13T>G | 0 | 0 | 0.000342 | 0 | 0.000207 | 0.003401 | ||
| c.2T>C | 0 | 1 | [28] (1/0) | 0 | 0 | 0 | 0 | |
| c.118C>T (p.Arg40 *) | 1 | [20] (1/0) | 1 | [28] (1/0) | 0 | 0 | 0.000050 | 0.000014 |
| c.169C>T (p.Gln57 *) | 0 | 1 | [28] (1/0) | 0 | 0 | 0 | 0 | |
| c.307T>C (p.Cys103Arg) | 0 | 0 | 0 | 0.000060 | 0 | 0 | ||
| c.309C>A (p.Cys103 *) | 0 | 2 | [29] (0/1) | 0 | 0 | 0 | 0 | |
| c.483dup (p.Lys162Glnfs*15) | 0 | 2 | [30] (2/0) | 0 | 0 | 0 | 0 | |
| c.546G>A (p.Thr182=) | 0 | 0 | 0 | 0.000179 | 0.000102 | 0.000030 | ||
| c.546G>T (p.Thr182=) | 2 | [22] (1/0), [25] (1/0) | 27 | [28] (6/5), [34] (0/2), [35] (2/1), [36] (1/0), [37] (0/1) | 0 | 0.000119 | 0 | 0 |
| c.547-1G>C | 0 | 1 | [28] (1/0) | 0 | 0 | 0 | 0 | |
| c.569G>A (p.Arg190His) | 0 | 1 | [28] (1/0) | 0 | 0.000060 | 0 | 0.000016 | |
| c.655G>A (p.Gly219Arg) | 0 | 1 | [28] (1/0) | 0 | 0 | 0 | 0.000018 | |
| c.670C>T (p.Arg224Trp) | 0 | 1 | [31] (1/0) | 0 | 0 | 0 | 0.000022 | |
| (c.752C>T; c.761C>T) ((p.Ser251Leu; p.Ser254Leu) | 0 | 2 | [28] (1/0) | 0.005476 | 0.001850 | 0.002759 | 0.000195 | |
| c.756_757insT (p.Pro253Serfs*77) | 0 | 1 | [28] (1/0) | 0 | 0 | 0 | 0 | |
| c.796C>T (p.Pro266Ser) | 1 | [26] (1/0) | 2 | [28] (2/0) | 0 | 0 | 0 | 0 |
| c.841C>T (p.Arg281Trp) | 0 | 0 | 0 | 0.000060 | 0 | 0.000205 | ||
| c.875A>G (p.Tyr292Cys) | 4 | [18] (1/0), [19] (1/0), [21] (1/0), [23] (1/0) | 0 | 0 | 0 | 0 | 0.000008 | |
| c.1156C>T (p.Gln386 *) | 1 | [23] (1/0) | 0 | 0 | 0 | 0 | 0 | |
| c.1225dup (p.Asp409Glyfs*97) | 1 | [20] (1/0) | 0 | 0 | 0 | 0 | 0 | |
| c.1309C>T (p.Arg437Cys) | 3 | [18] (1/0), [19] (1/0), [25] (1/0) | 12 | [28] (6/0), [33] (2/0), [38] (1/0), [39] (0/1), [40] (1/0) | 0 | 0.000060 | 0 | 0.000008 |
| c.1316T>A (p.Met439Lys) | 14 | [18] (4/0), [19] (1/0), [20] (1/0), [22] (1/0), [23] (1/0), [24] (1/0), [25] (3/0), [26] (2/0) | 3 | [28] (0/1), [35] (1/0) | 0.001027 | 0 | 0.000384 | 0.000028 |
| c.1322_1326+9del | 2 | [18] (1/0), [19] (1/0) | 0 | 0 | 0 | 0 | 0 | |
| c.1447G>A (p.Gly483Arg) | 0 | 0 | 0 | 0.000060 | 0 | 0.000008 | ||
| c.1579_1580del (p.Arg527Glyfs*3) | 2 | [19] (1/0), [20] (1/0) | 0 | 0 | 0 | 0 | 0.000004 | |
| c.1582_1583del (p.Gly528Leufs*2) | 1 | [18] (1/0) | 0 | 0 | 0 | 0 | 0 | |
| c.1585_1586delinsGT (p.Ser529Val) | 0 | 7 | [32] (2/2), [41] (1/0) | 0 | 0 | 0 | 0 | |
| c.1696T>C (p.Ser566Pro) | 0 | 4 | [28] (2/0), [30] (2/0) | 0 | 0 | 0 | 0 | |
| c.1735G>A (p.Glu579Lys) | 0 | 2 | [28] (1/0), [42] (1/0) | 0 | 0 | 0 | 0.000007 | |
| c.1798C>T (p.Arg600Cys) | 0 | 20 | [28] (7/0), [31] (1/0), [32] (7/1), [35] (2/0), [36] (1/0) | 0 | 0.000239 | 0 | 0.000004 | |
| c.1822C>T (p.Arg608 *) | 6 | [18] (2/0), [19] (2/0), [22] (1/0), [24] (1/0) | 4 | [28] (1/1), [35] (1/0) | 0 | 0 | 0.000051 | 0.000018 |
| c.1826dup (p.Tyr609 *) | 0 | 1 | [28] (1/0) | 0 | 0 | 0 | 0.000008 | |
| c.1857C>G (p.Ser619Arg) | 7 | [20] (1/0), [21] (1/0), [23] (2/0), [24] (1/0), [25] (1/0), [26] (1/0) | 15 | [28] (4/3), [31] (0/1), [33] (0/1), [42] (1/0) | 0.000342 | 0.000418 | 0 | 0 |
| c.1935C>A (p.Asp645Glu) | 0 | 3 | [32] (1/1) | 0 | 0 | 0.001729 | 0.000124 | |
| c.1979G>A (p.Arg660His) | 0 | 2 | [31] (2/0) | 0 | 0 | 0 | 0.000037 | |
| c.2014C>T (p.Arg672Trp) | 0 | 0 | 0 | 0.000060 | 0 | 0.000008 | ||
| c.2015G>A (p.Arg672Gln) | 5 | [20] (1/0), [24] (2/0), [25] (1/0), [26] (1/0) | 8 | [32] (2/2), [43] (0/1) | 0.000343 | 0 | 0.000111 | 0.000021 |
| c.2171C>A (p.Ala724Asp) | 3 | [18] (1/0), [19] (1/0), [25] (1/0) | 0 | 0 | 0 | 0 | 0 | |
| c.2177C>G (p.Pro726Arg) | 0 | 2 | [28] (1/0), [40] (1/0) | 0 | 0 | 0 | 0 | |
| c.2297A>G (p.Tyr766Cys) | 0 | 1 | [28] (1/0) | 0 | 0 | 0 | 0.000025 | |
| c.2238G>C (p.Trp746Cys) | 5 | [19] (1/0), [22] (1/0), [24] (1/0), [25] (2/0) | 0 | 0.000685 | 0 | 0.000351 | 0.000308 | |
| c.2238G>T (p.Trp746Cys) | 0 | 0 | 0 | 0.000119 | 0 | 0 | ||
| c.2326C>T (p.Gln776 *) | 0 | 2 | [33] (2/0) | 0 | 0 | 0 | 0 | |
| c.2407_2413del (p.Gln803 *) | 1 | [23] (1/0) | 0 | 0 | 0 | 0 | 0 | |
| c.2481+1G>A | 0 | 1 | [28] (1/0) | 0 | 0 | 0 | 0 | |
| c.2647-7G>A | 0 | 0 | 0.000342 | 0.000298 | 0 | 0.000018 | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, K.-S. Two Approaches for a Genetic Analysis of Pompe Disease: A Literature Review of Patients with Pompe Disease and Analysis Based on Genomic Data from the General Population. Children 2021, 8, 601. https://doi.org/10.3390/children8070601
Park K-S. Two Approaches for a Genetic Analysis of Pompe Disease: A Literature Review of Patients with Pompe Disease and Analysis Based on Genomic Data from the General Population. Children. 2021; 8(7):601. https://doi.org/10.3390/children8070601
Chicago/Turabian StylePark, Kyung-Sun. 2021. "Two Approaches for a Genetic Analysis of Pompe Disease: A Literature Review of Patients with Pompe Disease and Analysis Based on Genomic Data from the General Population" Children 8, no. 7: 601. https://doi.org/10.3390/children8070601
APA StylePark, K.-S. (2021). Two Approaches for a Genetic Analysis of Pompe Disease: A Literature Review of Patients with Pompe Disease and Analysis Based on Genomic Data from the General Population. Children, 8(7), 601. https://doi.org/10.3390/children8070601