
Epilepsy in Mitochondrial Diseases—Current State of Knowledge on Aetiology and Treatment


Abstract
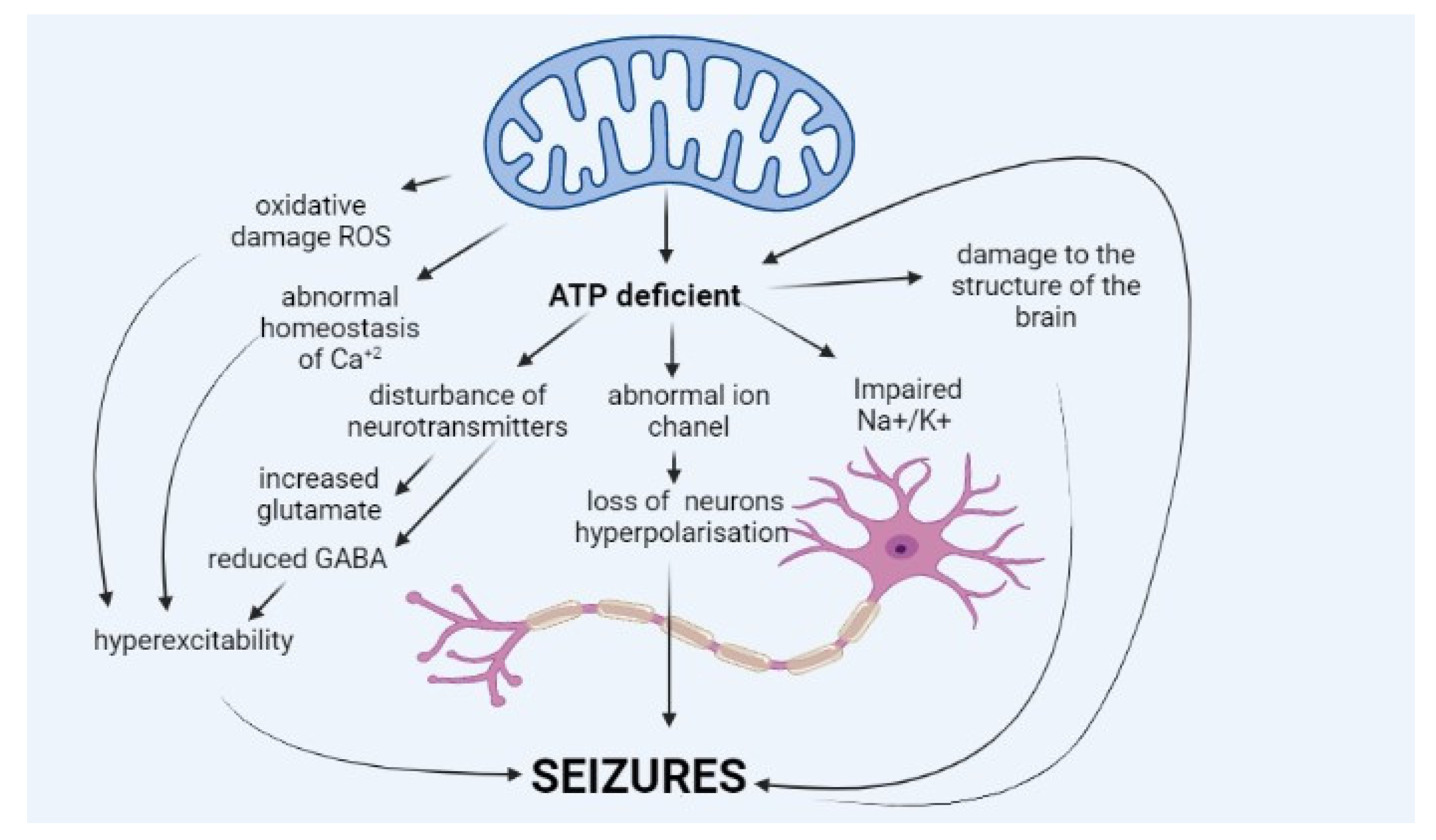
:1. Introduction
2. Pathophysiology of Epilepsy in Mitochondrial Diseases
3. Clinical Picture of Epilepsy in Mitochondrial Diseases
4. Diagnostics of Epilepsy in Mitochondrial Disease
4.1. Electroencephalography in Patients with Mitochondrial Disease
4.2. Neuroimaging
5. Pharmacological Treatment of Epilepsy in Patients with Mitochondrial Disease
6. Non-Pharmacological Treatment of Epilepsy in Patients with Mitochondrial Disease
7. Prognosis in Epilepsy in Patients with Mitochondrial Disease
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rahman, S. Pathophysiology of mitochondrial disease causing epilepsy and status epilepticus. Epilepsy Behav. 2015, 49, 71–75. [Google Scholar] [CrossRef]
- Stenton, S.L.; Prokisch, H. Genetics of mitochondrial diseases: Identifying mutations to help diagnosis. EBioMedicine 2020, 56, 102784. [Google Scholar] [CrossRef]
- Tan, J.; Wagner, M.; Stenton, S.L.; Strom, T.M.; Wortmann, S.B.; Prokisch, H.; Meitinger, T.; Oexle, K.; Klopstock, T. Lifetime risk of autosomal recessive mitochondrial disorders calculated from genetic databases. EBioMedicine 2020, 54, 102730. [Google Scholar] [CrossRef]
- Davison, J.E.; Rahman, S. Recognition, investigation and management of mitochondrial disease. Arch. Dis. Child. 2017, 102, 1082–1090. [Google Scholar] [CrossRef]
- Alston, C.L.; Rocha, M.C.; Lax, N.Z.; Turnbull, D.M.; Taylor, R.W. The genetics and pathology of mitochondrial disease. J. Pathol. 2017, 241, 236–250. [Google Scholar] [CrossRef]
- Lim, A.; Thomas, R.H. The mitochondrial epilepsies. Eur. J. Paediatr. Neurol. 2020, 24, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Coelho, M.P.; Martins, E.; Vilarinho, L. Diagnosis, management, and follow-up of mitochondrial disorders in childhood: A personalized medicine in the new era of genome sequence. Eur. J. Pediatr. 2019, 178, 21–32. [Google Scholar] [CrossRef] [PubMed]
- Matricardi, S.; Canafoglia, L.; Ardissone, A.; Moroni, I.; Ragona, F.; Ghezzi, D.; Lamantea, E.; Nardocci, N.; Franceschetti, S.; Granata, T. Epileptic phenotypes in children with early-onset mitochondrial diseases. Acta Neurol. Scand. 2019, 140, 184–193. [Google Scholar] [CrossRef]
- Khurana, D.S.; Salganicoff, L.; Melvin, J.J.; Hobdell, I.; Valencia, H.H.; Hardison, H.G.; Marks, W.D.; Grover, A.; Legido, A. Epilepsy and respiratory chain defects in children with mitochondrial encephalopathies. Neuropediatrics 2008, 39, 8–13. [Google Scholar] [CrossRef] [PubMed]
- Ticci, C.; Sicca, F.; Ardissone, A.; Bertini, E.; Carelli, V.; Diodato, D.; Di Vito, L.; Filosto, M.; La Morgia, C.; Lamperti, C.; et al. Mitochondrial epilepsy: A cross-sectional nationwide Italian survey. Neurogenetics 2020, 21, 87–96. [Google Scholar] [CrossRef]
- Fisher, R.S.; Cross, J.H.; French, J.A.; Higurashi, N.; Hirsch, E.; Jansen, F.E.; Lagae, L.; Moshé, S.L.; Peltola, J.; Roulet Perez, E.; et al. Operational classification of seizure types by the International League against Epilepsy: Position Paper of the ILAE Commission for Classification and Terminology. Epilepsia 2017, 58, 522–530. [Google Scholar] [CrossRef] [Green Version]
- Iizuka, T.; Saka, F.; Suzuki, N.; Hata, T.; Tsukahara, S.; Fukuda, M.; Takiyama, Y. Neuronal hyperexcitability in stroke-like episodes of MELAS syndrome. Neurology 2002, 59, 816–824. [Google Scholar] [CrossRef] [PubMed]
- Fei, Y.; Shi, R.; Song, Z.; Wu, J. Metabolic Control of Epilepsy: A Promising Therapeutic Target for Epilepsy. Front. Neurol. 2020, 11, 592514. [Google Scholar] [CrossRef] [PubMed]
- Lax, N.Z.; Grady, J.; Laude, A.; Chan, F.; Hepplewhite, P.D.; Gorman, G.; Whittaker, R.G.; Ng, Y.; Cunningham, M.O.; Turnbull, D.M. Extensive respiratory chain defects in inhibitory interneurones in patients with mitochondrial disease. Neuropathol. Appl. Neurobiol. 2016, 42, 180–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bindoff, L.A.; Engelsen, B.A. Mitochondrial diseases and epilepsy. Epilepsia 2012, 53 (Suppl. S4), 92–97. [Google Scholar] [CrossRef]
- Sanganahalli, B.G.; Herman, P.; Hyder, F.; Kannurpatti, S.S. Mitochondrial calcium uptake capacity modulates neocortical excitability. J. Cereb. Blood Flow Metab. 2013, 33, 1115–1126. [Google Scholar] [CrossRef] [Green Version]
- Hardie, D.G.; Ross, F.A.; Hawley, S.A. AMPK: A nutrient and energy sensor that maintains energy homeostasis. Nat. Rev. Mol. Cell Biol. 2012, 13, 251–262. [Google Scholar] [CrossRef] [Green Version]
- Bhandary, S.; Aguan, K. Pyruvate dehydrogenase complex deficiency and its relationship with epilepsy frequency-An overview. Epilepsy Res. 2015, 116, 40–52. [Google Scholar] [CrossRef]
- Tranah, G.J.; Katzman, S.M.; Lauterjung, K.; Yaffe, K.; Manini, T.M.; Kritchevsky, S.; Newman, A.B.; Harris, T.B.; Cummings, S.R. Mitochondrial DNA m.3243A>G heteroplasmy affects multiple aging phenotypes and risk of mortality. Sci. Rep. 2018, 8, 11887. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.; Baek, M.S.; Lee, Y.M. Lennox-Gastaut Syndrome in Mitochondrial Disease. Yonsei. Med. J. 2019, 60, 106–114. [Google Scholar] [CrossRef]
- Lee, Y.M.; Kang, H.C.; Lee, J.S.; Kim, S.H.; Kim, E.Y.; Lee, S.K.; Slama, A.; Kim, H.D. Mitochondrial respiratory chain defects: Underlying etiology in various epileptic conditions. Epilepsia 2008, 49, 685–690. [Google Scholar] [CrossRef] [PubMed]
- Whittaker, R.G.; Devine, H.E.; Gorman, G.S.; Schaefer, A.M.; Horvath, R.; Ng, Y.; Nesbitt, V.; Lax, N.Z.; McFarland, R.; Cunningham, M.O.; et al. Epilepsy in adults with mitochondrial disease: A cohort study. Ann. Neurol. 2015, 78, 949–957. [Google Scholar] [CrossRef] [Green Version]
- Rahman, S. Mitochondrial disease andepilepsy. Dev. Med. Child Neurol. 2012, 54, 397–406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pickett, S.J.; Grady, J.P.; Ng, Y.S.; Gorman, G.S.; Schaefer, A.M.; Wilson, I.J.; Cordell, H.J.; Turnbull, D.M.; Taylor, R.W.; McFarland, R. Phenotypic heterogeneity in m.3243A>G mitochondrial disease: The role of nuclear factors. Ann. Clin. Transl. Neurol. 2018, 5, 333–345. [Google Scholar] [CrossRef] [Green Version]
- Hikmat, O.; Naess, K.; Engvall, M.; Klingenberg, C.; Rasmussen, M.; Tallaksen, C.M.; Brodtkorb, E.; Ostergaard, E.; de Coo, I.F.M.; Pias-Peleteiro, L.; et al. Simplifying the clinical classification of polymerase gamma (POLG) disease based on age of onset; studies using a cohort of 155 cases. J. Inherit. Metab. Dis. 2020, 43, 726–736. [Google Scholar] [CrossRef] [Green Version]
- Rahman, S. Mitochondrial diseases and status epilepticus. Epilepsia 2018, 59 (Suppl. S2), 70–77. [Google Scholar] [CrossRef] [PubMed]
- Patel, K.P.; O’Brien, T.W.; Subramony, S.H.; Shuster, J.; Stacpoole, P.W. The spectrum of pyruvate dehydrogenase complex deficiency: Clinical, biochemical and genetic features in 371 patients. Mol. Genet. Metab. 2012, 105, 34–43. [Google Scholar] [CrossRef] [Green Version]
- Ciara, E.; Rokicki, D.; Halat, P.; Karkucińska-Więckowska, A.; Piekutowska-Abramczuk, D.; Mayr, J.; Trubicka, J.; Szymańska-Dębińska, T.; Pronicki, M.; Pajdowska, M.; et al. Difficulties in recognition of pyruvate dehydrogenase complex deficiency on the basis of clinical and biochemical features. The role of next-generation sequencing. Mol. Genet. Metab. Rep. 2016, 18, 70–76. [Google Scholar] [CrossRef]
- Chang, X.; Wu, Y.; Zhou, J.; Meng, H.; Zhang, W.; Guo, J. A meta-analysis and systematic review of Leigh syndrome: Clinical manifestations respiratory chain enzyme complex deficiency, and gene mutations. Medicine 2020, 99, e18634. [Google Scholar] [CrossRef]
- Horvath, R.; Chinnery, P.F. Diagnostic approach to mitochondrial diseases. In Diagnosis and Management of Mitochondria Disorders; Mancuso, M., Klostock, T., Eds.; Springer: Berlin/Heidelberg, Germany, 2019; pp. 281–287. [Google Scholar]
- Lamperti, C.; Zeviani, M. Myoclonus epilepsy in mitochondria disorders. Epileptic Disord. 2016, 18, 94–102. [Google Scholar]
- El-Hattab, A.W.; Adesina, A.M.; Jones, J.; Scaglia, F. MELAS syndrome: Clinical manifestations, pathogenesis and treatment options. Mol. Genet. Metabol. 2015, 116, 4–12. [Google Scholar] [CrossRef]
- El Sabbagh, S.; Lebre, A.S.; Bahi-Buisson, N.; Delonlay, P.; Soufflet, C.; Boddaert, N.; Rio, M.; Rötig, A.; Dulac, O.; Munnich, A.; et al. Epileptic phenotypes in children with respiratory chain disorders. Epilepsia 2010, 51, 1225–1235. [Google Scholar] [CrossRef]
- Surana, S.; Rossor, T.; Hassell, J.; Boyd, S.; D’Arco, F.; Aylett, S.; Bhate, S.; Carr, L.; Das, K.; DeVile, C.; et al. Diagnostic algorithm for children presenting with epilepsia partialis continua. Epilepsia 2020, 61, 2224–2233. [Google Scholar] [CrossRef]
- Finsterer, J. Pharmacotherapeutic management of epilepsy in MERRF syndrome. Expert Opin. Drug Metab. Toxicol. 2019, 20, 1289–1297. [Google Scholar] [CrossRef]
- Lee, S.; Na, J.H.; Lee, Y.M. Epilepsy in Leigh Syndrome with Mitochondrial DNA Mutations. Front. Neurol. 2019, 10, 496. [Google Scholar] [CrossRef]
- Chevallier, J.A.; Von Allmen, G.K.; Koenig, M.K. Seizure semiology and EEG findings in mitochondrial diseases. Epilepsia 2014, 55, 707–712. [Google Scholar] [CrossRef] [PubMed]
- Tzoulis, C.; Engelsen, B.A.; Telstad, W.; Aasly, J.; Zeviani, M.; Winterthun, S.; Ferrari, G.; Aarseth, J.H.; Bindoff, L.A. The spectrum of clinical disease caused by the A467T and W748S POLG mutations: A study of 26 cases. Brain 2006, 129, 1685–1692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parikh, S.; Goldstein, A.; Karaa, A.; Koenig, M.K.; Anselm, I.; Brunel-Guitton, C.; Christodoulou, J.; Cohen, B.H.; Dimmock, D.; Enns, G.M.; et al. Patient care standards for primary mitochondrial disease: A consensus statement from the Mitochondrial Medicine Society. Genet. Med. 2017, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garone, C.; Viscom, C. Towards a therapy for mitochondrial disease: An update. Biochem. Soc. Trans. 2018, 46, 1247–1261. [Google Scholar] [CrossRef] [Green Version]
- Almannai, M.; El-Hattab, A.W.; Ali, M.; Soler-Alfonso, C.; Scaglia, F. Clinical trials in mitochondrial disorders, an update. Mol. Genet. Metab. 2020, 131, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Tinker, R.; Lim, A.Z.; Stefanetti, R.; McFarland, R. Current and Emerging Clinical Treatment in Mitochondrial Disease. Mol. Diagn. Ther. 2021, 25, 181–206. [Google Scholar] [CrossRef] [PubMed]
- Mitochondrial Disease Care Recommendations. Physiotherapy Guidance for People with Mitochondrial Disease. Available online: www.newcastle-mitochondria.com (accessed on 26 June 2019).
- Rahman, S. Advances in the treatment of mitochondrial epilepsies. Epilepsy Behav. 2019, 101 Pt B, 106546. [Google Scholar] [CrossRef]
- Su, L.J.; Wang, Y.L.; Han, T.; Qiao, S.; Zang, K.J.; Wu, H.K.; Su, Y.X.; Liu, L.L.; Liu, X.W. Antimyoclonic effect of levetiracetam and clonazepam combined treatment on myoclonic epilepsy with ragged-red fiber syndrome with m.8344A>G Mutation. Chin. Med. J. 2018, 131, 2433–2438. [Google Scholar] [CrossRef] [PubMed]
- Santamarina, E.; Alpuente, A.; Maisterra, O.; Sueiras, M.; Sarria, S.; Guzman, L.; Abraira, L.; Salas-Puig, J.; Toledo, M. Perampanel: A therapeutic alternative in refractory status epilepticus associated with MELAS syndrome. Epilepsy Behav. Case Rep. 2019, 11, 92–95. [Google Scholar] [CrossRef] [PubMed]
- De Vries, M.C.; Brown, D.A.; Allen, M.E.; Bindoff, L.; Gorman, G.S.; Karaa, A.; Keshavan, N.; Lamperti, C.; McFarland, R.; Ng, Y.S.; et al. Safety of drug use in patients with a primary mitochondrial disease: An international Delphi-based consensus. J. Inherit. Metab. Dis. 2020, 43, 800–818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parikh, S.; Goldstein, A.; Koenig, M.K.; Scaglia, F.; Enns, G.M.; Saneto, R.; Anselm, I.; Cohent, B.H.; Falk, M.J.; Greene, C.; et al. Diagnosis and management of mitochondrial disease: A consensus statement from the Mitochondrial Medicine Society. Genet. Med. 2015, 17, 689–701. [Google Scholar] [CrossRef] [Green Version]
- Saneto, R.P.; Lee, I.C.; Koenig, M.K.; Bao, X.; Weng, S.W.; Naviaux, R.K.; Wong, L.J. POLG DNA testing as an emerging standard of care before instituting valproic acid therapy for pediatric seizure disorders. Seizure 2010, 19, 140–146. [Google Scholar] [CrossRef] [Green Version]
- Finsterer, J. Toxicity of Antiepileptic Drugs to Mitochondria. Handb. Exp. Pharmacol. 2017, 240, 473–488. [Google Scholar]
- Gruosso, F.; Montano, V.; Simoncini, C.; Siciliano, G.; Mancuso, M. Therapeutical Management and Drug Safety in Mitochondrial Diseases-Update 2020. J. Clin. Med. 2020, 10, 94. [Google Scholar] [CrossRef]
- Koga, Y.; Nishioka, Y.; Nishioka, J.; Yatsuga, S.; Povalko, N.; Katayama, K.; Matsuishi, T. MELAS and L-arginine therapy. Mitochondrion 2007, 7, 133–139. [Google Scholar] [CrossRef] [PubMed]
- Pineda, M.; Ormazabal, A.; Lopez-Gallardo, E.; Nascimento, A.; Solano, A.; Herrero, M.D.; Vilaseca, M.A.; Briones, P.; Ibáñez, L.; Montoya, J.; et al. Cerebral folate deficiency and leukoencephalopathy caused by a mitochondrial DNA deletion. Ann. Neurol. 2006, 59, 394–398. [Google Scholar] [CrossRef] [PubMed]
- Salviati, L.; Trevisson, E.; Doimo, M.; Navas, P. Primary Coenzyme Q10 Deficiency. In GeneReviews® [Internet], 1993–2020; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Mirzaa, G., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 2017. [Google Scholar]
- Repp, B.M.; Mastantuono, E.; Alston, C.L.; Schiff, M.; Haack, T.B.; Rötig, A.; Ardissone, A.; Lombès, A.; Catarino, C.B.; Diodato, D.; et al. Clinical, biochemical and genetic spectrum of 70 patients with ACAD9 deficiency: Is riboflavin supplementation effective? Orphanet J. Rare Dis. 2018, 13, 120. [Google Scholar] [CrossRef]
- Marcé-Grau, A.; Martí-Sánchez, L.; Baide-Mairena, H.; rtigoza-Escobar, J.D.; Pérez-Dueñas, B. Genetic defects of thiamine transport and metabolism: A review of clinical phenotypes, genetics, and functional studies. J. Inherit. Metab. Dis. 2019, 42, 581–597. [Google Scholar] [CrossRef]
- Dahlin, M.; Martin, D.A.; Hedlund, Z.; Jonsson, M.; von Döbeln, U.; Wedell, A. The ketogenic diet compensates for AGC1 deficiency and improves myelination. Epilepsia 2015, 56, e176–e181. [Google Scholar] [CrossRef] [Green Version]
- Viscomi, C.; Burlina, A.B.; Dweikat, I.; Savoiardo, M.; Lamperti, C.; Hildebrandt, T.; Tiranti, V.; Zeviani, M. Combined treatment with oral metronidazole and N-acetylcysteine is effective in ethylmalonic encephalopathy. Nat. Med. 2010, 16, 869–871. [Google Scholar] [CrossRef]
- Yamada, K.; Naiki, M.; Hoshino, S.; Kitaura, Y.; Kondo, Y.; Nomura, N.; Kimura, R.; Fukushi, D.; Yamada, Y.; Shimozawa, N.; et al. Clinical and biochemical characterization of 3-hydroxyisobutyryl-CoA hydrolase (HIBCH) deficiency that causes Leigh-like disease and ketoacidosis. Mol. Genet. Metab. Rep. 2014, 16, 455–460. [Google Scholar] [CrossRef] [PubMed]
- Wells, J.; Swaminathan, A.; Paseka, J.; Hanson, C. Efficacy and Safety of a Ketogenic Diet in Children and Adolescents with Refractory Epilepsy-A Review. Nutrients 2020, 17, 1809. [Google Scholar] [CrossRef]
- Kossoff, E.H.; Zupec-Kania, B.A.; Auvin, S.; Ballaban-Gil, K.R.; Christina Bergqvist, A.G.; Blackford, R.; Buchhalter, J.R.; Caraballo, R.H.; Cross, J.H.; Dahlin, M.G.; et al. Optimal clinical management of children receiving dietary therapies for epilepsy: Updated recommendations of the International Ketogenic Diet Study Group. Epilepsia Open. 2018, 3, 175–192. [Google Scholar] [CrossRef]
- van der Louw, E.; van den Hurk, D.; Neal, E.; Leiendecker, B.; Fitzsimmon, G.; Dority, L.; Thompson, L.; Marchió, M.; Dudzińska, M.; Dressler, A.; et al. Ketogenic diet guidelines for infants with refractory epilepsy. Eur. J. Paediatr. Neurol. 2016, 20, 798–809. [Google Scholar] [CrossRef] [PubMed]
- Martin-McGill, K.J.; Bresnahan, R.; Levy, R.G.; Cooper, P.N. Ketogenic diets for drug-resistant epilepsy. Cochrane Database Syst. Rev. 2020, 6, 1903. [Google Scholar] [CrossRef]
- Paleologou, E.; Ismayilova, N.; Kinali, M. Use of the Ketogenic Diet to Treat Intractable Epilepsy in Mitochondrial Disorders. J. Clin. Med. 2017, 6, 56. [Google Scholar] [CrossRef]
- Geffroy, G.; Benyahia, R.; Frey, S.; Desquiret-Dumas, V.; Gueguen, N.; Bris, C.; Belal, S.; Inisan, A.; Renaud, A.; Chevrollier, A.; et al. The accumulation of assembly intermediates of the mitochondrial complex I matrix arm is reduced by limiting glucose uptake in a neuronal-like model of MELAS syndrome. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 1596. [Google Scholar] [CrossRef]
- Santra, S.; Gilkerson, R.W.; Davidson, M.; Schon, E.A. Ketogenic treatment reduces deleted mitochondrial DNAs in cultured human cells. Ann. Neurol. 2004, 56, 662–669. [Google Scholar] [CrossRef] [PubMed]
- Ahola-Erkkila, S.; Carroll, C.J.; Peltola-Mjosund, K.; Tulkki, V.; Mattila, I.; Seppänen-Laakso, T.; Oresic, M.; Tyynismaa, H.; Suomalainen, A. Ketogenic diet slows down mitochondrial myopathy progression in mice. Hum. Mol. Genet. 2010, 19, 1974–1984. [Google Scholar] [CrossRef] [Green Version]
- Na, J.H.; Kim, H.D.; Lee, Y.M. Effective and safe diet therapies for Lennox-Gastaut syndrome with mitochondrial dysfunction. Ther. Adv. Neurol. Disord. 2020, 6, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.F.; Chi, C.S.; Tsai, C.R.; Chen, C.H. Epileptic seizures in infants and children with mitochondrial diseases. Pediatr. Neurol. 2011, 45, 169–174. [Google Scholar] [CrossRef] [PubMed]
- Inak, G.; Rybak-Wolf, A.; Lisowski, P.; Pentimalli, T.M.; Jüttner, R.; Glažar, P.; Uppal, K.; Bottani, E.; Brunetti, D.; Secker, C.; et al. Defective metabolic programming impairs early neuronal morphogenesis in neural cultures and an organoid model of Leigh syndrome. Nat. Commun. 2021, 12, 1929. [Google Scholar] [CrossRef]
- Khan, S.; Ince-Dunn, G.; Suomalainen, A.; Elo, L.L. Integrative omics approaches provide biological and clinical insights: Examples from mitochondrial diseases. J. Clin. Investig. 2020, 130, 20–28. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Disease | Gene | Clinical Picture | Treatment |
|---|---|---|---|
| Alpers–Huttenlocher syndrome (AHS) [25,26] | POLG (nDNA) | Progressive neurodegeneration, refractory seizures, movement disorder, neuropathy and hepatic failure, focal-onset seizures predominate, but seizure may also tonic-clonic, or myoclonic; 68% developed status epilepticus and 58% epilepsia partialis continua, status epilepticus is the leading cause of death in children with AHS | In case of refractory seizures, polytherapy is necessary (with no dedicated drug; however, valproic acid is absolutely contraindicated) |
| Pyruvate dehydrogenase complex deficiency (PDHc) [27,28] | PDHA, PDHB, LIAS, LIPT1, DLD, PDH, (nDNA) | Epilepsy begins in infancy with infantile spasms, clonic seizures or refractory focal epilepsy, developmental delay, ataxia, hypotonia, hypertonia, abnormal eye movements, dystonia, axonal neuropathy | The ketogenic diet is the treatment of choice; in some individuals, improvement after thiamine supply possible |
| Leigh syndrome (LS) [29] | More than 90 genes (nDNA and mtDNA) | Typical features include: (1) developmental regression or developmental delay, (2) specific basal ganglia/brain stem changes bilaterally, and (3) abnormal mitochondrial energy metabolism; epileptic seizures are frequent, both focal and generalised | Due to frequent drug-refractory seizures, polytherapy is often necessary |
| Myoclonic epilepsy with ragged red fibres (MERRF) [30,31] | The most common pathogenic variants in mtDNA, MTTL1 (80%): m.8344A>G; MTTK (10%): m.8356T>C, m.8363G>A, m.8361G>A | Onset usually in adults, 30% in childhood. Progressive myoclonic epilepsy is part of the phenotype, but seizures can be often generalised tonic, clonic or atonic. Seizure was reported in 33% to 100% of patients; co-occurs with cerebellar ataxia, cardiac arrhythmias, myopathy, diabetes, hearing loss, dementia | The combination of levetiracetam with carbamazepine may have the strongest beneficial effect on myoclonic seizures |
| Mitochondrial encephalopathy, lactic acidosis, and stroke-like episodes (MELAS) [32] | The most common pathogenic variants in mtDNA, MTTL1 gen: m.3243A >G m.3271T>C; MTND5 gen: m.13513G>A | Focal and generalised seizures are possible, preceded by or associated with migraine-like headache; the most typical are seizures in the course of a stroke-like episode, focal status epilepticus with a secondary encephalopathy is common | L-arginine and/or citrulline as prevention and treatment of stroke-like episodes |
| Mitochondria-Safe AEDs | AEDs to Use Carefully | AEDs Which Could Aggravate Myoclonus |
|---|---|---|
| Benzodiazepine [47,51] Gabapentin [47,51] Lacosamide [47,51] Lamotrigine [47,51] Levetiracetam [10,47,51] Oxcarbazepine [10,47,51] Peranpanel [46,47,51] Rufinamide [47,51] Stiripentol [10,47,51] Zonisamide [47,51] | Valproic acid—contraindicated in POLG mutations [25,39,51] Vigabatrin—may need to be avoided in patients with mtDNA depletion syndromes [39] Topiramate—may worsen acidosis [39] Phenytoin * [50] Carbamazepine * [50] Phenobarbital * [50] | Valproic acid [35] Phenobarbital [35] Lamotrigine [35] Phenytoin [35] Carbamazepine [35] Oxcarbazepine [35] Vigabatrin [35] Tiagabine [35] Gabapentin [35] Pregabalin [35] |
| Disease (Gene) | Clinical Features | Treatment |
|---|---|---|
| Primary co-enzyme Q10 deficiency (COQ2, COQ4, COQ5, COQ6, COQ7, COQ9, PDSS1, PDSS2) | Multisystem involvement with progressive neurological dysfunction, seizures, encephalopathy, stroke-like episodes, cerebellar ataxia, pyramidal dysfunction, cognitive impairment renal failure, and steroid-resistant nephrotic syndrome | High-dose oral CoQ10 supplementation (ranging from 5 to 50 mg/kg/day) [54] |
| Pyruvate dehydrogenase complex (PDHc) deficiency (PDHA1, PDHB, LIAS, PDP1, PDHX, DLAT) | Epilepsy, developmental delay, ataxia, hypotonia, hypertonia, abnormal eye movements, dystonia, ataxia, axonal neuropathy, and poor feeding | Ketogenic diet 3:1–4:1 and thiamine (50 mg/kg/day, max 300–900 mg/day) [18,27] |
| ACAD9 deficiency (ACAD9) | Hypertrophic cardiomyopathy, lactic acidosis, exercise intolerance, and occasional seizures | Riboflavin (vitamin B2) 20 mg/kg/day–max 400 mg/day [55] |
| Impairment of thiamine transport and metabolism (SLC19A3, SLC19A2, SLC25A19, TPK1) | Biotin–thiamine-responsive basal ganglia disease or Leigh syndrome; subacute encephalopathy with confusion, dysphagia, dysarthria, seizures, external ophthalmoplegia, and generalised stiffness following a history of febrile illness; progresses to severe quadriparesis, rigidity, dystonia, coma, and death if early treatment is not administered | Biotin (5–10 mg/kg/day) and thiamine (10–40 mg/kg/day, between 300 and 900 mg/day) [56] |
| AGC1 deficiency (aspartate–glutamate carrier isoform 1) (SLC25A12) | Severe hypotonia, arrested psychomotor development, and seizures from a few months of age, a global lack of myelination in the cerebral hemispheres | Ketogenic diet 3:1–4:1 [57] |
| Ethylmalonic encephalopathy (ETHE1) | Early onset, progressive disorder, developmental delay, generalised infantile hypotonia that evolves into hypertonia, spasticity and dystonia; generalised tonic–clonic seizures; and generalised microvascular damage | N-acetylcysteine in combination with metronidazole [58] |
| Beta-hydroxyisobutyryl-CoA deacylase deficiency (HIBCHD) | Progressive neurodegenerative disorder, associated with basal ganglia changes on brain magnetic resonance imaging; elevated hydroxy-C4-carnitine levels | Low-valine and high-carbohydrate diets, antioxidants (co-enzyme Q10, vitamin E, vitamin C), carnitine, and N-acetylcysteine [59] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wesół-Kucharska, D.; Rokicki, D.; Jezela-Stanek, A. Epilepsy in Mitochondrial Diseases—Current State of Knowledge on Aetiology and Treatment. Children 2021, 8, 532. https://doi.org/10.3390/children8070532
Wesół-Kucharska D, Rokicki D, Jezela-Stanek A. Epilepsy in Mitochondrial Diseases—Current State of Knowledge on Aetiology and Treatment. Children. 2021; 8(7):532. https://doi.org/10.3390/children8070532
Chicago/Turabian StyleWesół-Kucharska, Dorota, Dariusz Rokicki, and Aleksandra Jezela-Stanek. 2021. "Epilepsy in Mitochondrial Diseases—Current State of Knowledge on Aetiology and Treatment" Children 8, no. 7: 532. https://doi.org/10.3390/children8070532
APA StyleWesół-Kucharska, D., Rokicki, D., & Jezela-Stanek, A. (2021). Epilepsy in Mitochondrial Diseases—Current State of Knowledge on Aetiology and Treatment. Children, 8(7), 532. https://doi.org/10.3390/children8070532