
A Neonate with Autosomal Dominant Pseudohypoaldosteronism Type 1 Due to a Novel Microdeletion of the NR3C2 Gene at 4q31.23


{kind=link}
{kind=link}
Abstract
:1. Introduction
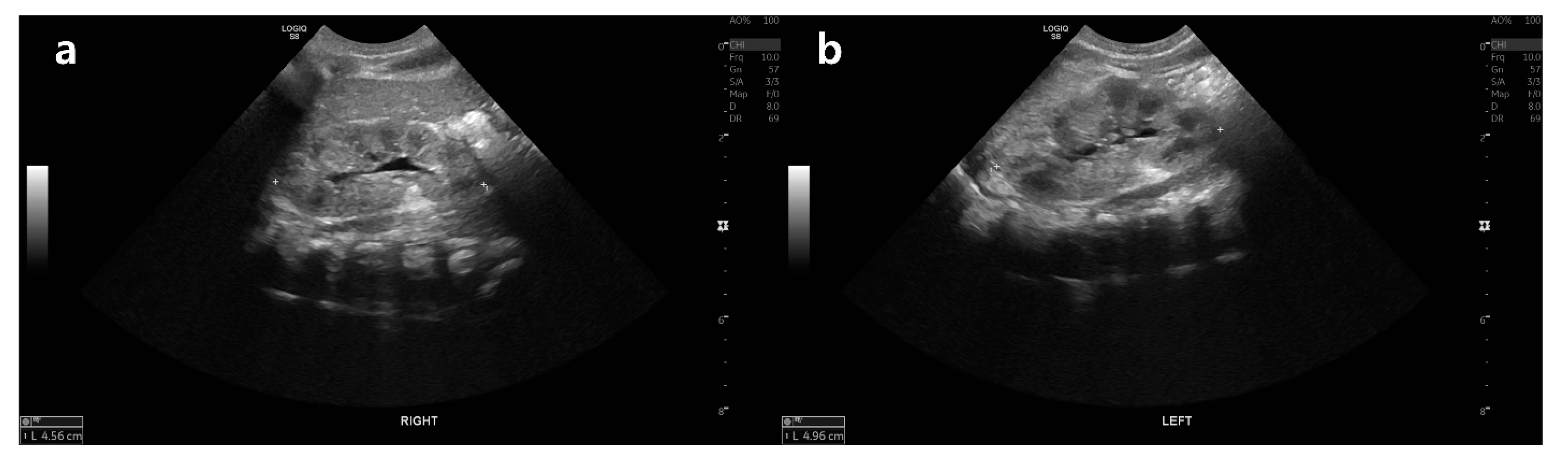
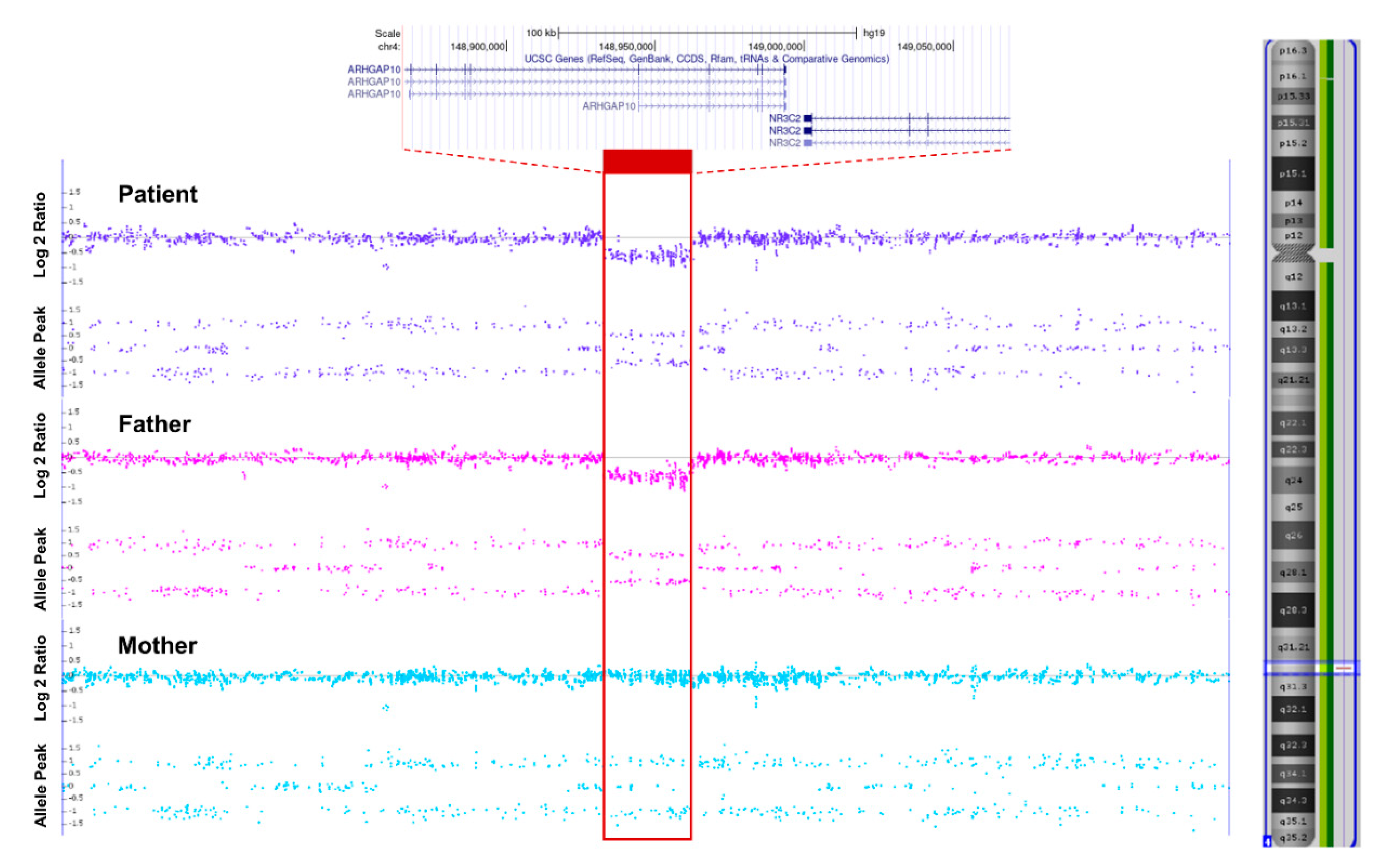
2. Case Presentation
3. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Amin, N.; Alvi, N.S.; Barth, J.H.; Field, H.P.; Finlay, E.; Tyerman, K.; Frazer, S.; Savill, G.; Wright, N.P.; Makaya, T.; et al. Pseudohypoaldosteronism type 1: Clinical features and management in infancy. Endocrinol. Diabetes Metab. Case Rep. 2013, 2013, 130010. [Google Scholar] [CrossRef] [PubMed]
- Riepe, F.G. Clinical and molecular features of type 1 pseudohypoaldosteronism. Horm. Res. 2009, 72, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Geller, D.S.; Rodriguez-Soriano, J.; Vallo Boado, A.; Schifter, S.; Bayer, M.; Chang, S.S.; Lifton, R.P. Mutations in the mineralocorticoid receptor gene cause autosomal dominant pseudohypoaldosteronism type I. Nat. Genet. 1998, 19, 279–281. [Google Scholar] [CrossRef] [PubMed]
- Geller, D.S.; Zhang, J.; Zennaro, M.C.; Vallo-Boado, A.; Rodriguez-Soriano, J.; Furu, L.; Haws, R.; Metzger, D.; Botelho, B.; Karaviti, L.; et al. Autosomal dominant pseudohypoaldosteronism type 1: Mechanisms, evidence for neonatal lethality, and phenotypic expression in adults. J. Am. Soc. Nephrol. 2006, 17, 1429–1436. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.S.; Grunder, S.; Hanukoglu, A.; Rosler, A.; Mathew, P.M.; Hanukoglu, I.; Schild, L.; Lu, Y.; Shimkets, R.A.; Nelson-Williams, C.; et al. Mutations in subunits of the epithelial sodium channel cause salt wasting with hyperkalaemic acidosis, pseudohypoaldosteronism type 1. Nat. Genet. 1996, 12, 248–253. [Google Scholar] [CrossRef] [PubMed]
- Saxena, A.; Hanukoglu, I.; Saxena, D.; Thompson, R.J.; Gardiner, R.M.; Hanukoglu, A. Novel mutations responsible for autosomal recessive multisystem pseudohypoaldosteronism and sequence variants in epithelial sodium channel alpha-, beta-, and gamma-subunit genes. J. Clin. Endocrinol. Metab. 2002, 87, 3344–3350. [Google Scholar] [CrossRef] [PubMed]
- Arai, K.; Papadopoulou-Marketou, N.; Chrousos, G.P. Aldosterone Deficiency and Resistance. In Endotext; Feingold, K.R., Anawalt, B., Boyce, A., Chrousos, G., de Herder, W.W., Dhatariya, K., Dungan, K., Grossman, A., Hershman, J.M., Hofland, J., et al., Eds.; MDText.com, Inc.: South Dartmouth, MA, USA, 2000. [Google Scholar]
- Kim, Y.M.; Choi, I.S.; Cheong, H.I.; Kim, C.J.; Yang, E.M. Pseudohypoaldosteronism Type 1 with a Novel Mutation in the NR3C2 Gene: A Case Report. Child. Kidney Dis. 2020, 24, 58–61. [Google Scholar] [CrossRef]
- Conversano, E.; Romano, S.; Taddio, A.; Faletra, F.; Zanon, D.; Barbi, E.; Pennesi, M. When salt is needed to grow: Answers. Pediatr. Nephrol. 2021, 36, 1131–1132. [Google Scholar] [CrossRef] [PubMed]
- Zennaro, M.C.; Fernandes-Rosa, F. 30 YEARS OF THE MINERALOCORTICOID RECEPTOR: Mineralocorticoid receptor mutations. J. Endocrinol. 2017, 234, T93–T106. [Google Scholar] [CrossRef]
- Pujo, L.; Fagart, J.; Gary, F.; Papadimitriou, D.T.; Claes, A.; Jeunemaitre, X.; Zennaro, M.C. Mineralocorticoid receptor mutations are the principal cause of renal type 1 pseudohypoaldosteronism. Hum. Mutat. 2007, 28, 33–40. [Google Scholar] [CrossRef]
- Bowden, S.A.; Cozzi, C.; Hickey, S.E.; Thrush, D.L.; Astbury, C.; Nuthakki, S. Autosomal dominant pseudohypoaldosteronism type 1 in an infant with salt wasting crisis associated with urinary tract infection and obstructive uropathy. Case Rep. Endocrinol. 2013, 2013, 524647. [Google Scholar] [CrossRef] [Green Version]
- Barone Pritchard, A.; Ritter, A.; Kearney, H.M.; Izumi, K. Interstitial 4q Deletion Syndrome Including NR3C2 Causing Pseudohypoaldosteronism. Mol. Syndromol. 2020, 10, 327–331. [Google Scholar] [CrossRef]
- Hanukoglu, A.; Vargas-Poussou, R.; Landau, Z.; Yosovich, K.; Hureaux, M.; Zennaro, M.C. Renin-aldosterone system evaluation over four decades in an extended family with autosomal dominant pseudohypoaldosteronism due to a deletion in the NR3C2 gene. J. Steroid. Biochem. Mol. Biol. 2020, 204, 105755. [Google Scholar] [CrossRef] [PubMed]
- O’Connell, S.M.; Johnson, S.R.; Lewis, B.D.; Staltari, L.; Peverall, J.; Ly, T.; Martin, A.C.; Jones, T.W.; Price, G.J.; Murch, A.; et al. Structural chromosome disruption of the NR3C2 gene causing pseudohypoaldosteronism type 1 presenting in infancy. J. Pediatr. Endocrinol. Metab. 2011, 24, 555–559. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, S.J.; Park, D.; Jang, W.; Lee, J. A Neonate with Autosomal Dominant Pseudohypoaldosteronism Type 1 Due to a Novel Microdeletion of the NR3C2 Gene at 4q31.23. Children 2021, 8, 1090. https://doi.org/10.3390/children8121090
Kim SJ, Park D, Jang W, Lee J. A Neonate with Autosomal Dominant Pseudohypoaldosteronism Type 1 Due to a Novel Microdeletion of the NR3C2 Gene at 4q31.23. Children. 2021; 8(12):1090. https://doi.org/10.3390/children8121090
Chicago/Turabian StyleKim, Su Jin, Dasom Park, Woori Jang, and Juyoung Lee. 2021. "A Neonate with Autosomal Dominant Pseudohypoaldosteronism Type 1 Due to a Novel Microdeletion of the NR3C2 Gene at 4q31.23" Children 8, no. 12: 1090. https://doi.org/10.3390/children8121090
APA StyleKim, S. J., Park, D., Jang, W., & Lee, J. (2021). A Neonate with Autosomal Dominant Pseudohypoaldosteronism Type 1 Due to a Novel Microdeletion of the NR3C2 Gene at 4q31.23. Children, 8(12), 1090. https://doi.org/10.3390/children8121090