
Genetic Variants of DMBT1 and SFTPD and Disease Severity in Paediatric Inflammatory Bowel Disease—A Polish Population-Based Study

 , , ,
, , ,
Abstract

1. Introduction
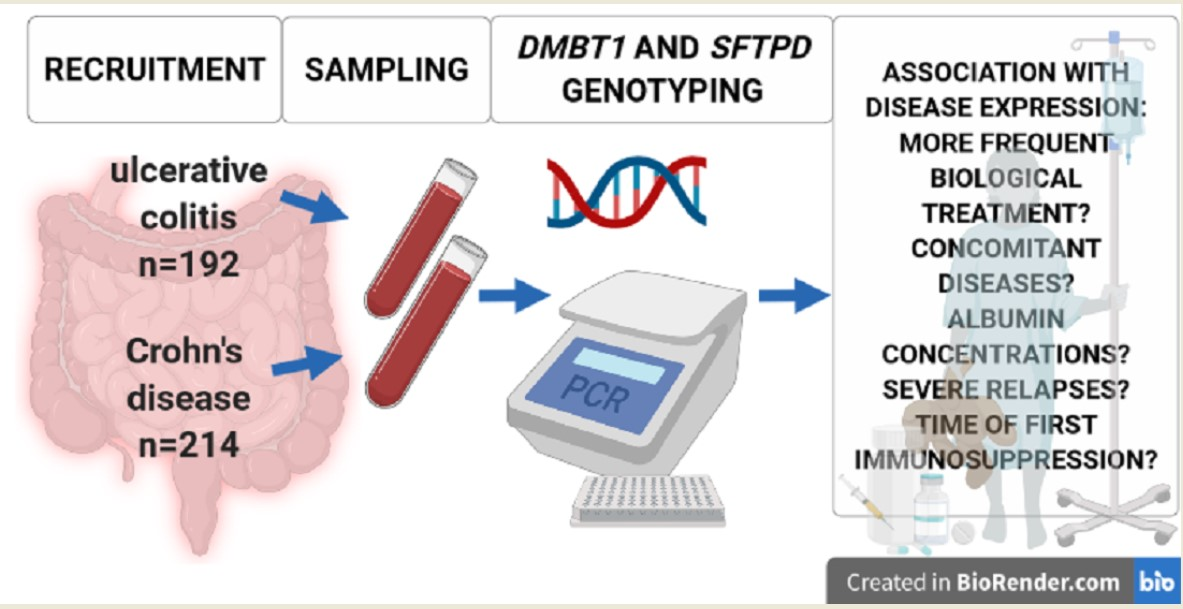
2. Materials and Methods
2.1. Patients
2.2. Genotyping
2.3. Clinical Phenotype
2.4. Disease Severity
2.5. Statistical Analysis
3. Results
3.1. Genotyping
3.2. Association with Disease Severity
3.2.1. DMBT1 rs2981804
3.2.2. DMBT1 rs2981745
3.2.3. SFTPD rs2243639
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ng, S.C.; Shi, H.Y.; Hamidi, N.; Underwood, F.E.; Tang, W.; Benchimol, E.I.; Panaccione, R.; Ghosh, S.; Wu, J.C.Y.; Chan, F.K.L.; et al. Worldwide Incidence and Prevalence of Inflammatory Bowel Disease in the 21st Century: A Systematic Review of Population-Based Studies. Lancet 2017, 390, 2769–2778. [Google Scholar] [CrossRef]
- Lichtenstein, G.R.; McGovern, D.P.B. Using Markers in IBD to Predict Disease and Treatment Outcomes: Rationale and a Review of Current Status. Am. J. Gastroenterol. Suppl. 2016, 3, 17–26. [Google Scholar] [CrossRef]
- Yarur, A.J.; Strobel, S.G.; Deshpande, A.R.; Abreu, M.T. Predictors of Aggressive Inflammatory Bowel Disease. Gastroenterol. Hepatol. 2011, 7, 652–659. [Google Scholar]
- Economou, M.; Trikalinos, T.A.; Loizou, K.T.; Tsianos, E.V.; Ioannidis, J.P.A. Differential Effects of NOD2 Variants on Crohn’s Disease Risk and Phenotype in Diverse Populations: A Metaanalysis. Am. J. Gastroenterol. 2004, 99, 2393–2404. [Google Scholar] [CrossRef]
- Henckaerts, L.; Van Steen, K.; Verstreken, I.; Cleynen, I.; Franke, A.; Schreiber, S.; Rutgeerts, P.; Vermeire, S. Genetic Risk Profiling and Prediction of Disease Course in Crohn’s Disease Patients. Clin. Gastroenterol. Hepatol. 2009, 7, 972–980.e2. [Google Scholar] [CrossRef]
- Hampe, J.; Grebe, J.; Nikolaus, S.; Solberg, C.; Croucher, P.J.P.; Mascheretti, S.; Jahnsen, J.; Moum, B.; Klump, B.; Krawczak, M.; et al. Association of NOD2 (CARD 15) Genotype with Clinical Course of Crohn’s Disease: A Cohort Study. Lancet 2002, 359, 1661–1665. [Google Scholar] [CrossRef]
- Ahmad, T.; Armuzzi, A.; Bunce, M.; Mulcahy-Hawes, K.; Marshall, S.E.; Orchard, T.R.; Crawshaw, J.; Large, O.; de Silva, A.; Cook, J.T.; et al. The Molecular Classification of the Clinical Manifestations of Crohn’s Disease. Gastroenterology 2002, 122, 854–866. [Google Scholar] [CrossRef]
- Seiderer, J.; Brand, S.; Herrmann, K.A.; Schnitzler, F.; Hatz, R.; Crispin, A.; Pfennig, S.; Schoenberg, S.O.; Göke, B.; Lohse, P.; et al. Predictive Value of the CARD15 Variant 1007fs for the Diagnosis of Intestinal Stenoses and the Need for Surgery in Crohn’s Disease in Clinical Practice: Results of a Prospective Study. Inflamm. Bowel Dis. 2006, 12, 1114–1121. [Google Scholar] [CrossRef]
- Abreu, M.T.; Taylor, K.D.; Lin, Y.-C.; Hang, T.; Gaiennie, J.; Landers, C.J.; Vasiliauskas, E.A.; Kam, L.Y.; Rojany, M.; Papadakis, K.A.; et al. Mutations in NOD2 Are Associated with Fibrostenosing Disease in Patients with Crohn’s Disease. Gastroenterology 2002, 123, 679–688. [Google Scholar] [CrossRef]
- Weersma, R.K.; Stokkers, P.C.F.; van Bodegraven, A.A.; van Hogezand, R.A.; Verspaget, H.W.; de Jong, D.J.; van der Woude, C.J.; Oldenburg, B.; Linskens, R.K.; Festen, E.A.M.; et al. Molecular Prediction of Disease Risk and Severity in a Large Dutch Crohn’s Disease Cohort. Gut 2009, 58, 388–395. [Google Scholar] [CrossRef] [PubMed]
- Solon, J.G.; Burke, J.P.; Walsh, S.R.; Coffey, J.C. The Effect of NOD2 Polymorphism on Postsurgical Recurrence in Crohn’s Disease: A Systematic Review and Meta-Analysis of Available Literature. Inflamm. Bowel Dis. 2013, 19, 1099–1105. [Google Scholar] [CrossRef]
- Adler, J.; Rangwalla, S.C.; Dwamena, B.A.; Higgins, P.D.R. The Prognostic Power of the NOD2 Genotype for Complicated Crohn’s Disease: A Meta-Analysis. Am. J. Gastroenterol. 2011, 106, 699–712. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.C.; Espéli, M.; Anderson, C.A.; Linterman, M.A.; Pocock, J.M.; Williams, N.J.; Roberts, R.; Viatte, S.; Fu, B.; Peshu, N.; et al. Human SNP Links Differential Outcomes in Inflammatory and Infectious Disease to a FOXO3-Regulated Pathway. Cell 2013, 155, 57–69. [Google Scholar] [CrossRef] [PubMed]
- Potocnik, U.; Ferkolj, I.; Glavac, D.; Dean, M. Polymorphisms in Multidrug Resistance 1 (MDR1) Gene Are Associated with Refractory Crohn Disease and Ulcerative Colitis. Genes Immun. 2004, 5, 530–539. [Google Scholar] [CrossRef] [PubMed]
- Siegel, C.A.; Melmed, G.Y. Predicting Response to Anti-TNF Agents for the Treatment of Crohn’s Disease. Therap. Adv. Gastroenterol. 2009, 2, 245–251. [Google Scholar] [CrossRef]
- Goyette, P.; Boucher, G.; Mallon, D.; Ellinghaus, E.; Jostins, L.; Huang, H.; Ripke, S.; Gusareva, E.S.; Annese, V.; Hauser, S.L.; et al. High-Density Mapping of the MHC Identifies a Shared Role for HLA-DRB1*01:03 in Inflammatory Bowel Diseases and Heterozygous Advantage in Ulcerative Colitis. Nat. Genet. 2015, 47, 172–179. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, T.; Armuzzi, A.; Neville, M.; Bunce, M.; Ling, K.-L.; Welsh, K.I.; Marshall, S.E.; Jewell, D.P. The Contribution of Human Leucocyte Antigen Complex Genes to Disease Phenotype in Ulcerative Colitis. Tissue Antigens 2003, 62, 527–535. [Google Scholar] [CrossRef]
- Renner, M.; Bergmann, G.; Krebs, I.; End, C.; Lyer, S.; Hilberg, F.; Helmke, B.; Gassler, N.; Autschbach, F.; Bikker, F.; et al. DMBT1 Confers Mucosal Protection In Vivo and a Deletion Variant Is Associated With Crohn’s Disease. Gastroenterology 2007, 133, 1499–1509. [Google Scholar] [CrossRef]
- Holmskov, U.; Mollenhauer, J.; Madsen, J.; Vitved, L.; Gronlund, J.; Tornoe, I.; Kliem, A.; Reid, K.B.M.; Poustka, A.; Skjodt, K. Cloning of Gp-340, a Putative Opsonin Receptor for Lung Surfactant Protein D. Proc. Natl. Acad. Sci. USA 1999, 96, 10794–10799. [Google Scholar] [CrossRef]
- Mollenhauer, J.; Wiemann, S.; Scheurlen, W.; Korn, B.; Hayashi, Y.; Wilgenbus, K.K.; von Deimling, A.; Poustka, A. DMBT1, a New Member of the SRCR Superfamily, on Chromosome 10q25.3-26.1 Is Deleted in Malignant Brain Tumours. Nat. Genet. 1997, 17, 32–39. [Google Scholar] [CrossRef] [PubMed]
- Holmskov, U.; Lawson, P.; Teisner, B.; Tornoe, I.; Willis, A.C.; Morgan, C.; Koch, C.; Reid, K.B. Isolation and Characterization of a New Member of the Scavenger Receptor Superfamily, Glycoprotein-340 (Gp-340), as a Lung Surfactant Protein-D Binding Molecule. J. Biol. Chem. 1997, 272, 13743–13749. [Google Scholar] [CrossRef]
- Mollenhauer, J.; Herbertz, S.; Holmskov, U.; Tolnay, M.; Krebs, I.; Merlo, A.; Schrøder, H.D.; Maier, D.; Breitling, F.; Wiemann, S.; et al. DMBT1 Encodes a Protein Involved in the Immune Defense and in Epithelial Differentiation and Is Highly Unstable in Cancer. Cancer Res. 2000, 60, 1704–1710. [Google Scholar]
- Cayatte, C.; Joyce-Shaikh, B.; Vega, F.; Boniface, K.; Grein, J.; Murphy, E.; Blumenschein, W.M.; Chen, S.; Malinao, M.-C.; Basham, B.; et al. Biomarkers of Therapeutic Response in the IL-23 Pathway in Inflammatory Bowel Disease. Clin. Transl. Gastroenterol. 2012, 3, e10. [Google Scholar] [CrossRef] [PubMed]
- Diegelmann, J.; Czamara, D.; Le Bras, E.; Zimmermann, E.; Olszak, T.; Bedynek, A.; Göke, B.; Franke, A.; Glas, J.; Brand, S. Intestinal DMBT1 Expression Is Modulated by Crohn’s Disease-Associated IL23R Variants and by a DMBT1 Variant Which Influences Binding of the Transcription Factors CREB1 and ATF-2. PLoS ONE 2013, 8, e77773. [Google Scholar] [CrossRef] [PubMed]
- Madsen, J.; Mollenhauer, J.; Holmskov, U. Review: Gp-340/DMBT1 in Mucosal Innate Immunity. Innate Immun. 2010, 16, 160–167. [Google Scholar] [CrossRef] [PubMed]
- Leth-Larsen, R.; Garred, P.; Jensenius, H.; Meschi, J.; Hartshorn, K.; Madsen, J.; Tornoe, I.; Madsen, H.O.; Sørensen, G.; Crouch, E.; et al. A Common Polymorphism in the SFTPD Gene Influences Assembly, Function, and Concentration of Surfactant Protein D. J. Immunol. 2005, 174, 1532–1538. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Arimura, Y.; Goto, A.; Hosokawa, M.; Nagaishi, K.; Yamashita, K.; Yamamoto, H.; Sonoda, T.; Nomura, M.; Motoya, S.; et al. Genetic Variants in Surfactant, Pulmonary-Associated Protein D (SFTPD) and Japanese Susceptibility to Ulcerative Colitis. Inflamm. Bowel Dis. 2009, 15, 918–925. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.; John, G.; Hegarty, J.P.; Berg, A.; Yu, W.; Wang, Y.; Kelly, A.A.; Peterson, B.Z.; Poritz, L.S.; Floros, J.; et al. Genetic Variants and Monoallelic Expression of Surfactant Protein-D in Inflammatory Bowel Disease: Monoallelic Expression of SP-D in IBD. Annals Hum. Genet. 2011, 75, 559–568. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Jamka, M.; Kaczmarek, N.; Mądry, E.; Krzyżanowska-Jankowska, P.; Bajerska, J.; Kręgielska-Narożna, M.; Bogdański, P.; Walkowiak, J. Metabolic Health in Obese Subjects-Is There a Link to Lactoferrin and Lactoferrin Receptor-Related Gene Polymorphisms? Nutrients 2020, 12, 2843. [Google Scholar] [CrossRef]
- Palczewska, I.; Niedzwiedzka, Z. Somatic development indices in children and youth of Warsaw. Med. Wieku Rozwoj. 2001, 5, 18–118. [Google Scholar]
- Assa, A.; Rinawi, F.; Shamir, R. The Long-Term Predictive Properties of the Paris Classification in Paediatric Inflammatory Bowel Disease Patients. J. Crohns Colitis 2018, 12, 39–47. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Xu, W.; Wang, H.; Cao, M.; Li, M.; Zhao, J.; Hu, Y.; Wang, Y.; Li, S.; Xie, Y.; et al. Inhibition of CREB-Mediated ZO-1 and Activation of NF-ΚB-Induced IL-6 by Colonic Epithelial MCT4 Destroys Intestinal Barrier Function. Cell Prolif. 2019, 52, e12673. [Google Scholar] [CrossRef]
- Hughes-Fulford, M.; Sugano, E.; Schopper, T.; Li, C.-F.; Boonyaratanakornkit, J.B.; Cogoli, A. Early Immune Response and Regulation of IL-2 Receptor Subunits. Cell. Signal. 2005, 17, 1111–1124. [Google Scholar] [CrossRef]
- Boonstra, A.; Rajsbaum, R.; Holman, M.; Marques, R.; Asselin-Paturel, C.; Pereira, J.P.; Bates, E.E.M.; Akira, S.; Vieira, P.; Liu, Y.-J.; et al. Macrophages and Myeloid Dendritic Cells, but Not Plasmacytoid Dendritic Cells, Produce IL-10 in Response to MyD88- and TRIF-Dependent TLR Signals, and TLR-Independent Signals. J. Immunol. 2006, 177, 7551–7558. [Google Scholar] [CrossRef] [PubMed]
- Parry, G.C.; Mackman, N. Role of Cyclic AMP Response Element-Binding Protein in Cyclic AMP Inhibition of NF-KappaB-Mediated Transcription. J. Immunol. 1997, 159, 5450–5456. [Google Scholar]
- Park, J.M.; Greten, F.R.; Wong, A.; Westrick, R.J.; Arthur, J.S.C.; Otsu, K.; Hoffmann, A.; Montminy, M.; Karin, M. Signaling Pathways and Genes That Inhibit Pathogen-Induced Macrophage Apoptosis--CREB and NF-KappaB as Key Regulators. Immunity 2005, 23, 319–329. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Rincon, M.; Flavell, R.A.; Aune, T.M. Defective Th Function Induced by a Dominant-Negative CAMP Response Element Binding Protein Mutation Is Reversed by Bcl-2. J. Immunol. 2000, 165, 1762–1770. [Google Scholar] [CrossRef]
- Pasquinelli, V.; Townsend, J.C.; Jurado, J.O.; Alvarez, I.B.; Quiroga, M.F.; Barnes, P.F.; Samten, B.; García, V.E. IFN-Gamma Production during Active Tuberculosis Is Regulated by Mechanisms That Involve IL-17, SLAM, and CREB. J. Infect. Dis. 2009, 199, 661–665. [Google Scholar] [CrossRef]
- Miyata, Y.; Fukuhara, A.; Otsuki, M.; Shimomura, I. Expression of Activating Transcription Factor 2 in Inflammatory Macrophages in Obese Adipose Tissue. Obesity (Silver Spring) 2013, 21, 731–736. [Google Scholar] [CrossRef]
- Reimold, A.M.; Kim, J.; Finberg, R.; Glimcher, L.H. Decreased Immediate Inflammatory Gene Induction in Activating Transcription Factor-2 Mutant Mice. Int. Immunol. 2001, 13, 241–248. [Google Scholar] [CrossRef]
- Iskandar, H.N.; Ciorba, M.A. Biomarkers in Inflammatory Bowel Disease: Current Practices and Recent Advances. Transl. Res. 2012, 159, 313–325. [Google Scholar] [CrossRef] [PubMed]
- Biasci, D.; Lee, J.C.; Noor, N.M.; Pombal, D.R.; Hou, M.; Lewis, N.; Ahmad, T.; Hart, A.; Parkes, M.; McKinney, E.F.; et al. A Blood Based Prognostic Biomarker in IBD. Gut 2019, 68, 1386–1395. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| UC n = 192 | CD n = 214 | p Value * | OR (95% CI) | |
|---|---|---|---|---|
| rs2981804 | ||||
| AA | 58 (30.2) | 70 (32.7) | 0.595 | 0.89 (0.59–1.36) |
| AG | 100 (52.1) | 98 (45.8) | 0.233 | 1.29 (0.87–1.90) |
| GG | 34 (17.7) | 46 (21.5) | 0.382 | 0.79 (0.48–1.29) |
| rs2981745 | ||||
| TT | 14 (7.3) | 22 (10.3) | 0.342 | 0.69 (0.34–1.39) |
| CT | 87 (45.3) | 87 (40.7) | 0.366 | 1.22 (0.82–1.81) |
| CC | 91 (47.4) | 105 (49.0) | 0.766 | 0.94 (0.63–1.38) |
| rs2243639 | ||||
| TT | 28 (14.6) | 34 (15.9) | 0.783 | 0.90 (0.53–1.56) |
| CT | 98 (51.0) | 105 (49.1) | 0.766 | 1.08 (0.73–1.60) |
| CC | 66 (34.4) | 75 (35.0) | 0.917 | 0.97 (0.65–1.46) |
| Variables Median (IQR) or n (%) | AA | AG | GG | p Value * |
|---|---|---|---|---|
| Selected biochemical parameters | ||||
| CRP at diagnosis (mg/L) | 2.5 (0.6–9.3) | 1.7 (0.5–9.6) | 5.6 (0.7–16.2) | 0.173 |
| CRP at worst flare (mg/L) | 2.7 (0.6–14.2) | 2.4 (0.5–11.2) | 3.9 (1.0–27.8) | 0.445 |
| Albumin level at diagnosis (g/dL) | 4.0 (3.5–4.3) | 4.2 (4.0–4.5) | 4.0 (3.4–4.4) | 0.0091 |
| Albumin level at worst flare (g/dL) | 4.0 (3.7–4.4) | 4.2 (3.9–4.5) | 4.3 (3.8–4.4) | 0.307 |
| Disease activity scales | ||||
| PUCAI at diagnosis | 50 (40–65) | 45 (25–60) | 41 (28–53) | 0.125 |
| PUCAI at worst flare | 55 (40–65) | 50 (33–65) | 53 (35–65) | 0.707 |
| Treatment | ||||
| Systemic steroids | 45 (78) | 72 (72) | 21 (62) | 0.265 |
| Number of courses of steroid treatment | 1 (1–2) | 1 (1–2) | 1 (0–2) | 0.631 |
| Immunosuppressive treatment | 36 (63) | 60 (60) | 16 (47) | 0.296 |
| Number of immunosuppressants | 1 (0–1) | 1 (0–1) | 0 (0–1) | 0.449 |
| Time-to-first dose of immunosuppressive treatment (months) | 4.0 (1.0–10.0) | 3.0 (0.3–10.0) | 1.9 (0.0–10.1) | 0.682 |
| Age at first intake of immunosuppressive treatment (years) | 10.2 (6.7–14.0) | 11.6 (7.8–14.7) | 13.2 (8.9–14.7) | 0.460 |
| Biological therapy | 21 (36) | 23 (23) | 5 (15) | 0.052 |
| Time-to-first dose of biological treatment (months) | 18.4 (10.0–28.1) | 12.5 (7.2–25.1) | 12.2 (4.0–29.0) | 0.570 |
| Age at first biological treatment | 11.7 (6.6–15.3) | 13.1 (7.9–15.6) | 10.7 (5.3–11.8) | 0.271 |
| Operative treatment | 1 (2) | 3 (3) | 0 (0) | 0.557 |
| Age at first surgery (years) | 15.6 | 10.0 (6.8–14.8) | - | 1.000 |
| Time-to-first surgery (months) | 0.9 | 22.7 (10.9–33.0) | - | 1.000 |
| Hospitalisations for relapse (per 1 year of the disease) | 0.6 (0.3–0.9) | 0.6 (0.3–1.3) | 1.0 (0.4–1.6) | 0.335 |
| Days of hospitalisation for relapse (per 1 year of the disease) | 4.0 (1.5–7.1) | 5.0 (1.3–9.7) | 5.4 (2.5–10.3) | 0.721 |
| Relapses from diagnosis (per 1 year of the disease) | 0.5 (0.3–0.7) | 0.6 (0.3–1.3) | 1.0 (0.3–1.8) | 0.378 |
| Severe relapses from diagnosis (per 1 year of the disease) | 0.1 (0.0–0.4) | 0.0 (0.0–0.6) | 0.3 (0.0–0.4) | 0.900 |
| Concomitant diseases | 24 (41) | 37 (37) | 9 (26) | 0.353 |
| Extraintestinal manifestations | 11 (19) | 23 (23) | 3 (9) | 0.194 |
| Variables Median (IQR) or n (%) | AA | AG | GG | p Value * |
|---|---|---|---|---|
| Selected biochemical parameters | ||||
| CRP at diagnosis (mg/L) | 14.4 (2.4–37.8) | 13.2 (2.1–25.0) | 9.4 (1.9–24.9) | 0.395 |
| CRP at worst flare (mg/L) | 14.7 (3.0–34.0) | 12.7 (2.6–30.0) | 14.2 (3.1–39.1) | 0.829 |
| Albumin level at diagnosis (g/dL) | 3.8 (3.4–4.2) | 3.9 (3.5–4.3) | 4.0 (3.7–4.5) | 0.271 |
| Albumin level at worst flare (g/dL) | 3.9 (3.5–4.2) | 4.0 (3.5–4.3) | 3.8 (3.6–4.1) | 0.638 |
| Disease activity scales | ||||
| PCDAI at diagnosis | 30 (23–50) | 35 (23–50) | 30 (23–43) | 0.677 |
| PCDAI at worst flare | 41 (30–53) | 40 (25–53) | 48 (30–53) | 0.629 |
| Treatment | ||||
| Systemic steroids | 35 (50) | 52 (53) | 28 (61) | 0.509 |
| Number of courses of steroid treatment | 1 (0–2) | 1 (0–1) | 1 (1–2) | 0.523 |
| Immunosuppressive treatment | 55 (79) | 76 (78) | 37 (80) | 0.926 |
| Number of immunosuppressants | 1 (1–1) | 1 (1–1) | 1 (1–1) | 0.636 |
| Time-to-first dose of immunosuppressive treatment (months) | 2.5 (0.0–12.0) | 1.0 (0.0–6.8) | 0.7 (0.0–15.0) | 0.377 |
| Age at first intake of immunosuppressive treatment (years) | 13.1 (11.3–14.9) | 12.2 (9.4–14.9) | 13.0 (9.2–13.9) | 0.135 |
| Biological therapy | 40 (57) | 45 (46) | 22 (48) | 0.338 |
| Number of biological agents | 1 (0–1) | 0 (0–1) | 0 (0–1) | 0.538 |
| Time-to-first dose of biological treatment (months) | 11.4 (4.0–21.0) | 13.0 (6.0–28.3) | 25.1 (12.0–44.0) | 0.074 |
| Age at first biological treatment | 13.5 (12.5–14.8) | 13.9 (10.8–15.4) | 14.1 (11.3–15.1) | 0.969 |
| Operative treatment | 12 (17) | 12 (12) | 5 (11) | 0.558 |
| Age at first surgery (years) | 14.5 (13.1–16.3) | 13.8 (11.7–15.6) | 14.9 (14.5–15.7) | 0.675 |
| Time-to-first surgery (months) | 8.0 (0.0–41.1) | 35.0 (23.1–56.8) | 11.5 (3.0–27.0) | 0.259 |
| Hospitalisations (if duration ≥1 years) | ||||
| Hospitalisations for relapse (per 1 year of the disease) | 0.4 (0.3–0.9) | 0.5 (0.2–0.9) | 0.5 (0.3–0.9) | 0.925 |
| Days of hospitalisation for relapse (per 1 year of the disease) | 3.7 (1.6–8.5) | 4.5 (1.2–7.2) | 3.5 (1.0–6.5) | 0.949 |
| Relapses from diagnosis (per 1 year of the disease) | 0.5 (0.3–0.9) | 0.5 (0.2–0.9) | 0.4 (0.2–1.1) | 0.916 |
| Severe relapses from diagnosis (per 1 year of the disease) | 0.3 (0.0–0.5) | 0.2 (0.0–0.5) | 0.0 (0.0–0.3) | 0.116 |
| Concomitant diseases | 28 (40) | 28 (29) | 8 (17) | 0.0311 |
| Extraintestinal manifestations | 18 (26) | 28 (29) | 7 (15) | 0.218 |
| Variables Median (IQR) or n (%) | CC | CT | TT | p Value * |
|---|---|---|---|---|
| Selected biochemical parameters | ||||
| CRP at diagnosis (mg/L) | 13.6 (2.6–31.1) | 13.0 (2.0–28.1) | 9.8 (2.0–18.3) | 0.518 |
| CRP at worst flare (mg/L) | 11.9 (2.1–32.0) | 14.2 (3.2–39.4) | 14.7 (7.1–30.2) | 0.776 |
| Albumin level at diagnosis (g/dL) | 3.8 (3.5–4.2) | 3.9 (3.5–4.3) | 4.0 (3.7–4.4) | 0.675 |
| Albumin level at worst flare (g/dL) | 3.9 (3.5–4.3) | 3.9 (3.5–4.2) | 3.9 (3.5–4.2) | 0.949 |
| Disease activity scales | ||||
| PCDAI at diagnosis | 30 (23–48) | 35 (25–50) | 25 (25–39) | 0.262 |
| PCDAI at worst flare | 40 (30–53) | 43 (30–53) | 35 (28–53) | 0.801 |
| Treatment | ||||
| Systemic steroids | 55 (52) | 45 (52) | 15 (68) | 0.358 |
| Number of courses of steroid treatment | 1 (0–2) | 1 (0–2) | 1 (1–2) | 0.875 |
| Immunosuppressive treatment | 81 (77) | 69 (79) | 18 (82) | 0.865 |
| Number of immunosuppressants | 1 (1–1) | 1 (1–1) | 1 (1–1) | 0.634 |
| Time-to-first dose of immunosuppressive treatment (months) | 1.6 (0.0–7.0) | 2.0 (0.0–7.8) | 0.5 (0.0–17.3) | 0.981 |
| Age at first intake of immunosuppressive treatment (years) | 13.1 (10.7–14.7) | 12.3 (9.2–14.9) | 13.0 (9.7–14.2) | 0.315 |
| Biological therapy | 57 (54) | 38 (44) | 12 (55) | 0.311 |
| Number of biological agents | 1 (0–1) | 0 (0–1) | 1 (0–1) | 0.379 |
| Time-to-first dose of biological treatment (months) | 11.0 (4.1–20.8) | 20.4 (9.6–35.0) | 20.3 (7.6–40.2) | 0.055 |
| Age at first biological treatment | 13.6 (12.5–15.1) | 13.8 (10.9–15.5) | 14.2 (10.8–14.8) | 0.810 |
| Operative treatment | 18 (17) | 7 (8) | 4 (18) | 0.150 |
| Age at first surgery (years) | 14.0 (11.6–15.6) | 15.2 (14.0–15.7) | 14.7 (14.2–15.3) | 0.475 |
| Time-to-first surgery (months) | 10.0 (2.2–41.6) | 29.1 (27.0–56.8) | 7.3 (2.3–15.5) | 0.154 |
| Hospitalisations (if duration ≥1 years) | ||||
| Hospitalisations for relapse (per 1 year of the disease) | 0.5 (0.3–0.9) | 0.5 (0.2–0.7) | 0.5 (0.3–0.9) | 0.588 |
| Days of hospitalisation for relapse (per 1 year of the disease) | 4.6 (1.8–7.6) | 3.5 (1.2–7.1) | 2.7 (0.9–6.5) | 0.504 |
| Relapses from diagnosis (per 1 year of the disease) | 0.6 (0.3–0.9) | 0.4 (0.2–0.8) | 0.4 (0.0–1.1) | 0.208 |
| Severe relapses from diagnosis (per 1 year of the disease) | 0.3 (0.0–0.6) | 0.2 (0.0–0.4) | 0.0 (0.0–0.2) | 0.0201 |
| Concomitant diseases | 36 (34) | 24 (28) | 4 (18) | 0.269 |
| Extraintestinal manifestations | 30 (29) | 20 (23) | 3 (14) | 0.297 |
| Variables Median (IQR) or n (%) | CC | CT | TT | p Value * |
|---|---|---|---|---|
| Selected biochemical parameters | ||||
| CRP at diagnosis (mg/L) | 2.2 (0.6–10.2) | 1.9 (0.4–7.8) | 9.2 (3.7–21.5) | 0.057 |
| CRP at worst flare (mg/L) | 2.6 (0.6–13.3) | 2.2 (0.5–7.1) | 7.9 (3.6–28.4) | 0.120 |
| Albumin level at diagnosis (g/dL) | 4.1 (3.6–4.4) | 4.2 (3.8–4.5) | 4.0 (3.5–4.4) | 0.504 |
| Albumin level at worst flare (g/dL) | 4.1 (3.7–4.4) | 4.2 (3.9–4.5) | 4.2 (3.8–4.4) | 0.309 |
| Disease activity scales | ||||
| PUCAI at diagnosis | 50 (30–60) | 45 (25–60) | 40 (33–50) | 0.373 |
| PUCAI at worst flare | 55 (36–65) | 50 (35–65) | 50 (43–65) | 0.956 |
| Treatment | ||||
| Systemic steroids | 71 (78) | 60 (69) | 7 (50) | 0.069 |
| Number of courses of steroid treatment | 1 (1–2) | 1 (0–2) | 1 (0–2) | 0.250 |
| Immunosuppressive treatment | 56 (62) | 52 (60) | 4 (29) | 0.058 |
| Number of immunosuppressants | 1 (0–1) | 1 (0–1) | 0 (0–1) | 0.084 |
| Time-to-first dose of immunosuppressive treatment (months) | 4.1 (1.0–10.0) | 1.5 (0.0–6.1) | 20.5 (8.0–32.4) | 0.0451 |
| Age at first intake of immunosuppressive treatment (years) | 11.5 (6.8–14.4) | 11.9 (9.2–14.6) | 13.7 (12.0–14.6) | 0.632 |
| Biological therapy | 28 (31) | 20 (23) | 1 (7) | 0.130 |
| Time-to-first dose of biological treatment (months) | 17.0 (10.0–26.9) | 12.0 (5.6–23.5) | 29.0 | 0.247 |
| Age at first biological treatment | 13.1 (6.6–15.3) | 11.9 (10.0–14.7) | 10.7 | 0.632 |
| Operative treatment | 2 (2) | 2 (2) | 0 (0) | 0.851 |
| Age at first surgery (years) | 11.2 (9.0–13.4) | 10.4 (9.6–14.8) | 1.000 | |
| Time-to-first surgery (months) | 8.8 (4.8–12.7) | 28.7 (16.8–33.0) | 0.248 | |
| Hospitalisations (if duration ≥1 years) | ||||
| Hospitalisations for relapse (per 1 year of the disease) | 0.6 (0.3–1.2) | 0.7 (0.3–1.4) | 0.7 (0.5–1.0) | 0.972 |
| Days of hospitalisation for relapse (per 1 year of the disease) | 5.2 (1.6–9.5) | 4.6 (1.7–9.0) | 5.4 (3.1–6.5) | 0.897 |
| Relapses from diagnosis (per 1 year of the disease) | 0.6 (0.3–1.1) | 0.6 (0.3–1.3) | 0.6 (0.3–1.4) | 0.944 |
| Severe relapses from diagnosis (per 1 year of the disease) | 0.1 (0.0–0.4) | 0.0 (0.0–0.6) | 0.0 (0.0–0.2) | 0.709 |
| Concomitant diseases | 38 (42) | 29 (33) | 3 (21) | 0.242 |
| Extraintestinal manifestations | 18 (20) | 18 (21) | 1 (7) | 0.484 |
| Variables Median (IQR) or n (%) | CC | CT | TT | p Value * |
|---|---|---|---|---|
| Selected biochemical parameters | ||||
| CRP at diagnosis (mg/L) | 2.2 (0.5–9.1) | 2.1 (0.4–10.5) | 2.9 (1.0–18.0) | 0.672 |
| CRP at worst flare (mg/L) | 2.1 (0.6–7.5) | 2.9 (0.6–14.7) | 2.7 (1.0–19.0) | 0.716 |
| Albumin level at diagnosis (g/dL) | 4.2 (3.8–4.5) | 4.1 (3.5–4.4) | 4.1 (3.8–4.3) | 0.247 |
| Albumin level at worst flare (g/dL) | 4.2 (3.8–4.4) | 4.1 (3.7–4.5) | 4.1 (3.9–4.3) | 0.594 |
| Disease activity scales | ||||
| PUCAI at diagnosis | 45 (30–55) | 45 (25–58) | 45 (28–68) | 0.813 |
| PUCAI at worst flare | 50 (35–65) | 53 (35–65) | 60 (38–65) | 0.846 |
| Treatment | ||||
| Systemic steroids | 41 (62) | 77 (79) | 20 (71) | 0.072 |
| Number of courses of steroid treatment | 1 (0–2) | 1 (1–3) | 1 (1–3) | 0.0371 |
| Immunosuppressive treatment | 35 (53) | 59 (61) | 18 (64) | 0.495 |
| Number of immunosuppressants | 1 (0–1) | 1 (0–1) | 1 (0–1) | 0.706 |
| Time-to-first dose of immunosuppressive treatment (months) | 2.1 (0.0–8.4) | 3.5 (0.1–10.1) | 5.2 (1.8–11.0) | 0.392 |
| Age at first intake of immunosuppressive treatment (years) | 13.0 (7.9–15.6) | 11.3 (7.5–13.7) | 12.8 (9.1–14.9) | 0.334 |
| Biological therapy | 18 (27) | 23 (23) | 8 (29) | 0.795 |
| Number of biological agents | 0 (0–1) | 0 (0–0) | 0 (0–1) | 0.736 |
| Time-to-first dose of biological treatment (months) | 13.5 (4.5–20.8) | 20.1 (10.2–29.1) | 12.1 (6.0–23.3) | 0.258 |
| Age at first biological treatment | 12.5 (8.7–15.3) | 11.0 (6.9–14.5) | 12.1 (7.1–15.6) | 0.710 |
| Operative treatment | 3 (5) | 0 (0) | 1 (4) | 0.115 |
| Age at first surgery (years) | 12.6 (9.8–15.4) | 6.8 | 0.317 | |
| Time-to-first surgery (months) | 16.8 (4.0–30.8) | 16.7 | 1.000 | |
| Hospitalisations (if duration ≥1 years) | ||||
| Hospitalisations for relapse (per 1 year of the disease) | 0.7 (0.2–1.4) | 0.6 (0.3–1.1) | 0.9 (0.4–1.5) | 0.854 |
| Days of hospitalisation for relapse (per 1 year of the disease) | 3.6 (1.2–9.2) | 5.2 (2.3–7.7) | 7.7 (2.7–13.8) | 0.492 |
| Relapses from diagnosis (per 1 year of the disease) | 0.6 (0.2–1.1) | 0.6 (0.3–1.1) | 0.9 (0.3–1.3) | 0.925 |
| Severe relapses from diagnosis (per 1 year of the disease) | 0.1 (0.0–0.4) | 0.1 (0.0–0.5) | 0.1 (0.0–0.5) | 0.889 |
| Concomitant diseases | 20 (30) | 41 (42) | 9 (32) | 0.283 |
| Extraintestinal manifestations | 16 (24) | 14 (14) | 7 (25) | 0.201 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Glapa-Nowak, A.; Szczepanik, M.; Banaszkiewicz, A.; Iwańczak, B.; Kwiecień, J.; Szaflarska-Popławska, A.; Grzybowska-Chlebowczyk, U.; Osiecki, M.; Kierkuś, J.; Banasiuk, M.; et al. Genetic Variants of DMBT1 and SFTPD and Disease Severity in Paediatric Inflammatory Bowel Disease—A Polish Population-Based Study. Children 2021, 8, 946. https://doi.org/10.3390/children8110946
Glapa-Nowak A, Szczepanik M, Banaszkiewicz A, Iwańczak B, Kwiecień J, Szaflarska-Popławska A, Grzybowska-Chlebowczyk U, Osiecki M, Kierkuś J, Banasiuk M, et al. Genetic Variants of DMBT1 and SFTPD and Disease Severity in Paediatric Inflammatory Bowel Disease—A Polish Population-Based Study. Children. 2021; 8(11):946. https://doi.org/10.3390/children8110946
Chicago/Turabian StyleGlapa-Nowak, Aleksandra, Mariusz Szczepanik, Aleksandra Banaszkiewicz, Barbara Iwańczak, Jarosław Kwiecień, Anna Szaflarska-Popławska, Urszula Grzybowska-Chlebowczyk, Marcin Osiecki, Jarosław Kierkuś, Marcin Banasiuk, and et al. 2021. "Genetic Variants of DMBT1 and SFTPD and Disease Severity in Paediatric Inflammatory Bowel Disease—A Polish Population-Based Study" Children 8, no. 11: 946. https://doi.org/10.3390/children8110946
APA StyleGlapa-Nowak, A., Szczepanik, M., Banaszkiewicz, A., Iwańczak, B., Kwiecień, J., Szaflarska-Popławska, A., Grzybowska-Chlebowczyk, U., Osiecki, M., Kierkuś, J., Banasiuk, M., Banasiewicz, T., Madsen, J., & Walkowiak, J. (2021). Genetic Variants of DMBT1 and SFTPD and Disease Severity in Paediatric Inflammatory Bowel Disease—A Polish Population-Based Study. Children, 8(11), 946. https://doi.org/10.3390/children8110946